

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disease characterized by the degeneration of motor neurons. Currently, there is no effective therapy for ALS. Stem cell transplantation is a potential therapeutic strategy for ALS, and the reprogramming of adult somatic cells into induced pluripotent stem cells (iPSCs) represents a novel cell source. In this study, we isolated a specific neural stem cell (NSC) population from human iPSCs based on high aldehyde dehydrogenase activity, low side scatter and integrin VLA4 positivity. We assessed the therapeutic effects of these NSCs on the phenotype of ALS mice after intrathecal or intravenous injections. Transplanted NSCs migrated and engrafted into the central nervous system via both routes of injection. Compared with control ALS, treated ALS mice exhibited improved neuromuscular function and motor unit pathology and significantly increased life span, in particular with the systemic administration of NSCs (15%). These positive effects are linked to multiple mechanisms, including production of neurotrophic factors and reduction of micro- and macrogliosis. NSCs induced a decrease in astrocyte number through the activation of the vanilloid receptor TRPV1. We conclude that minimally invasive injections of iPSC-derived NSCs can exert a therapeutic effect in ALS. This study contributes to advancements in iPSC-mediated approaches for treating ALS and other neurodegenerative diseases.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that is characterized by clinical and pathological signs of upper and lower motor neuron degeneration, progressive paralysis and precocious death (1). Currently, there are no effective treatments. The majority of cases are sporadic, and ∼10% are familial (1). Of the familial cases, ∼20% are linked to mutations in the superoxide dismutase 1 (SOD1) gene (2). Although several other causative genes have been identified in recent years, with C9ORF72 and TARDBP among the most common, almost 50% of familial ALS cases have unidentified genetic aetiology, although this proportion may vary according to different genetic backgrounds (1). The aethiopathogenesis of sporadic ALS is largely unknown (3), which hinders the development of clinically meaningful treatment (1,4,5).
Expression of mutant SOD1 in rodents causes progressive degeneration of motor neurons, leading to progressive paralysis that resembles the manifestations of human ALS (6,7). Although the molecular mechanisms leading to progressive neuronal degeneration are likely multi-factorial, increasing evidence implies a role for non-autonomous cell death as a key event in the disease pathology (3). In particular, environmental non-neuronal cells such as astrocytes and microglia are involved in the death of motor neurons (8).
Cell transplantation therapy could be a promising therapeutic strategy for a number of disease processes. There is accumulating experimental evidence of non-cell-autonomous contributions of cells other than motor neurons to ALS pathology. In addition, the disease is characterized by a complex interplay of several pathogenic events, such as protein aggregation, glutamate toxicity, RNA dysfunction and inflammatory events. Stem cell transplantation can be therapeutic via multiple mechanisms, including replacement of micro-environmental cells such as astrocytes and neuronal precursors. Furthermore, stem cell therapy can reduce inflammation and neuropathological hallmarks such as neuronal tangles/aggregates, protect motor neurons and neuronal circuitry and ultimately replace degenerated cells (9).
Our group and others have demonstrated these positive effects of stem cell transplantation (neural and non-neural cells) (9–15). In 2009, the US Food and Drug Administration approved the first phase I safety trial of direct intraspinal transplantation of neural stem cells (NSCs) into patients with ALS; the study is still in progress (9). Recently, transplanted NSCs were demonstrated to effectively slow the disease and prolong survival in ALS mice, and in some cases (25% of treated mice), the phenotype was completely rescued (survival increased to over a full year versus 4 months in untreated animals) (10). These data represent an unprecedented therapeutic success in motor neuron disease models compared with other pharmacological/molecular treatments. However, 40% of mice injected with NSCs show a very mild increase in survival. The reason for this variability is unknown and may be related to the type of cell transplanted and our incomplete understanding of the key variables important for therapeutic success. Human NSCs are derived from primary central nervous system (CNS) tissue (fetal brain), which is a limited source of cells. Furthermore, multiple direct injections of stem cells into the spinal cord will require major invasive surgery if this strategy is translated to humans.
The method of cell delivery represents a major hurdle in developing a cell-mediated approach. The cells must be distributed along the CNS, targeting both lower and upper motor neurons, using a non-invasive method that leads to sufficient engraftment. The method presented here progresses beyond the current limits of cell therapy, which is based on direct injection into the spinal cord. We utilize a cell population that can efficiently migrate into the CNS from the cerebrospinal fluid (CSF) or from the blood after systemic injection. We previously described that the selection of a specific subset of NSCs can provide a major advantage in therapeutic efficacy with respect to mixed NSC/neuronal precursor populations (11–13). We previously demonstrated positive therapeutic effects of a subset of self-renewing, multipotent rodent NSCs selected for their aldehyde dehydrogenase (ALDH) activity. When intrathecally transplanted into the SMA and SMARD1 mouse models of motor neuron disease, the cells migrate into the spinal cord and ameliorate the disease phenotype (11,13).
In this work, we sought to test the therapeutic potential of human NSCs that were selected for their high ALDH activity, low orthogonal light scattering (ALDHhiSSClo) and expression of integrin VLA4. The presence of VLA4 in NSCs allows them to cross the blood-brain barrier (BBB), particularly in the presence of inflammation or a weakened BBB, as in ALS animal models and human patients (16,17).
In addition, we wanted to explore minimally invasive administration protocols, such as intrathecal and systemic intravenous transplantation, in an ALS mouse model. This strategy allowed us to repeatedly deliver the cells, achieving a widespread distribution of a significant amount of cells in the CNS. We hypothesized that a progressive escalation in cell dosage might lead to a progressively enhanced therapeutic effect. Thus, we initiated a regimen in which cell dosage was amplified by gradually increasing the number of injections over time. We derived the NSCs from non-virally generated induced pluripotent stem cells (IPSCs) that were reprogrammed from skin fibroblasts of healthy subjects. This cell source is attractive due to availability, similar pluripotency as embryonic stem cells (ESCs) and no associated ethical controversy. With this strategy, we achieved a significant improvement in the disease phenotype, even when treating early-mid symptomatic animals.
RESULTS
Isolation and characterization of an ALDHhiSSCloVLA4+ NSC population from human iPSCs
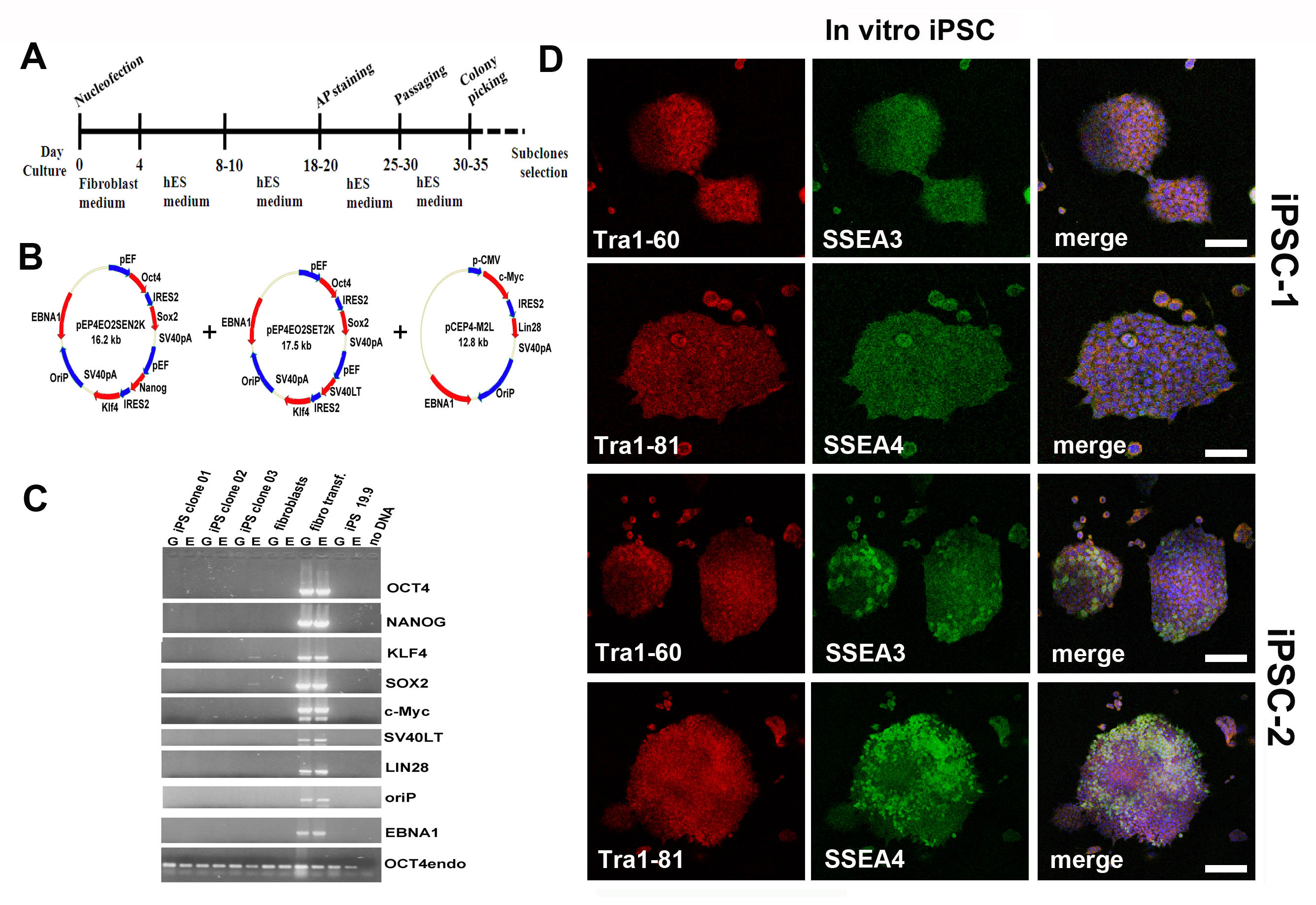
We used an iPSC line generated in our lab with a non-viral, non-integrating method and fully characterized its pluripotent features (Supplementary Material, Fig. S1). We differentiated iPSCs into NSCs with a multistage differentiation procedure described previously for human ESCs and iPSCs (15,18) (Fig. 1A). This protocol generates >90% PAX6-positive neuroepithelial cells. Then, we used FACS analysis to isolate a specific cell fraction based on ALDHhiSSClo properties, as previously described for murine NSCs (Fig. 1B) (11,13). This fraction was further selected for the expression of VLA4+ (Fig. 1B and C). ALDHhiSSCloVLA4+ cells are self-renewing and multipotent, and they also express neural precursor markers such as SOX1, SOX2 and nestin (Fig. 1D). Moreover, these cells can differentiate into the three major neuroectodermal lineages (Fig. 1D–G); they differentiate into motor neurons when grown in the presence of retinoic acid (RA) and sonic hedgehog (SHH) (15) (Supplementary Material, Fig. S2).
Figure 1.

Differentiation of iPSCs into NSCs and selection of the ALDHhiSSCloVLA4+ fraction. (A) Scheme of the differentiation protocol of NSCs from iPSCs. Initially, the iPSCs are positive for OCT4. After detaching from the plate, the iPSCs form aggregates that are cultured for 7 days in neural medium. The differentiated cells show features of neuroepithelial (NE) rosettes. The primitive NE cells were positive for PAX6. These cells were then selected by FACS for their ALDHhiSSClo and VLA4 positivity and expanded in vitro. The cells were cultured with RA and SHH and differentiated into OLIG2-expressing motor neurons. They could be further matured into their neuronal phenotype in the presence of neurotrophic factors and reduced concentrations of SHH and RA. (B) Representative flow cytometric analysis of ALDH activity and VLA4+ positivity on iPSC-derived NSCs. NSCs were selected according to forward and side scatter properties. Cells incubated with Aldefluor substrate and the specific inhibitor of ALDH (DEAB) were used to establish the baseline fluorescence and to define the ALDHhi region. After sorting, this cell population is enriched as shown in the right panel. The ALDHhiSSClo fraction was further selected by FACS for VLA4 expression (left panel) and seeded in culture. Scale bar: 70 µm. (C) iPSC cells positive for VLA4 (red; nuclei stained with DAPI, blue signal). (D) Sorted ALDHhiSSCloVLA4+ cells grown at clonal density in NEP medium form epithelial-like clones positive for NSC markers such as SOX1, SOX2 and nestin. Scale bar: 100 µm. After in vitro differentiation, ALDHhiSSClo cells could give rise to cells of the three neuroectodermal lineages including GFAP-positive astrocytes (red), O4-positive oligodendrocytes (red) and motor neurons (ChAT red, TuJ1, green). Nuclei are counterstained with DAPI (blue signal). Scale bar: GFAP, 100 µm; O4, 50 µm; and TuJ1 and ChAT, 100 µm. (e) Quantification of cells positive for NSC markers. (F) Quantification of cells showing positive staining for astrocyte and oligodendrocyte markers after appropriate differentiation. (G) Quantification of cells that are double-positive for motor neuron markers TuJ1 and ChAT.
Both intrathecally and systemically transplanted ALDHhiSSCloVLA4+ NSCs engraft in the spinal cord of SOD1G93A mice
To assess the therapeutic potential of ALDHhiSSCloVLA4+ NSCs, we evaluated whether the NSCs reached the CNS following intrathecal injection or systemic administration. For these experiments, we used the SOD1G93A mouse model of ALS (Figs 2 and 3). ALDHhiSSCloVLA4+ NSCs (1 × 106 cells) were delivered into the CSF by lumbar puncture in SOD1 mice at 90 days of age, with additional injections at 105 and 120 days of age (Fig. 2A). For the systemic injection, cells were delivered beginning at 90 days of age with weekly injections into the tail vein until the end stage (Fig. 3A). To trace the transplanted cells in the host spinal cord, we used ALDHhiSSCloVLA4+ cells expressing green fluorescent protein (GFP). To assess donor cell distribution and engraftment, the brain and spinal cord were collected at 140 days and at the end stage of the disease. Visualization of ALDHhiSSCloVLA4+ cells in the spinal cord revealed single cells in the host parenchyma and cells distributed in clusters (intrathecal model: Fig. 2B; systemic model: Fig. 3B). When ALDHhiSSCloVLA4+ donor cells were intrathecally or systemically administered, they engrafted in both grey and white matter regions of the mouse spinal cord. A quantitative analysis of donor-derived cells showed a significantly higher density of donor cells in the anterior horn, the site of active degeneration. In particular, the anterior horns in the cervical and lumbar regions had higher cell densities relative to the thoracic zone (intrathecal model: Fig. 2C; systemic model: Fig. 3C).
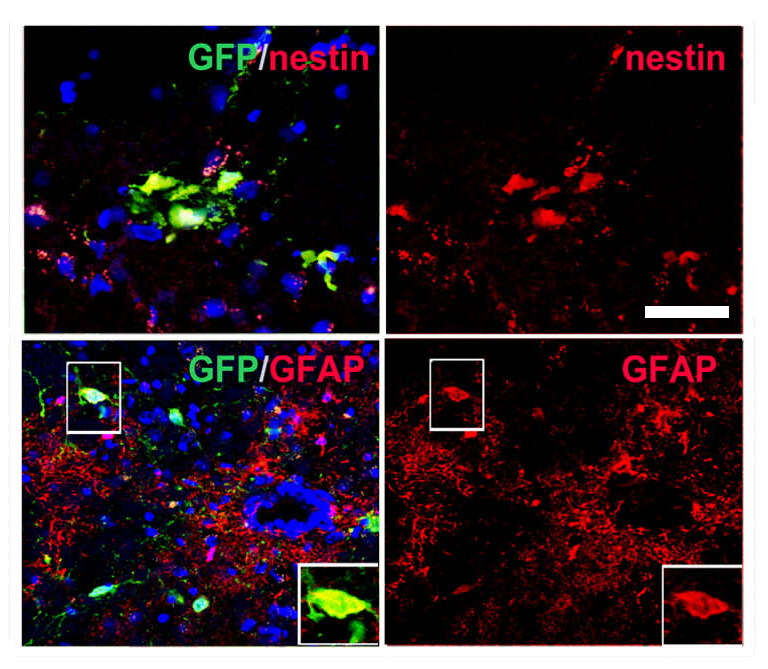
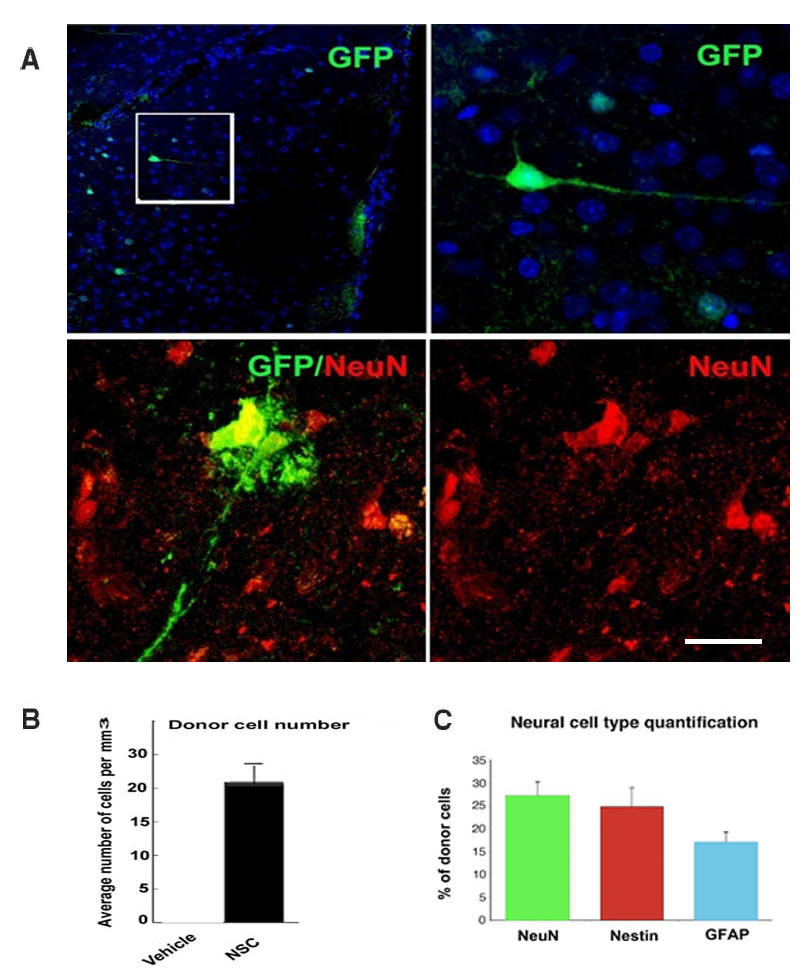
Figure 2.

iPSC-derived NSCs migrate and engraft into the spinal cords of SOD1G93A mice after intrathecal and systemic transplantation. (A) Experimental design: GFP-NSCs (1 × 106 cells) were delivered by intrathecal injection into SOD1G93A mice at 90 days of age, with two additional injections at 105 and 120 days. (B) Donor GFP+ cells were detected in the spinal cord, particularly in the anterior horns. (C) Quantification of GFP-donor cells in the cervical, thoracic and lumbar spinal cord. Error bars indicate the SD. (D) Quantification of the phenotype acquired by the donor cells revealed the presence of cells with an undifferentiated phenotype (nestin), neuronal (NeuN) phenotype and glial cells (GFAP). Error bars indicate the SD. (E) Representative images of cells acquiring a neuronal phenotype, as indicated by positivity for NeuN (red) and GFP (green). Nuclei are counterstained with DAPI (blue signal). Scale bars: (B) 150 µm and (E) 50 µm.
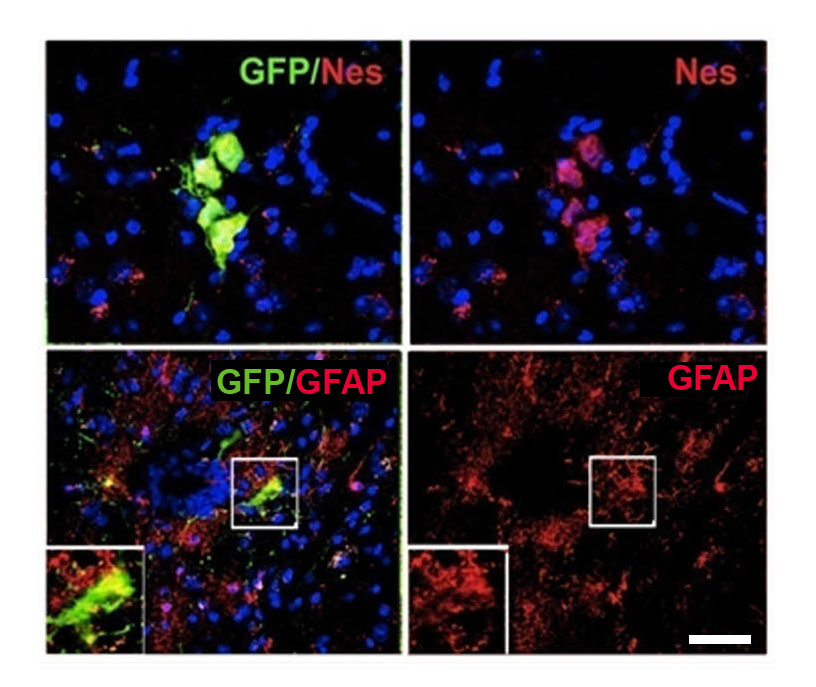
Figure 3.

iPSC-derived NSCs migrate and engraft into the spinal cords of SOD1G93A mice after intravenous transplantation. (A) Experimental design: GFP-NSC cells (1 × 106 cells) were delivered by weekly intravenous injection into SOD1G93A mice starting at 90 days of age. (B and C) Donor GFP+ cells were detected in the spinal cord, particularly in the anterior horns. (C) Quantification of GFP-donor cells in the cervical, thoracic and lumbar spinal cord. Error bars indicate the SD. (D) Quantification of the phenotype acquired by the donor cells revealed the presence of cells with an undifferentiated phenotype (nestin), a neuronal (NeuN) phenotype and a glial (GFAP) phenotype. Error bars indicate the SD. (E) Representative images of cells acquiring a neuronal phenotype that are positive for NeuN (red) and GFP (green). Scale bars: (B) 150 µm right, 120 µm left; (E) 50 µm upper panel, 75 µm lower panel.
Quantitative measurements of donor-derived cells in the spinal cord showed that an average of 6.49 ± 0.55 cells per section in the cervical and 6.7 ± 0.41 cells per section in the lumbar spinal cord were present following intrathecal administration (Fig. 2C). A significant proportion of donor cells maintained an undifferentiated phenotype, while the rest differentiated into neuronal and glia cells (Fig. 2D and E, Supplementary Material, Fig. S3). The vast majority of cells express GABA, supportive of a neuronal precursor phenotype or of an interneuronal phenotype. Few cells display a cholinergic phenotype, supported by their positivity for ChAT. Glial cells are almost all astrocytes, and only a minor proportion acquired an oligodendrocytic phenotype (<5%) as quantified in Figure 2. As expected for their ectodermal embryonic layer commitment, NSCs do not differentiated into microglia. Donor cells also reached the brain and were detected in the cortical and subcortical brain areas (Supplementary Material, Fig. S4). No adverse effects, including tumour formation, abnormal cell growth or inflammation, were detected in the recipient animals.
In the animals treated with systemic injections, quantification of donor-derived cells in the spinal cord revealed the presence of 8.66 ± 0.49 cells per section in the cervical and 8.93 ± 0.38 cells per section in the lumbar spinal cord (Fig. 3C). We found IgG deposits from blood vessels in the spinal cord of SOD1 mice but not in wild-type mice (Supplementary Material, Fig. S5). These data confirmed the weakness of the BBB in SOD1G93A mice (16,17,19), which allowed transplanted cells to reach the spinal cord after the systemic injection. The phenotype of the cells transplanted by intravenous injection was similar to the intrathecal administration; the engrafted cells maintained an undifferentiated phenotype or differentiated into neuronal and glia cells (Fig. 3D and E, Supplementary Material, Fig. S6). With systemic administration, we also detected the presence of donor cells in the cortical and subcortical brain areas of ALS mice (Supplementary Material, Fig. S7). Neuronal undifferentiated cells survived and were detectable until the late stage of the disease, when the animals were sacrificed. No adverse effects were observed after systemic transplantation.
Cell transplantation improved disease phenotype and extended survival of ALS animals
We first evaluated whether both intrathecal and systemic delivery of ALDHhiSSCloVLA4+ cells could ameliorate the phenotype and extend survival in SOD1G93A mice (Fig. 4). For the phenotypic assessment, mice were tested on the rotarod, and the survival was recorded.
Figure 4.

Transplantation of ALDHhiSSCloVLA4+ NSCs improves neuromuscular function, increases survival and reduces motor neuron and axon loss in ALS mice. (A and C) Transplantation of NSCs significantly improved motor performance in SOD1 mice, as demonstrated by the rotarod test both in the intrathecally transplanted group (A) and in systemically injected mice (C) (4 weeks after transplantation, P < 0.001, ANOVA). (B and D) Kaplan–Meier survival curves for mutant SOD1 mice treated intrathecally (B) or systemically (D) with ALDHhiSSCloVLA4+ NSCs or with vehicle. Survival was significantly extended for NSC-transplanted mice compared with vehicle-treated mice for both treatment groups (P < 0.05, log-rank test). (E) The motor neuron count (n = 6 for each group) in the lumbar spinal cord of NSC-transplanted, vehicle-treated SOD1 mice and wild-type mice (data represent the mean ± SD of the number of motor neurons per section) at 140 days of age. The evaluation revealed significantly increased numbers of surviving motor neurons in treated SOD1G93A mice (P < 0.001, ANOVA). (F) Quantification of axons (data represent the mean ± SD) at 140 days of age (n = 6 for each group) demonstrated that transplanted SOD1G93A mice showed a significantly increased number of axons (P < 0.001, ANOVA).
Intrathecal transplantation of ALDHhiSSCloVLA4+ cells into SOD1G93A mice (n = 24) significantly extended survival by 10 days compared with vehicle-treated animals (n = 25) (vehicle, 143 ± 6 days mean survival; ALDHhiSSCloVLA4+, 153 ± 5 days; χ2 = 6.1, P = 0.0135; Fig. 4B). Systemic transplantation of ALDHhiSSCloVLA4+ cells into SOD1G93A mice (n = 16) also significantly extended survival by 23 days compared with vehicle-treated animals (n = 24) (vehicle, 148 ± 7 days mean survival; ALDHhiSSCloVLA4+, 171 ± 6 days; χ2 = 11.81, P = 0.0006; Fig. 4D). The survival difference between the intrathecal and systemic group was statistically significant (P = 0,0001). The rotarod test was used to assess neuromuscular function in transplanted and untreated animals. At 4 weeks after starting transplantation, the cell-treated animals presented a significant improvement in rotarod performance relative to untreated (P < 0.001) and fibroblast-treated animals (P < 0.001) (Fig. 4A and C). Rotarod functional outcome was similar between the two groups and the difference is not statistically significant.
To determine whether ALDHhiSSCloVLA4+ transplantation protects against the loss of motor neurons, we quantified the number of motor neurons in spinal cord sections and ventral spinal nerve roots. At 140 days of age, motor neuron loss was significantly reduced by cell transplantation via both intrathecal and systemic administration. Mice transplanted with stem cells had 40% more motor neurons than vehicle-treated SOD1G93A mice (P < 0.001); this increase likely accounted for the observed functional improvement (Fig. 4E). Transplanted animals also presented with a preservation of axonal density in the L4 ventral root, while control mice showed a substantial reduction in axons compared with wild-type mice. Quantitative assessment revealed that in both transplanted SOD1G93A groups, significantly fewer axons were lost with untreated SOD1 mice (treated versus untreated P < 0.001). Axons in adult L4 ventral nerve roots were preserved, showing an increase of 50% with respect to vehicle-treated animals (P < 0.001) (Fig. 4F).
We detected donor GFP cells in the muscle located in the proximity of the sarcolemma of the muscle fibres in mice transplanted systemically at 140 days of age. We hypothesized that neuromuscular junction (NMJ) preservation may have been involved in the improved neuromuscular function in the systematically delivered group (Supplementary Material, Fig. S8).
Changes in growth factor levels and a reduction in micro- and macrogliosis after cell transplantation
We next investigated the molecular mechanisms that may underlie the observed therapeutic effect. Using an enzyme-linked immunosorbent assay (ELISA), we determined that iPSC-derived ALDHhiSSCloVLA4+ NSCs produced a spectrum of neurotrophic and neurite outgrowth-promoting factors (Fig. 5) (14,15). ALDHhiSSCloVLA4+ NSCs secreted significant amounts of glial cell-derived growth factor (GDNF) (713.8 ± 69.75 pg/ml; P < 0.001 versus fibroblasts), brain-derived neurotrophic factor (BDNF) (469.67 ± 52.76 pg/ml; P < 0.001), neurotrophin-3 (NT3) (158.46 ± 21.68 pg/ml; P < 0.001) and transforming growth factor alpha (TGF-α) (123.51 ± 11.42; P < 0.001) (Fig. 5A–D).
Figure 5.

NSCs produce neurotrophins and reduce micro- and macrogliosis in SOD1G93A mice. The amounts of (A) GDNF, (B) BDNF, (C) NT3 and (D) TGF-α secreted by the NSCs were determined by ELISA, which demonstrated a significant production of these neuroprotective substances with respect to fibroblasts. (E) Expression of GFAP (red) was reduced in the spinal cord of transplanted SOD1 mice. Nuclei are stained with DAPI (blue). Scale bar: 70 µm. NSC transplantation significantly reduced the presence of microglial cells (F) and astrocytes (G) in the spinal cord of intrathecally and systemically injected mice (data represent the mean ± SD, *significant, P < 0.001, ANOVA).
In our neuropathological analysis, the CNS tissues of SOD1G93A ALS mice showed the presence of a significant inflammatory (microglial) reaction that may play an important role in the disease pathogenesis (1,10). Interestingly, we demonstrated a significant reduction in macrophage/microglia cell populations in both lumbar and cervical spinal cord in treated mice compared with vehicle-treated and untreated SOD1G93A mice. The number of activated microglia in the ventral horn was 146.8 ± 13.4 per section in vehicle-treated SOD1G93A mice versus 65.4 ± 7.9 in the intrathecally treated SOD1G93A mice (n = 6 per group/condition; P < 0.001) (Fig. 5F). Similar results were obtained with systemic injection, with 54.9 ± 9.6 microglia per section in the treated SOD1G93A mice versus 153.4 ± 14.9 in vehicle-treated animals (n = 6 per group/condition; P < 0.001) (Fig. 5F).
Astrogliosis also plays an important role in ALS pathogenesis, particularly in non-autonomous motor neuron death (1,10). Staining for the astrocytic marker glial fibrillary acidic protein (GFAP) suggested diminished astrogliosis in the spinal cord (particularly in the critical cervical/lumbar regions) of NSC-engrafted SOD1G93A mice (Fig. 5E). The number of hypertrophic reactive astrocytes in the ventral horn was 254.9 ± 24.6 per section in vehicle-treated SOD1G93A mice versus 163.9 ± 12.4 in intrathecally treated SOD1G93A mice (n = 6 per group; P < 0.001) (Fig. 5G). In the systemic injection group, we observed a mean of 145.6 ± 22.3 GFAP-positive cells compared with 243.7 ± 21.8 in the vehicle-treated group (n = 6 per group; P < 0.001) (Fig. 5G).
NSCs inhibit ALS astrocyte activation through TRPV1 receptor activation
We next analysed the possible molecular mechanisms involved in the reduction of astrocyte activation/proliferation by NSC transplantation (Fig. 6). In a series of in vitro experiments, we demonstrated that stimulating mouse astrocytes with factors released from iPSC-derived NSCs, but not with factors released from fibroblasts, significantly reduced the viability of SOD1G93A astrocytes. NSCs reduce astrocytic tumour expansion by releasing endovanilloids that activate the vanilloid receptor (transient receptor potential vanilloid subfamily member-1, TRPV1) on astrocytic cells (20). Therefore, we investigated whether similar events occurred during ALS. First, we demonstrated that astrocytes from the CNS of SOD1 mice express TRPV1 (Fig. 6A). We found that NSCs are toxic for astrocytes when grown in co-culture (Fig. 6B). We further showed that blocking TRPV1 with the selective antagonists iodoresiniferatoxin (I-RTX, 10 nm) or capsazepine (CZP, 1 μm) decreased the cytotoxicity of NSCs on astrocytes (Fig. 6C). The toxic effect of NSCs on astrocytes was selectively restricted on the SOD1 background, while non-transgenic and ALDHhiSSCloVLA4+ NSC-derived astrocytes were not affected (Fig. 6D). Thus, we conclude that donor NSCs reduce reactive astrocytes in the CNS of SOD1 mice by activating TRPV1.
Figure 6.
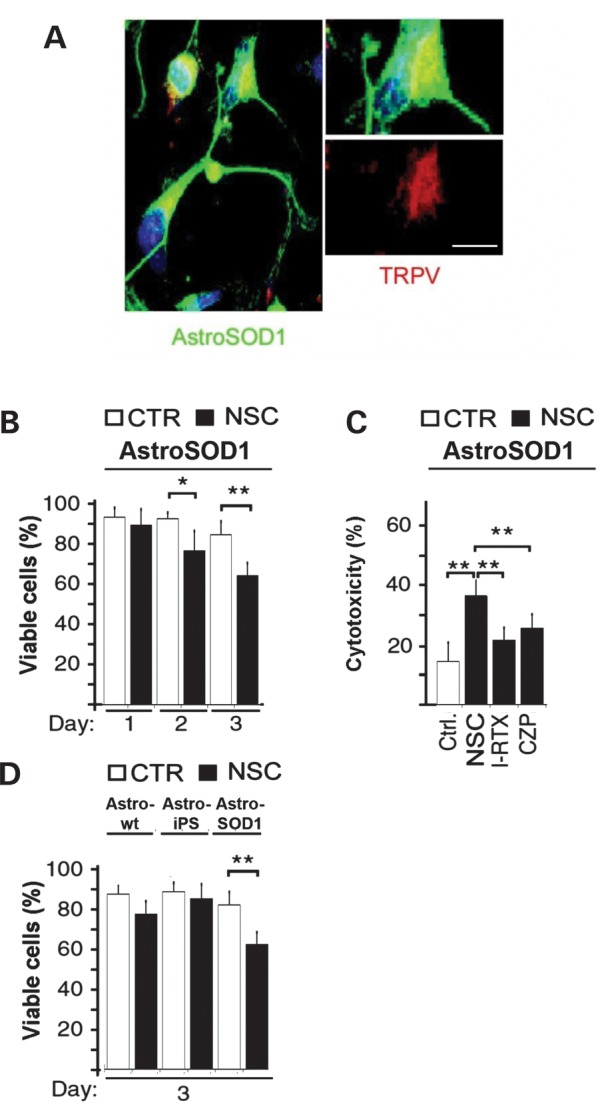
NSC-mediated astrocyte suppression by activating endovanilloid receptor TRPV1. (A) SOD1G93A astrocytes (GFAP, green) are immunopositive for TRPV1 (red). A single cell is magnified on the left, and pixels with co-localized staining in a single optical section are shown. Scale bars: 40 μm on the left, 10 μm in the higher magnification panel. (B) The viability of SOD1G93A astrocytes is reduced after stimulation with NSC-conditioned medium (NSC) but not after stimulation with non-conditioned medium (Ctrl). (C) The NSC-induced cytotoxicity of astrocytes was blocked by the TRPV1 selective antagonists I-RTX and CZP. (D) The toxic effect of NSCs on astrocytes was present on SOD1 astrocytes, while non-transgenic mouse astrocytes and on ALDHhiSSCloVLA4+ NSC-derived astrocytes were not significantly affected. All data are mean ± SD. *P < 0.05, **P < 0.005 by Student's t-test.
DISCUSSION
In this study, we found that repeated intrathecal or systemic injection of a specific NSC subpopulation ameliorates the ALS disease phenotype in a mouse model. These results, combined with the minimally invasive nature of the delivery method, support the rationale and utility of this approach for patients with ALS. The beneficial effects of engrafted ALDHhiSSCloVLA4+ NSCs include improved neuromuscular function and survival, neuroprotection and positive host-environment modifications at the neuropathological level.
Stem cell transplantation is gaining increased interest as a therapeutic strategy because its multifaceted impact can counteract a number of processes that underlie disease pathogenesis. Molecular and pharmacological strategies that focus on a single or few targets have been minimally successful in ALS clinical trials, likely because they address only one aspect of this complex neurodegenerative disease. In contrast, several experimental approaches of NSC transplantation have produced positive effects on motor neuron disease in animal models (9–15).
Among the potential cell sources, our population of ALDHhiSSCloVLA4+ stem cells/precursors is an attractive choice for both reproducibility and the lack of controversy regarding their collection; they can be derived from autologous or donor fibroblasts, reprogrammed into iPSCs, and grown in vitro. In addition, they can migrate from the CSF or blood to the spinal cord parenchyma. This property allows non-invasive transplantation, which is important for clinical translation to patients. Furthermore, these cells present robust survival in the host CNS and do not trigger side effects such as tumours or abnormal cell growth. Indeed, we showed that the donor cells appropriately migrate and localize throughout the entire spinal cord, particularly in the specific areas that are affected by the disease: the cervical/lumbar enlargement, the anterior horns and the areas around endogenous motor neurons. The donor cells were also located in cortical brain areas, thus positively influencing the upper motor neurons. Of importance, donor ALDHhiSSCloVLA4+ cells maintained their stem cell/precursor phenotype or differentiated into neuronal and glial cells, changing the spectrum of cell phenotypes in the host tissue microenvironment.
Regarding the mechanisms of cell migration from the CSF and blood toward the parenchyma, the neuronal death that occurs in ALS could be a signal that promotes NSC attraction into the CNS. Indeed, the presence of an altered BBB in ALS mice and patients can promote cell migration (17). VLA4 is a surface protein that mediates transendothelial migration. It is expressed on some haematopoietic cell fractions, in particular on T lymphocytes, and mediates their ability to cross the BBB (21). A humanized monoclonal antibody against VLA4, natalizumab, is used to treat multiple sclerosis because it blocks the migration of T lymphocytes into the CNS (21). In contrast, the presence of VLA4 on NSCs gives them the ability to cross the BBB, particularly in the presence of inflammation or weakened BBB, as demonstrated in humans and animal models of ALS (16,17).
ALDHhiSSCloVLA4+ NSC-treated SOD1 mice presented an amelioration of the motor neuron phenotype, demonstrated by neuromuscular function tests and increased survival. Transplantation of ALDHhiSSCloVLA4+ cells significantly prolonged survival compared with vehicle-treated animals. The increased survival time, although limited, is relevant considering previous reports of NSC transplantation in rodent models of ALS that also showed a transient improvement in motor performance and a modest increase in survival (22). Recently, the Teng group reported a remarkable survival increase of 200 days in 40% or up to a full year in 20% of SOD1 animals injected with NSCs at four sites of the spinal cord during the presymptomatic stage of disease (10). However, the remaining 40% of mice showed a very mild increase in survival, demonstrating a large variability in outcome with unknown cause. The transplantation method used by Teng et al. (10) required a direct intraparenchymal injection at multiple sites. This procedure is more invasive than the intrathecal and systemic injections that were used in this study. One possibility for the difference in benefit obtained with the more invasive cell therapy by Teng et al. (10) can lie in the number of NSCs that successfully engrafted the parenchyma. However, a direct comparison cannot be performed because the authors do not present a quantitative analysis of engrafted donor cell number, even if we can assume indirectly from other data, such as images of the treated mouse spinal cord, that the number of cells was higher than what we achieved in our experiments. Indeed, both our group and Teng's group observed a positive correlation between the number of engrafted cells and the positive outcome.
The strategy of repeated delivery of the cells allowed achieving a widespread distribution of a significant amount of cells in the CNS. We hypothesized that also in clinical translation, the therapeutic effect would be proportional to the number of engrafted cells that can be achieved with multiple injections. Regarding the risks of raising an immunoresponse with this strategy, differentiated cells derived from iPSCs, including NSCs, present a low immunogenic profile (23,24). Moreover, the CNS is a low responding immunological site. In any case, immunoresponse stimulated by repeated injection of NSCs can be controlled by immunosuppressant agents, as we achieved here in mice; no signs of immunorejection were observed. VLA4 is a physiological antigen expressed in human cells, so it will not initiate an immune response in humans.
We demonstrated an increased therapeutic effect when the cells were administered by systemic injection compared with intrathecal injection. We hypothesize that, with a systemic injection, donor NSCs can exert a positive role both locally and peripherally. After entering the CNS via cell adhesion molecules (such as VLA4) and chemokine receptors, systemically injected NSCs accumulate in perivascular CNS areas and in the areas of active degeneration. As a consequence, transplanted NSCs survive and promote neuroprotection by releasing neurotrophic factors in situ. There is further evidence suggesting a peripheral immunomodulatory effect of the NSCs (25). Systemically injected NSCs also enter peripheral organs (e.g. draining lymph nodes and spleen), where they accumulate at the boundaries of blood vessels. Here, they interact closely with lymphocytes and professional antigen-presenting cells, impairing their maturation and functional activation (25). Indeed, we hypothesize a peripheral effect of NSCs, which secrete neuroprotective factors at the skeletal muscle level, on the maintenance of NMJ integrity; however, experimental demonstration is still needed. Accumulating evidence suggests that clinical ALS is possibly a multi-organ disease. Although motor neurons are selectively affected in ALS, the possibility that ALS might be part of a systemic disease in which motor neurons are especially vulnerable has been hypothesized. In fact, a number of well-established abnormalities in ALS occur in the immune system, skin, skeletal muscle tissue and lipid metabolism (see for review26). Moreover, we have to consider that the systemic delivery relies on multiple injections respect to the intrathecal delivery over the same period of time and the maintenance of the NSCs presence may have been more continuous for the systemic route than for the other one, even if at 140 days and at the end stage of the disease the difference in NSC integration between the two routes of delivery was only slightly different.
We hypothesize that the therapeutic effect of ALDHhiSSCloVLA4+ cells in our study relies on multiple mechanisms that include both neuroprotection and reduction of macro- and microglia. NSCs constitutively secrete various neurotrophic and neuroprotective molecules that can exert a synergistic effect superior to the administration of a single molecule. These factors seem to protect host motor neurons and functional neuromuscular units, reduce macrophage/microglial infiltration and decrease endogenous astrogliosis (10,22). In particular, we observed an increase in the production of GDNF, which can exert a direct neuroprotective effect on endogenous cells. The modest effects of neurotrophic factor treatment on lifespan in previous clinical studies of ALS suggest several explanations including that they are physiologically effective but limited by pharmacokinetic constraints (27).
Indeed, NSCs constitutively secrete more than one neurotrophic and neuroprotective factor. We also demonstrated a reduction in the levels of activated macro- and microglia after cell transplantation, suggesting a potential anti-inflammatory effect of ALDHhiSSCloVLA4+ cells. Even if wild-type astrocytes can exert a positive effect on motor neurons, we and others (10) have demonstrated that the reduction in astrocytosis plays a part in the therapeutic effect achieved by cell transplantation. Therefore, a reduction in astrocytes can be considered a shift of the ALS neuropathological features towards a physiological wild-type pattern. Our results suggest a novel mechanism in which NSCs reduce macrogliosis by activating the vanilloid receptor. Thus, NSCs not only provide cells of the three neuroectodermal lineages to the host cell environmental, but they can also influence the behaviour of endogenous cells. Identifying the full range of variables underlying the success of cell transplantation is essential for confirming the reproducibility of the therapeutic response for clinical application.
In the present work, we described a feasible and efficient protocol for the generation of NSC cells from iPSCs that are scalable for producing highly pure populations of functional iPSC-derived NSCs sufficient for clinical translation. Converting the cell dose used in a mouse to a dose based on surface area (28), we can estimate that for humans we would have to transplant an approximate dose of 1 × 108 cells/m2 that for a man of 60 kg (with a body surface area of 1.6 m2) would be 1.6 × 108. Given their indefinite self-renewal, with a doubling time between 24 and 48 h, human iPSCs have the potential to yield an unlimited supply of NSCs.
Our results provide evidence that ALDHhiSSCloVLA4+ NSCs derived from iPSCs may represent an effective approach for the treatment of ALS. We demonstrated that the beneficial effects observed after stem cell transplantation arise from multiple events that counteract multiple aspects of the disease; this feature is crucial for multifaceted disease such as ALS. A combination of therapeutic approaches that target different pathogenic mechanisms of the disorder, including pharmacology, molecular therapy and cell transplantation, will increase the chances of a clinically successful therapy for ALS.
MATERIALS AND METHODS
Generation of iPSCs
Human skin fibroblasts were reprogrammed as previously described (15) using oriP/EBNA1-based episomal vectors encoding the human genes OCT4, SOX2, NANOG, LIN28, c-Myc and KLF4. After nucleofection, fibroblasts were plated onto 3 × 10 cm dishes covered with Matrigel (BD Biosciences) in fibroblast culture medium, which was changed every other day. Four days after transfection, the fibroblast culture medium was replaced with human ESC culture medium (mTeSR, Stemcell Technologies Inc.) for 8–10 days. At day 18 after transfection, the first colonies with an iPSC-like morphology could be identified. We then picked the iPSC colonies that were morphologically similar to ESCs for further analysis and expansion, as previously described (15).
Differentiation of iPSCs and selection of the ALDHhiSSCloVLA4+ fraction
We differentiated iPSCs into neuroepithelial cells using a previously described differentiation protocol (15). iPSCs were plated in neuronal medium composed of Dulbecco's modified Eagle's medium/F12 (Gibco, Invitrogen). The medium was supplemented with MEM non-essential amino acids solution, N2 and heparin (2 µg/ml; Sigma-Aldrich) to induce the neuroepithelial commitment. Cells were then selected by FACS for their ALDHhiSSClo properties, as previously described (11,13). The isolated cells were further selected by FACS for their VLA4 positivity (anti-human VLA4, BD) using an appropriate negative control with cells labelled only with the same isotypic Ig.
For cell expansion and clonal culture, ALDHhiSSCloVLA4+ cells were plated in a previously described growth medium (NEP medium) containing FGF with or without EGF (13). Differentiation of cultured ALDHhiSSCloVLA4+ cells was induced by plating on poly-lysine/laminin-coated dishes (poly-lysine at 20 µg/ml in DPBS; Sigma) and reducing the FGF concentration with the addition of RA (1 µm). To induce the motor neuronal phenotype, NSCs were exposed to additional RA (0.1 µm; Sigma-Aldrich) for neural caudalization. After 7 days, we collected the posteriorized neuroectodermal cells and suspended them for a week in the same medium with RA (0.1 µm) and SHH (100–200 ng/ml; R&D Systems Inc.). On day 24, we added the growth factors BDNF, GDNF and insulin-like growth factor-1 (10 ng/ml each; PeproTech).
Immunocytochemistry of iPSCs and their derivatives
Cells were fixed in 4% paraformaldehyde (PFA) for 10 min, permeabilized with 0.5% Tween-20 in phosphate-buffered saline (PBS) and exposed to 0.1% Tween-20 with 10% horse serum. We incubated the cells with primary antibodies overnight and with secondary antibodies for 1 h (Alexa Fluor, Invitrogen). We used the following primary antibodies: SSEA-3 (1:100, Chemicon), SSEA-4 (1:500, Chemicon), TRA1-60 (1:500, Chemicon), TRA1-81 (1:500, Chemicon), Pax6 (1:200, Millipore), nestin (1:200, Millipore), Sox1 (1:1000, R&D,), SOX2 (1:1000, Millipore), TuJ1 (1:200, Millipore), GFAP (1:300, Sigma), Islet1/2 (1:200, Millipore), HB9 (1:200 Millipore), ChAT (1:200, Millipore), MAP2 (mouse monoclonal, 1:100; Sigma), O4 (10–20 μg/ml, Millipore) and Musashi (1:200, Novus Biological). The HB9::GFP gene reporter was used to identify motor neuron cells, as previously described (29). For all imaging, we used a confocal LEICA LCS2 microscope.
Animal models
We used transgenic mice of the strain B6.Cg-Tg(SOD1-G93A)1Gur/J, which carries a high copy number of the mutant human SOD1 allele containing a Gly93Ala (G93A) substitution. Progeny for experimental analyses was obtained by breeding SOD1G93A transgenics with C57BL/6 wild-type mice. Transgenic mice were identified by polymerase chain reaction, as previously described (14). All animal experiments were approved by the University of Milan and Italian Ministry of Health review boards and are in compliance with the US National Institutes of Health guidelines (14).
Cell transplantation
Ninety-day-old transgenic SOD1G93A mice were transplanted with ALDHhiSSCloVLA4+ cells (1 × 106 cells) or vehicle only (saline) by intrathecal injection (5 µl) (three injections) or by injection through the tail vein (100 µl, weekly injections) (14). These cells were also genetically engineered to express GFP driven by the cytomegalovirus promoter (15). The cells were harvested and prepared for transplantation as previously described (15). The intrathecal group included 24 mice (12 males) for transplantation and 25 mice (12 males) in the vehicle control group. The intravenous group included 16 transplantation mice (8 males) and the vehicle group of 24 mice (12 males). The animals were evaluated up to the end stage for rotarod performance, survival and histological evaluation of donor cell phenotype.
The study was designed such that littermates were distributed equally among the transplanted and non-transplanted groups. Three groups of animals (each composed of six mice per time point per condition) were euthanized at 140 days of age for histological analysis, quantification of motor neurons and axons and growth factor evaluation via ELISA.
Tissue analysis
Animals were euthanized, perfused and fixed with 4% PFA in PBS (pH 7.4). The spinal cord was isolated, immersed in PFA for 1 h and then soaked overnight in 20% sucrose in PBS (pH 7.4). The fixed tissue was frozen in Tissue Tek OCT compound (Sakura) with liquid nitrogen. Tibialis anterior muscles were removed prior flash-frozen in supercooled isopentane. The tissues were cryosectioned and mounted on gelatinized glass slides. Every tenth 20 µm section was collected. All sections were blocked with 1% fetal calf serum in PBS and permeabilized with 0.25% Triton X-100. Sections were processed for multiple markers to determine the cellular phenotype of GFP-labelled cells. Primary antibodies were added overnight at 4°C at dilutions of 1:200 for NeuN (mouse monoclonal antibody, Chemicon), 1:200 for nestin (mouse monoclonal, Chemicon) and 1:200 for GFAP (mouse monoclonal Cy3 conjugate, Sigma) and laminin (rabbit polyclonal, 1: 500, Sigma-Aldrich).
Mouse and rabbit secondary antibodies conjugated with FITC, RPE, CY3 or biotin (1:200; Jackson ImmunoResearch Laboratories, Inc., and Dako) were applied for 1 h at room temperature when unconjugated primary antibodies were used. Anti-GFP antibody rabbit serum Alexa 488 (1:400 dilution; Molecular Probes) was used to reveal GFP expression. Co-expression of GFP and tissue-specific markers was evaluated by conventional fluorescence microscopy (Zeiss Axiophot) and laser confocal scanning microscopy (Leica TCS SP2 AOBS). Optical dissectors and random sampling were used to obtain an unbiased stereological estimate of the number of GFP-positive cells. To quantify donor cells, we used every tenth coronal section (20 µm) throughout the entire spinal cord (a mean of 25 sections per animal). The numerical density of cells was estimated using the optical dissector method (14). Optical dissectors sized 100 × 70 × 14 µm were randomly sampled, and the number of positive cells in each dissector was quantified. The density was calculated by dividing the total number of donor cells by the total volume of optical dissectors. The total volume of tissue per specimen (Vcord) containing donor cells was calculated using the Cavalieri method (14). This total volume of tissue, multiplied by the number of donor cells per cubic micrometre (Nv), yielded the total number of donor cells per specimen (n = Nv × Vcord) (14).
Assessment of motor function and survival
Vehicle- and cell-injected SOD1G93A mice were monitored daily following transplantation for clinical signs of disease onset. Motor performance was assessed using an accelerating rotarod device (4–40 rpm Rota-Rod 7650; Ugo Basile, Comerio, Italy). The length of time that the mice remained on the rotarod was recorded. The investigators performing the behavioural assessment were blind to the treatment. Mortality was scored as the age at which the mouse was unable to right itself within 30 s when placed on its back, as previously described (14).
Motor neuron and axon count
Paraffin serial sections (12.5 µm) of lumbar spinal cord were Nissl-stained to quantify motor neuron numbers via light microscopy, as previously described (14). For the axon count, the tissue was dissected, immersed in 2.5% glutaraldehyde overnight and post-fixed in 2% osmium tetroxide. Samples were then dehydrated in ethanol and embedded in Epon resin. Semi-thin transverse sections (1 µm) were stained with toluidine blue. L4 roots were examined for axon counting on an optic microscope.
Enzyme-linked immunosorbent assay
BDNF, GDNF, NT3 and TGF-α secretion by NSCs was measured by ELISA as previously described (14). The data were analysed using Student's t-tests.
Analysis of macro- and microgliosis
Histological analysis for quantification of microglia and astrocytes was performed as described (10).
TRPV1 evaluation and cytotoxicity assay
Detection of the TRPV1 receptor, conditioned medium experiments and the cytotoxicity assay were performed as previously described (20).
Statistical analysis
Kaplan–Meier survival analysis and the log-rank test were used for survival comparisons. The rotarod test data were analysed using an ANOVA followed by a Tukey post hoc analysis for multiple comparisons. The numbers of motor neurons and axons were statistically evaluated by one-way ANOVA followed by a Tukey post hoc analysis. ELISA results were analysed using Student's t-tests. In culture assays, differences between mean values were analysed using a two-tailed Student's t-test. We used StatsDirect for Windows (version 2.6.4) for all statistical analyses, rejecting the null hypothesis at the 0.05 level.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the ARISLA Foundation to G.P.C. ‘iPSALS grant’ and MIUR (FIRB RBFR08RV86) to S.C. Funding to pay the Open Access publication charges for this article was provided by ARISLA (Fondazione Italiana di Ricerca per la SLA - Sclerosi Laterale Amiotrofica).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the ‘Associazione Amici del Centro Dino Ferrari’ for their support.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Ludolph A.C., Brettschneider J., Weishaupt J.H. Amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2012;25:530–535. doi: 10.1097/WCO.0b013e328356d328. [DOI] [PubMed] [Google Scholar]
- 2.Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J.P., Deng H.X., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Ferraiuolo L., Kirby J., Grierson A.J., Sendtner M., Shaw P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- 4.Morren J.A., Galvez-Jimenez N. Current and prospective disease-modifying therapies for amyotrophic lateral sclerosis. Expert. Opin. Investig. Drugs. 2012;21:297–320. doi: 10.1517/13543784.2012.657303. [DOI] [PubMed] [Google Scholar]
- 5.Riboldi G., Nizzardo M., Simone C., Falcone M., Bresolin N., Comi G.P., Corti S. ALS Genetic modifiers that increase survival of SOD1 mice and are suitable for therapeutic development. Prog. Neurobiol. 2011;95:133–148. doi: 10.1016/j.pneurobio.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Gurney M.E., Pu H., Chiu A.Y., Dal Canto M.C., Polchow C.Y., Alexander D.D., Caliendo J., Hentati A., Kwon Y.W., Deng H.X., et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 7.Nagai M., Aoki M., Miyoshi I., Kato M., Pasinelli P., Kasai N., Brown R.H., Jr, Itoyama Y. Rats expressing human cytosolic copper–zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J. Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamanaka K., Boillee S., Roberts E.A., Garcia M.L., McAlonis-Downes M., Mikse O.R., Cleveland D.W., Goldstein L.S. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl Acad. Sci. USA. 2008;105:7594–7599. doi: 10.1073/pnas.0802556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boulis N.M., Federici T., Glass J.D., Lunn J.S., Sakowski S.A., Feldman E.L. Translational stem cell therapy for amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011;8:172–176. doi: 10.1038/nrneurol.2011.191. [DOI] [PubMed] [Google Scholar]
- 10.Teng Y.D., Benn S.C., Kalkanis S.N., Shefner J.M., Onario R.C., Cheng B., Lachyankar M.B., Marconi M., Li J., Yu D., et al. Multimodal actions of neural stem cells in a mouse model of ALS: a meta-analysis. Sci. Transl. Med. 2012;4:165–164. doi: 10.1126/scitranslmed.3004579. [DOI] [PubMed] [Google Scholar]
- 11.Corti S., Locatelli F., Papadimitriou D., Donadoni C., Del Bo R., Crimi M., Bordoni A., Fortunato F., Strazzer S., Menozzi G., et al. Transplanted ALDHhiSSClo neural stem cells generate motor neurons and delay disease progression of nmd mice, an animal model of SMARD1. Hum. Mol. Genet. 2006;15:167–187. doi: 10.1093/hmg/ddi446. [DOI] [PubMed] [Google Scholar]
- 12.Corti S., Locatelli F., Papadimitriou D., Del Bo R., Nizzardo M., Nardini M., Donadoni C., Salani S., Fortunato F., Strazzer S., et al. Neural stem cells LewisX+ CXCR4+ modify disease progression in an amyotrophic lateral sclerosis model. Brain. 2007;130:1289–1305. doi: 10.1093/brain/awm043. [DOI] [PubMed] [Google Scholar]
- 13.Corti S., Nizzardo M., Nardini M., Donadoni C., Salani S., Ronchi D., Saladino F., Bordoni A., Fortunato F., Del Bo R., et al. Neural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophy. J. Clin. Invest. 2008;118:3316–3330. doi: 10.1172/JCI35432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corti S., Nizzardo M., Nardini M., Donadoni C., Salani S., Simone C., Falcone M., Riboldi G., Govoni A., Bresolin N., et al. Systemic transplantation of c-kit+ cells exerts a therapeutic effect in a model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2010;19:3782–3796. doi: 10.1093/hmg/ddq293. [DOI] [PubMed] [Google Scholar]
- 15.Corti S., Nizzardo M., Simone C., Falcone M., Nardini M., Ronchi D., Donadoni C., Salani S., Riboldi G., Magri F., et al. Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci. Transl. Med. 2012;4:165–162. doi: 10.1126/scitranslmed.3004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pluchino S., Zanotti L., Rossi B., Brambilla E., Ottoboni L., Salani G., Martinello M., Cattalini A., Bergami A., Furlan R., et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;438:266–271. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- 17.Winkler E.A., Sengillo J.D., Sullivan J.S., Henkel J.S., Appel S.H., Zlokovic B.V. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:111–120. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu B.Y., Zhang S.C. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat. Protoc. 2009;4:1295–1304. doi: 10.1038/nprot.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong Z., Deane R., Alim Z., Parisi M., Shapovalov Y., O'Banion M.K., Stojanovic K., Sagare A., Boillee S., Cleveland D.W., et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stock K., Kumar J., Synowitz M., Petrosino S., Imperatore R., Smith E.S., Wend P., Purfürst B., Nuber U.A., Gurok U., et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012;18:1232–1238. doi: 10.1038/nm.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat. Rev. Immunol. 2009;9:440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- 22.Gowing G., Svendsen C.N. Stem cell transplantation for motor neuron disease: current approaches and future perspectives. Neurotherapeutics. 2011;8:591–606. doi: 10.1007/s13311-011-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araki R., Uda M., Hoki Y., Sunayama M., Nakamura M., Ando S., Sugiura M., Ideno H., Shimada A., Nifuji A., et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature. 2013;494:100–104. doi: 10.1038/nature11807. [DOI] [PubMed] [Google Scholar]
- 24.Liu P., Chen S., Li X., Qin L., Huang K., Wang L., Huang W., Li S., Jia B., Zhong M., et al. Low immunogenicity of neural progenitor cells differentiated from induced pluripotent stem cells derived from less immunogenic somatic cells. PLoS ONE. 2013;8:e69617. doi: 10.1371/journal.pone.0069617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donegà M., Giusto E., Cossetti C., Pluchino S. Neural Stem Cells—New Perspectives. 2013. Systemic neural stem cell-based therapeutic interventions for inflammatory CNS disorders. Chapter 11, Springer, New York. [Google Scholar]
- 26.D'Amico E., Factor-Litvak P., Santella R.M., Mitsumoto H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013;65:509–527. doi: 10.1016/j.freeradbiomed.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gould T.W., Oppenheim R.W. Motor neuron trophic factors: therapeutic use in ALS? Brain Res. Rev. 2011;67:1–39. doi: 10.1016/j.brainresrev.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reagan-Shaw S., Nihal M., Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 29.Marchetto M.C., Muotri A.R., Mu Y., Smith A.M., Cezar G.G., Gage F.H. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}