

Abstract
CHARGE syndrome is a multiple congenital anomaly disorder that leads to life-threatening birth defects, such as choanal atresia and cardiac malformations as well as multiple sensory impairments, that affect hearing, vision, olfaction and balance. CHARGE is caused by heterozygous mutations in CHD7, which encodes an ATP-dependent chromatin remodeling enzyme. Identification of the mechanisms underlying neurological and sensory defects in CHARGE is a first step toward developing treatments for CHARGE individuals. Here, we used mouse models of Chd7 deficiency to explore the function of CHD7 in the development of the subventricular zone (SVZ) neural stem cell niche and inner ear, structures that are important for olfactory bulb neurogenesis and hearing and balance, respectively. We found that loss of Chd7 results in cell-autonomous proliferative, neurogenic and self-renewal defects in the perinatal and mature mouse SVZ stem cell niche. Modulation of retinoic acid (RA) signaling prevented in vivo inner ear and in vitro neural stem cell defects caused by Chd7 deficiency. Our findings demonstrate critical, cooperative roles for RA and CHD7 in SVZ neural stem cell function and inner ear development, suggesting that altered RA signaling may be an effective method for treating Chd7 deficiency.
INTRODUCTION
CHARGE syndrome is a multiple anomaly disorder occurring in 1:10 000 live births and characterized by ear and heart defects, choanal atresia, retardation of growth and/or development and delayed puberty (1,2). In addition, olfactory bulb (OB) hypoplasia and hyposmia/anosmia are highly penetrant in CHARGE individuals; strikingly, arrhinencephaly and ventriculomegaly have been reported in CHARGE fetuses (3–6). Heterozygous mutations in CHD7, which encodes an ATP-dependent chromatin remodeling enzyme, account for over 90% of CHARGE syndrome cases, yet no clear genotype–phenotype correlations have emerged (5,7,8). Beyond supportive treatments and surgical intervention, there are no curative therapies for CHARGE-related defects.
Mouse models of CHARGE syndrome recapitulate most phenotypes observed in humans, including inner ear defects (semicircular canal and cochlear hypoplasia/dysplasia), OB hypoplasia and hyposmia/anosmia (4,9–14). The underlying causes of inner ear defects and OB hypoplasia in Chd7-deficiency states are unknown, but there is evidence for disrupted neural stem cell proliferation in the otocyst and olfactory epithelium (4,10). In the olfactory system, OB interneurons (granule and periglomerular cells) arise from neural stem cells in the subventricular zone (SVZ) of the lateral ventricles (LVs), which give rise to neuroblasts that migrate through the rostral migratory stream (RMS) over a 2- to 3-week period (15). SVZ-derived neuroblasts then terminally differentiate and integrate into the granule cell (GCL) and glomerular (GL) layers of the OB, where they mature into inhibitory interneurons that modulate mitral and tufted cells, which relay olfactory sensory signals to the olfactory cortex (15). In rodents, OB neurogenesis begins around embryonic day (E) 15 (E15) and continues throughout life (16,17). Thus, OB neurogenesis is an excellent system for studying CHD7 function in neural development and maintenance.
The development and maintenance of the inner ear, SVZ, RMS and OB are regulated in part by retinoic acid (RA) signaling (18–20). Notably, the treatment of SVZ neural stem cell cultures with RA promotes neurogenesis (21), whereas the reduction or absence of RA signaling results in proliferative and neurogenic defects in both the adult and developing SVZ (22). Interestingly, deficiency in RA receptors or synthetic enzymes leads to phenotypes similar to those observed in CHARGE syndrome, including eye, heart and brain abnormalities (8,23). Despite having highly similar developmental effects in mammalian tissues, potential interactions between CHD7 and RA signaling pathways have not been explored.
The highly penetrant OB hypoplasia/aplasia and olfaction deficits in CHARGE individuals and Chd7-deficient mice suggest that Chd7 may have a role in the maintenance and differentiation of neural stem cells in the SVZ. Recently, CHD7 was shown to be expressed in the adult SVZ and in the subgranular zone (SGZ) of the dentate gyrus in the hippocampus (24). Additionally, OB and hippocampal neurogenesis were impaired in adult Tlx-CreERT2 Chd7 conditional knockout mice (24). These findings demonstrate that CHD7 activity is required for ongoing neurogenesis in the mature SVZ; however, it is not known whether similar defects are present earlier in development or with heterozygous Chd7 deletion. To investigate these questions, we generated conditional Chd7-deficient adult and neonatal mice and monitored changes in SVZ proliferation, self-renewal and neuronal differentiation. We also examined potential links between CHD7 and RA signaling by modulating RA levels in neurosphere assays and during inner ear development. Our findings demonstrate that proper Chd7 dosage is required for the optimal function of SVZ neural stem cells during development and throughout adulthood, while modulation of RA signaling can attenuate inner ear and neural stem cell defects that arise from Chd7 deficiency.
RESULTS
Chd7-deficient mice display neuronal defects in the adult OB and RMS
We sought to determine the cell type specificity of Chd7 in the mature mouse OB and to clarify whether Chd7 is required for proper formation and maintenance of OB interneurons. In the adult OB, CHD7 co-localized with markers of immature (doublecortin, DCX) and mature interneurons (calbindin, CALB; calretinin, CALR and tyrosine hydroxylase, TH; Supplementary Material, Fig. S1), consistent with recently published observations (24). In the OB GL of Chd7Gt/+ mice, there were 35% fewer CALB+ cells, 48% fewer TH+ cells and no change in the number of CALR+ cells compared with wild type (Fig. 1C–K). In the OB GCL of Chd7Gt/+ mice, there were 15% fewer CALR+ cells compared with wild type (Fig. 1F–H). Our data are consistent with a recent report showing reduced numbers of olfactory interneurons in adult Tlx-CreERT2 Chd7 conditional knockout mice (24) and suggest that haploinsufficiency of Chd7 also disrupts mature OB interneurons.
Figure 1.

Adult heterozygous Chd7 mice have fewer mature OB interneurons. (A) Schematic sagittal view of an adult mouse brain showing the location of coronal sections through the OB in (C, D, F, G, I and J) (dotted line, OB). (B) Schematic of a DAPI-stained coronal section through the OB denoting the location of panels (C, D, F, G, I and J) in the GL and GCL (orange box). Immunolabeling with anti-CALB (C and D), anti-CALR (F and G) and anti-TH (I and J) antibodies in wild-type and Chd7 heterozygous null (Chd7Gt/+) mice shows 35 and 48% reductions in the number of CALB+ and TH+ interneurons in the GL, respectively, and 15% reduction in the number of CALR+ interneurons in the GCL of Chd7Gt/+ mice compared with wild type (E, H and K). Scale bars in (C and D), (F and G), (I and J): 75 μm. Error bars in (E, H and K) indicate standard error of the mean (SEM) (n = 3 per genotype). *P < 0.05, ***P < 0.001 by unpaired Student's t-test. EPL: external plexiform layer
There are no reported cases of humans with homozygous CHD7 mutations, and mice with complete loss of Chd7 die by embryonic day 11 (E11), presumably from cardiovascular defects (9,14). To study the postnatal effects of complete Chd7 deficiency, we used Chd7flox/flox and Ubc-CreERT2 mice, which ubiquitously express a tamoxifen-inducible Cre (25). Five-week-old control (Chd7flox/+ or Chd7flox/flox), Ubc conditional heterozygous (Ubc-CreERT2; Chd7flox/+, Ubc Cond Het) and Ubc conditional knockout (Ubc-CreERT2; Chd7flox/flox, Ubc Cond KO) mice were treated with tamoxifen by daily intraperitoneal injection for 5 days, then allowed to recover for 14 days (Fig. 2A). Immunofluorescence and fluorescence intensity (FI) quantification on coronal cryosections of the OB from control, Ubc Cond Het and Ubc Cond KO adult mice were performed to characterize DCX+ migrating neuroblasts and glial fibrillary acidic protein+ (GFAP) glia in the RMS and OB. In the RMS, there was a reduction in DCX-FI and a corresponding increase in GFAP-FI in Ubc Cond Het and Ubc Cond KO mice compared with controls (Fig. 2C–E, I and J). Interestingly, GFAP-FI was also increased in the GL of the OB in Ubc Cond KO mice compared to controls (Fig. 2F, H and K), whereas Ubc Cond Het mice were unaffected (Fig. 2F, G and K). Thus, adult-onset reduction in Chd7 dosage leads to fewer immature and mature OB neurons and a visible expansion of glia, consistent with ongoing postnatal requirements for Chd7.
Figure 2.

Chd7 deficiency results in fewer immature neurons with glial expansion in the adult OB. (A) Schematic showing the tamoxifen dosing regimen for adult mice. At 5 weeks of age, mice were treated with a single dose of tamoxifen (0.2 mg/gbw) by daily intraperitoneal injection for 5 days, allowed to recover for 14 days then processed for frozen sectioning. (B) Schematic of a DAPI-stained coronal section through the OB denoting the location of panels (C–E) in the RMS (yellow box) and panels (F–H) in the GL (orange box). (C–E) Immunofluorescence on coronal sections through the OB showing reduced DCX+ neuroblasts in the RMS of Ubc Cond Het (D) and Ubc Cond KO (E) mice compared with control (C, quantified in I). Immunofluorescence on coronal sections through the OB showing increased glia in the RMS (C–E) and GL (F–H) of Ubc Cond Het (D and G) and Ubc Cond KO (E and H) mice compared with control (C and F, quantified in J and K). Scale bars in (C–E), (F–H): 75 μm. Error bars in (I–K) indicate SEM (n = 3 per genotype). *P < 0.05, **P < 0.01 by unpaired Student's t-test.
Reduced Chd7 dosage leads to neurogenic and proliferative defects in the adult mouse SVZ
Reduction of neurons in the OB could be due to abnormalities in SVZ-derived neuronal production, migration, differentiation or survival. In the SVZ, CHD7 was present in a subset of neural stem and progenitor cells marked by GFAP and SRY-box2 (SOX2) (Supplementary Material, Fig. S2). In the RMS, CHD7 co-localized with DCX+ neuroblasts (progeny of SOX2+ SVZ cells) and was absent in GFAP+ glia (Supplementary Material, Fig. S2). Therefore, Chd7 is expressed in SVZ neural stem and progenitor cells and continues to be expressed in DCX+ neuroblasts as they migrate through the RMS into the OB where they differentiate into mature interneurons. These data are in agreement with co-localization studies by Feng et al. (24), which showed CHD7 present in a subset of GFAP+ neural stem cells as well as the majority of Mash1+ neural progenitors and DCX+ neuroblasts.
Next, we examined whether loss of Chd7 leads to abnormalities in the SVZ and RMS of adult mice. Hematoxylin and eosin (H&E) staining of SVZ tissue from Ubc Cond KO adult mice revealed ventriculomegaly, consistent with hydrocephalus (Fig. 3A and C), whereas Ubc Cond Het mice were unaffected (Fig. 3B). Immunofluorescence with anti-CHD7 confirmed deletion of CHD7+ cells in the SVZ of Ubc Cond Het and Ubc Cond KO mice compared with controls (Fig. 3D–F). As observed in the OB (Fig. 2J), there was decreased DCX-FI, disorganization of the RMS and increased GFAP-FI in the RMS of Ubc Cond Het and Ubc Cond KO adult mice when compared with controls (Fig. 3G–I). The trend of decreased DCX-FI and increased GFAP-FI were also observed in adult Ubc Cond KO SVZ compared with controls (Supplementary Material, Fig. S3). These observations suggest that Chd7 is required for proper maintenance of the SVZ stem cell niche and the RMS. The number of proliferating Ki67+ cells was reduced by 11% in the SVZ of Ubc Cond KO compared with controls and was unchanged in Ubc Cond Het mice (Fig. 3J–M), consistent with a minor role for Chd7 in the positive regulation of mature SVZ neural stem and progenitor cell proliferation. Our results differ slightly from Feng et al. (24), who showed no abnormalities in proliferating cells in the SVZ of adult Tlx-CreERT2 Chd7 conditional knockout mice. This difference may reflect the timing of tamoxifen-induced deletion, assay sensitivity or the specificity/efficiency of the Cre line.
Figure 3.

Loss of Chd7 leads to ventriculomegaly and mild proliferative defects in the adult mouse SVZ. (A–I) H&E staining and immunofluorescence analysis of adult SVZ in control, Ubc Cond Het and Ubc Cond KO mice. (A–C) Reduced Chd7 dosage results in ventriculomegaly in Ubc Cond KO (C) mice compared with control and Ubc Cond Het (A and B). (D–F) CHD7 immunofluorescence is decreased in Ubc Cond Het (E) and Ubc Cond KO SVZ (F) compared with control (D), confirming gene deletion. (G–I) DCX-positive neuroblasts are reduced in the RMS in Ubc Cond Het (H) and Ubc Cond KO (I) mice compared with control (G). (J–L) and (N–P) Representative coronal sections through the SVZ of adult control (J and N), Ubc Cond Het (K), Ubc Cond KO (L), Nestin Cond Het (O) and Nestin Cond KO (P) mice processed for immunofluorescence with anti-Ki67 antibody and counterstained with DAPI. (M and Q) Quantification showing the percentage of Ki67+ cells relative to the total number of DAPI-stained cells in control, Ubc Cond Het and Ubc Cond KO and control, Nestin Cond Het and Nestin Cond KO. The percentage of Ki67+ cells in the SVZ of Ubc Cond KO and Nestin Cond KO adult mice is decreased by 11 and 15%, respectively, compared with controls. Scale bars in (A–I), (J–L), (N–P): 75 μm. Error bars in (M and Q) indicate SEM (n = 3 per genotype). *P < 0.05, **P < 0.01 by unpaired Student's t-test.
Considering that Ubc-CreERT2 deletes Chd7 in all cells, ventriculomegaly and SVZ deficits may be influenced by secondary, non-cell-autonomous deletion of Chd7 in tissues or cells that interact with the SVZ. To test this, we used Nestin-Cre mice which express a neural stem cell-specific Cre that is active by E11 in the central nervous system (26,27). Ki67+ cells in the SVZ of adult Nestin Cond KO were reduced by 15% compared with controls and were unchanged in Nestin Cond Het (Fig. 3N–Q), consistent with mild, lineage-specific proliferative defects.
Since E11–15 is a period of active cortical neurogenesis (28), Nestin Cond KO mice were tested for defects in forebrain structures, including the cortex (Ctx), corpus callosum (CC) and LVs. H&E staining of forebrains revealed no differences in thickness of the Ctx or CC between adult control and Nestin Cond KO mice (Supplementary Material, Fig. S4). Similar to adult Ubc Cond KO mice, Nestin Cond KO also displayed ventriculomegaly. Thus, Nestin-Cre-mediated loss of Chd7 in the brain results in ventriculomegaly without major abnormalities in the Ctx or CC.
SVZ neural stem and progenitor cells exhibit impaired proliferation, self-renewal and neurogenesis in Chd7-deficient adult mice
To test whether Chd7 is necessary for maintenance of proliferation, self-renewal and multipotency of SVZ neural stem and progenitor cells, we generated and analyzed neurospheres from the SVZ of adult mice (29,30). The neurosphere assay can be used to determine the in vitro potential of a stem cell population and to explore the regulatory mechanisms underlying self-renewal and differentiation (Fig. 4A–C). Ubc Cond Het and Ubc Cond KO primary neurospheres (1°NSs) were smaller than controls, but there were no differences in 1°NS frequency (Fig. 4D and E). To test the self-renewal potential of neural stem and progenitor cells over time, we generated secondary neurospheres (2°NSs) by dissociating and re-plating 1°NSs under non-adherent conditions (Supplementary Material, Fig. S5). Ubc Cond KO 2°NSs were smaller than controls, whereas Ubc Cond Het 2°NS size was unchanged (Fig. 4G). There were fewer Ubc Cond Het and Ubc Cond KO 2°NSs compared with controls (Fig. 4H). Both 1°NS and 2°NS from Nestin Cond Het and Nestin Cond KO mice were reduced in size and frequency in a Chd7 dosage-dependent and cell-autonomous manner (Fig. 4J, K, M and N).
Figure 4.

Neural stem cells derived from adult Chd7 conditional knockout mice exhibit impaired proliferation, self-renewal and neuronal potential. (A–C) Representative images of neurospheres derived from SVZ tissue of adult control (A), Ubc Cond Het (B) and Ubc Cond KO (C) mice. (D–O) show quantification of 1°NS and 2°NS size (μm), 1°NS frequency, 2°NS self-renewal (expressed as the number of 2°NSs formed from a single 1°NS) and percentage of neuronal lineage-positive (TUJ1+) differentiated adherent neurospheres from control, Ubc Cond Het, Ubc Cond KO, Nestin Cond Het and Nestin Cond KO adult mice. Neurospheres derived from Ubc Cond Het and Ubc Cond KO adult mice exhibit Chd7 dosage-dependent reductions in 1°NS size (D), as well as 2°NS size (G), and self-renewal (H) compared with controls. Neurospheres generated from Nestin Cond Het and Nestin Cond KO adult mice show Chd7 dosage-dependent reductions in 1°NS size (J) and frequency (K), and 2°NS size (M) and self-renewal (N), compared with controls. (E) There is no change in Ubc Cond Het or Ubc Cond KO 1°NS frequency compared with controls. (I, L and O) Neuronal potential is reduced by 61% in Ubc Cond KO 2°NSs (I), 42% in Nestin Cond KO 1°NSs (L) and 44% in Nestin Cond KO 2°NSs (O) compared with controls. There is no change in neuronal potential of Ubc Cond Het or Ubc Cond KO 1°NSs compared with controls (F). Scale bars in (A–C): 100 μm. Error bars in (D–O) indicate SEM (n = 3 per genotype). *P < 0.05, **P < 0.01, ***P < 0.001 by unpaired Student's t-test. 1°NS: primary neurosphere, 2°NS: secondary neurosphere.
A hallmark of stem cells derived from neural tissue is their ability to differentiate in vitro into the three major cell types present in the central nervous system: glia (GFAP+), oligodendrocytes (O4+) and neuronal class III beta-tubulin (TUJ1+) (31). TUJ1+ colonies derived from Ubc Cond Het, Ubc Cond KO and Nestin Cond KO adult 2°NSs, and Nestin Cond KO 1°NSs, were significantly reduced compared with controls (Fig. 4I, L and O). Alternatively, neuronal potentials of Ubc Cond Het, Ubc Cond KO and Nestin Cond Het 1°NSs were unaffected compared with controls (Fig. 4F and L). Oligodendrocyte and glial potentials were normal for all Ubc and Nestin Cond Het and Cond KO neurospheres (data not shown). Due to the plating of whole, undissociated neurospheres for differentiation, we were unable to quantify specific lineages beyond the presence or absence in the culture dish; thus, experimentally induced increases in glial cells would not be detected in this assay. We conclude that Chd7-deficient neurospheres exhibit cell-autonomous reductions in neuronal potential, with preserved or potentially enhanced differentiation into both oligodendrocytes and astrocytes.
Perinatal mice with reduced Chd7 dosage have impaired proliferation, self-renewal and neurogenesis of SVZ neural stem and progenitor cells
In the rodent SVZ, OB neurogenesis is a lifelong process that begins during late embryogenesis and continually populates the OB with new interneurons (16,17). Considering that Nestin-Cre is active in utero before E11, defects in SVZ neural stem cell proliferation in Nestin-Cre Chd7 CKO mice could be secondary to early Chd7 deletion. To determine whether late embryonic Chd7 deletion impacts perinatal SVZ function, we induced deletion of Chd7 with Ubc-CreERT2 and Nestin-CreERT2 (32) at E15.5 (Fig. 5A), when mouse SVZ neural stem cells begin the transition from cortical neurogenesis to OB neurogenesis (33). In the SVZ of postnatal day 1 (P1) Ubc Cond KO and Nestin-ERT2 Cond KO mice, there were 38 and 37% fewer Ki67+ cells than controls, whereas Ki67+ cells in Ubc Cond Het and Nestin-ERT2 Cond Het mice were unchanged (Fig. 5B–I), consistent with impaired proliferation. Additionally, there was no change in apoptosis frequency in the SVZ of Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO perinatal mice compared with controls (Fig. 5K–N).
Figure 5.

P1 Chd7 conditional knockout mice display reduced SVZ proliferation. (A) Schematic showing the tamoxifen dosing regimen where pregnant females received a single intraperitoneal injection of tamoxifen (0.2 mg/gbw) at E15.5. (B–D), (F–H) and (K–M) Representative coronal sections showing immunofluorescence for the proliferative marker Ki67 or apoptosis marker Casp3 with DAPI counterstain in the SVZ of P1 control (B, F and J), Ubc Cond Het (C), Ubc Cond KO (D), Nestin-ERT2 Cond Het (G and K) and Nestin-ERT2 Cond KO (H and L) mice. (E and I) Quantification showing the percentage of Ki67+ cells relative to the total number of DAPI-stained cells in control, Ubc Cond Het and Ubc Cond KO (E), and Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO (I) P1 SVZ. The percentage of Ki67+ cells are reduced by 37 and 38% in the SVZ of Ubc Cond KO and Nestin-ERT2 Cond KO P1 mice, respectively, compared with controls. (J) Quantitative PCR shows that 45S pre-rRNA is decreased by 26% in Ubc Cond KO SVZ compared with controls. (K–N) There is no change in the number of Casp3+ cells per section in Nestin-ERT2 Cond Het (L) and Nestin-ERT2 Cond KO (M) P1 SVZ compared with control (K). Scale bars in (B–D), (F–H): 75 μm, (K–M): 50 μm. Error bars in (E, I, J and N) indicate SEM (n = 3 per genotype). *P < 0.05, ***P < 0.001 by unpaired Student's t-test.
To explore potential mechanisms for the moderate decrease in SVZ proliferation with loss of Chd7, we performed quantitative real-time polymerase chain reaction (qRT-PCR) for genes involved in proliferation and cell cycle regulation on RNA derived from the SVZ of control and Ubc Cond KO perinatal mice (Supplementary Material, Fig. S6). There were no significant changes in expression of the positive cell cycle regulators Ccnd1 (cyclinD1), Ccnd2 (cyclinD2) and Cdk4, or the negative regulators Cdkn1a (p21Cip1) and Cdkn1b (p27Kip1) (34). Previous studies in mouse embryonic stem cells have demonstrated that CHD7 directly binds to precursor ribosomal RNA (rRNA) genes and regulates their transcription, resulting in global reductions in cell proliferation and protein levels upon loss of Chd7 (35). We observed a 26% reduction in 45S pre-rRNA transcripts in RNA isolated from Ubc Cond KO SVZ compared with controls (Fig. 5J). This is consistent with the 20–40% decrease in rRNA in Chd7-deficient mouse embryonic stem cells, inner ears and eyes reported by Zentner et al. (35). These data suggest that CHD7 effects on SVZ cell proliferation may be caused by global reductions in protein translation and rRNA. Importantly, our studies do not rule out the possibility of smaller effects of CHD7 on other downstream genes that, in combination, may exert robust effects on the cell cycle.
We then tested the in vitro proliferation, self-renewal and differentiation capacity of neonatal Chd7-deficient SVZ neural stem and progenitor cells by generating 1°NSs from control, Ubc Cond Het, Ubc Cond KO, Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO P1 mice (Supplementary Material, Figs S7 and S8). As in adult mice, 1°NS and 2°NSs from Ubc Cond KO and Nestin-ERT2 Cond KO mice were reduced in size, frequency, self-renewal and neuronal potential compared with controls. Neurospheres from Ubc Cond Het and Nestin-ERT2 Cond Het mice exhibited intermediate phenotypes, with milder reductions compared with Cond KO mice. Taken together, the in vivo proliferation and in vitro neurosphere data from neonatal mice indicate that Chd7 deficiency contributes to altered SVZ neural stem and progenitor cell function as early as P1.
Modulation of RA signaling attenuates defects in Chd7-deficient neurospheres and semicircular canals
RA signaling is necessary for the proper development and maintenance of the forebrain and OBs (36). RA treatment of neurospheres promotes self-renewal and neuronal differentiation (21), while inhibition of RA signaling leads to fewer, but larger neurospheres (22). We hypothesized that alteration of RA signaling by addition of RA or citral (inhibitor of RA synthesis) (37) in SVZ neurosphere cultures may influence the size, self-renewal and/or neuronal differentiation of Chd7-deficient neurospheres. The treatment of 2°NSs with RA produced smaller and more abundant neurospheres from control, Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO mice (Fig. 6A and B). Interestingly, RA increased neuronal potential by 46% in Nestin-ERT2 Cond KO 2°NSs, suggesting partial rescue of the observed neuronal differentiation defect (Fig. 6E).
Figure 6.

Modified RA signaling influences defects in Chd7-deficient neurospheres and semicircular canal development. (A–F) 2°NSs were derived from P1 control, Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO mice then treated with varying concentrations of all-trans RA (A, B and E) or the RA signaling inhibitor citral (C, D and F), and assayed for changes in size, self-renewal and neuronal potential. (A and B) Treatment with RA (0.1 or 1 μM) results in smaller neurospheres (A) and increased self-renewal (B) for all three genotypes. (C and D) Treatment with citral (1 μM or 10 μM) in non-adherent neurosphere cultures causes increased Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO neurosphere size (C), and reduced self-renewal (D) for all three genotypes. (E) Treatment of differentiating, adherent Nestin-ERT2 Cond KO neurospheres with 1 μM RA resulted in a 33% increase (from 39 to 72%) in the proportion of Nestin-ERT2 Cond KO TUJ1+ colonies compared with control TUJ1+ colonies. (F) Neuronal potentials of Nestin-ERT2 Cond KO differentiating neurospheres were unaffected by citral treatment. (G–L) Pregnant females were untreated or treated with either RA by oral gavage or citral by intraperitoneal injection at E7.5, then embryos were extracted at E14.5 and processed for inner ear paint filling. (G–I) Wild-type embryos are unaffected by treatment with either RA or citral. Chd7Gt/+ embryos consistently display LC truncation (denoted by white asterisk) in untreated conditions (J) which worsen with RA treatment (K), while treatment with citral results in rescue of the LC truncation in 29% (8 of 28 ears) of mutant inner ears (denoted by white arrowhead, L). Error bars in (A–F) indicate SEM (n = 3 per genotype). ED: endolymphatic duct; AC: anterior semicircular canal; SAC: saccule; COC: cochlea.
Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO 2°NSs increased in size by 19 and 39%, respectively, with 10 µM citral treatment, whereas the control was unchanged (Fig. 6C). Self-renewal of Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO 2°NSs were reduced by 15 and 27%, respectively, with 10 µM citral exposure, whereas those of control neurospheres was decreased by 58% (Fig. 6D). These data indicate that reduced Chd7 dosage may confer protection against the reduction in self-renewal that occurs with citral treatment. Citral treatment did not alter the neuronal potential of control, Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO 2°NSs (Fig. 6F). We conclude that decreased RA signaling does not further inhibit neuronal differentiation in Chd7-deficient neurospheres. Interestingly, glial and oligodendrocyte potential were unaffected for all genotypes regardless of RA or citral exposure (data not shown). Our results provide evidence that increased RA signaling partially attenuates neuronal differentiation defects in Chd7-deficient neurospheres, without affecting size or self-renewal. Conversely, inhibition of RA signaling with citral (i) moderately corrects size deficits in Chd7 Cond Het and Cond KO neurospheres, (ii) partially protects against further reduction in self-renewal capacity and (iii) has no effect on neuronal differentiation.
To test the in vivo effects of RA modulation in Chd7-deficient mice, we chose to investigate the inner ear, an organ that displays well characterized, highly penetrant defects with loss of Chd7 and is amenable to pharmacological manipulation (10,11). Inner ears from Chd7Gt/+ mice displayed truncation or agenesis of the E14.5 lateral semicircular canal (LC; Fig. 6G and J), and worsened semicircular canal defects after in utero exposure to RA (Fig. 6H and K). Interestingly, intrauterine citral treatment had no effect on wild-type ears (18 of 18) and on most Chd7Gt/+ inner ears (20 of 28), whereas in roughly one-third of Chd7Gt/+ inner ears (8 of 28), citral fully prevented the semicircular canal defects (Fig. 6L, Supplementary Material, Fig. S9). This rescue or prevention of inner ear defects in Chd7-deficient ears with modulation of RA levels suggests a mechanistic interaction between CHD7 function and RA signaling.
Chd7 directly regulates RA receptor and neuronal differentiation genes in neural stem and progenitor cells
Given that CHD7 regulates transcription through chromatin remodeling (10,38,39), we predicted that loss of Chd7 may disrupt expression of genes coding for transcription factors and RA signaling mediators involved in neuronal differentiation. To test this, we selected three genes (Rarb, Rxrg and Neurod1) known to be involved in either SVZ neurogenesis or neuronal differentiation by qRT-PCR. Rarb and Rxrg encode receptors that facilitate RA signaling (40) and Neurod1 encodes a basic helix-loop-helix transcription factor that promotes neuronal differentiation (41). In Ubc Cond KO P1 SVZ, expression of Rarb, Rxrg and Neurod1 were reduced by 58, 57, and 55%, respectively, relative to controls (Fig. 7A). Consistent with prior studies showing that CHD7 and SOX2 interact in neural stem cells (42), Sox2 expression was unchanged in the SVZ of P1 Ubc Cond KO mice, relative to control. Therefore, loss of Chd7 results in down-regulation of key RA and neuronal differentiation genes, but does not affect global expression of neural stem cell-specific genes.
Figure 7.

CHD7 binds to the promoters of RA receptor and pro-neuronal transcription factor genes and regulates their expression. (A) qRT-PCR analysis of total RNA from P1 control and Ubc Cond KO microdissected SVZ shows expression levels of Rarb, Rxrg, Neurod1 and Sox2. Threshold cycles are normalized to Gapdh. In Ubc Cond KO P1 SVZ, expression levels of Rarb, Rxrg and Neurod1 are decreased by 55–58% compared with controls. Sox2 expression is unchanged in Ubc Cond KO SVZ compared with controls. Black dashed line denotes control gene expression levels. (B–D) By ChIP-qPCR, CHD7 is enriched relative to IgG at conserved regions 1, 2, 4, 6 and 8 for Rarb (B), conserved regions 1, 3, 4 and 6 for Rxrg (C) and conserved regions 1–4 for Neurod1 (D). Non-conserved regions are not enriched for CHD7 relative to IgG. (E) Proposed model showing CHD7 function in neural progenitors, including regulation of Rarb, Rxrg and pro-neuronal gene transcription and promotion of differentiation into neuroblasts. Chd7 deficiency results in decreased Rarb, Rxrg and pro-neuronal Neurod1 gene transcription leading to fewer neuroblasts and a shift toward glial fates. Error bars in (A–D) indicate SEM. *P < 0.05.
To determine whether CHD7 participates in a complex that binds directly to Rarb, Rxrg and Neurod1 promoter regions, we performed chromatin immunoprecipitation (ChIP) on chromatin isolated from P19 mouse embryonal carcinoma cells, followed by qRT-PCR at evolutionarily conserved promoter regions. P19 cells are known to form neural stem cell-like neurospheres and differentiate in response to RA (43,44). We observed enrichment for CHD7 at a subset of conserved promoter regions for Rarb, Rxrg and Neurod1 (Fig. 7B–D). Additionally, CHD7 was not bound to non-conserved regions in these genes. Taken together, our findings provide new insights into interactions between RA signaling pathways and CHD7 in neural stem cells and the inner ear.
DISCUSSION
Here, we show that Chd7 is expressed throughout the SVZ, RMS and OB in adult mice. In both adult and neonatal mice, conditional loss of Chd7 leads to proliferative, self-renewal and neurogenic deficits both in vivo and in vitro with preserved or enhanced glial potential. Modulation of RA signaling by treatment with RA increases neuronal potential of Chd7-deficient neurospheres, whereas inhibition of RA signaling attenuates size and self-renewal deficits. Additionally, inhibition of RA signaling in vivo leads to the correction of semicircular canal defects in the inner ears of Chd7-deficient mouse embryos. Expression of key RA receptor genes (Rarb and Rxrg) and at least one neurogenic transcription factor gene (Neurod1) are significantly reduced in the Chd7 knockout SVZ; moreover, these genes are directly bound by CHD7-containing complexes in P19 neurospheres. Taken together, these findings indicate that Chd7 and RA signaling promote proliferation, self-renewal and neurogenesis during late embryogenesis and into adulthood, thereby suggesting a therapeutic avenue for intervention during pregnancy.
We propose a model whereby CHD7 regulates RA signaling and pro-neuronal genes to facilitate the transition from neural progenitor cell to neuroblast (Fig. 7E). Chd7 deficiency also leads to decreased progenitor cell differentiation into neuroblasts and a shift in cell fate toward the glial lineage. Interestingly, in vitro modification of RA signaling partially compensates for loss of Chd7, but does not fully correct the observed deficits in size, self-renewal and neuronal differentiation. Similar to RA, CHD7 has been shown to participate in several signaling pathways, including Wnt (45), BMP (12), fibroblast growth factor (FGF) (10,12,38) and Notch (12). Loss of Chd7 in the SVZ and inner ear may cause deficits in these pathways, potentially explaining the partial rescue of neurosphere and inner ear defects by RA modulators.
Disrupted RA signaling leads to a variety of organ defects, many of which also occur with Chd7 deficiency including abnormalities of the eye, heart, urogenital system, brain and skeleton (8,23). The treatment of Nestin-lineage Chd7-deficient neurospheres with RA increased neuronal potential, whereas citral treatment restored neurosphere size to control levels. Two RA receptor genes, Rarb and Rxrg, were also down-regulated in the Ubc Cond KO SVZ. Interestingly, CHD7 was present in a complex that was directly bound to the highly conserved promoter regions of Rarb and Rxrg in P19 neurospheres. In the SVZ, reduced or absent RA signaling results in proliferative and neurogenic defects both in vivo and in vitro during development and in the adult, whereas increased RA leads to increased neurogenesis (21,22). Consistent with these studies, our data imply a direct relationship between CHD7 and RA signaling that may contribute to the defects associated with CHARGE syndrome.
Our findings also imply that decreased numbers of OB interneurons derived from the SVZ may contribute to OB hypoplasia in Chd7-deficient mice, in addition to the influence of reduced olfactory sensory neurons, as previously reported (4). This result is in agreement with recently published data showing reduced OB interneurons in adult mice with Tlx-CreERT2-mediated conditional deletion of Chd7 (24). Interestingly, the magnitude of OB interneuron reductions was similar (40–60%) in our study and that of Feng et al., suggesting that reduction in Chd7 gene function <50% may not lead to further impairments in OB development. Additionally, we observed ventriculomegaly in Chd7-deficient adult mice, consistent with a recent study of 40 CHD7 mutation positive human fetuses that demonstrated a variety of brain malformations including arrhinencephaly and ventriculomegaly (6). Similar to our data, disruption of SVZ neural stem cell function results in ventriculomegaly in mouse models of Bardet-Biedl syndrome and Sox2 deficiency (46,47). CHD7 has been shown to regulate neurogenesis in the adult dentate gyrus of the hippocampus (24) and is also implicated in autism spectrum disorder (48). Therefore, our findings add to a growing body of work supporting a specific role for CHD7 function in SVZ, SGZ and olfactory neural stem cells, and perhaps in other regions of the nervous system that are affected in CHARGE. Notably, CHARGE individuals can also present with hindbrain and cerebellar abnormalities (6,49–51), and future studies should help clarify whether CHD7 also regulates neural stem and progenitor cells in these regions. Our work opens new avenues for investigation of CHD7 activity in stem cell populations affected by CHARGE syndrome and should help guide future work toward the development of diagnostic and therapeutic treatments.
MATERIALS AND METHODS
Mouse strains, breeding and genotyping
Chd7Gt/+ and Chd7flox/flox mice were maintained as previously described (9,10). Ubc-CreERT2, Nestin-Cre and Nestin-CreERT2 mice were maintained on a C57BL/6 background and mated with Chd7flox/flox mice to generate Cre;Chd7flox/+ mice. Cre;Chd7flox/+mice were crossed with Chd7flox/flox to produce control (Chd7flox/+ and Chd7flox/flox), Chd7 conditional heterozygous (Cre;Chd7flox/+) and conditional knockout (Cre;Chd7flox/flox) mice. Genomic DNA was isolated from tail and brain tissue and PCR genotyped as previously described (10). All procedures were approved by The University of Michigan University Committee on the Use and Care of Animals (UCUCA).
Tamoxifen administration
Tamoxifen (Sigma-Aldritch, St Louis, MO, USA) was dissolved in sterile corn oil at a concentration of 10 mg/ml. For adult studies, tamoxifen (0.2 mg/gbw) was administered by intraperitoneal injection to 5-week-old littermate mice once daily for 5 days. Mice were allowed to recover for 14 days, then euthanized and processed for further analysis. For perinatal studies, a single dose of tamoxifen (0.2 mg/gbw) was administered by intraperitoneal injection to pregnant females at E15.5.
Histology and embedding
P1 mice were decapitated, skin was dissected away and heads were placed in 4% paraformaldehyde for 4 h, then cryoprotected in 30% sucrose, embedded in OCT (Tissue Tek, Torrance, CA, USA), frozen and sectioned at 14 µm. Adult 7-week-old mice underwent terminal cardiac perfusion with 4% paraformaldehyde, then heads were removed, post-fixed with 4% paraformaldehyde, cryoprotected in 30% sucrose and embedded in OCT (Tissue Tek), then frozen and sectioned at 14 µm. For histological analyses, sections were stained with H&E (Sigma), imaged with bright field microscopy on a Leica upright DMRB microscope (Leica Camera AG, Solms, Germany) and processed in Photoshop CS5.1 (Adobe, San Jose, CA, USA).
Immunofluorescence
Cryosections from 7-week-old and P1 mice (n = 3 per genotype) were processed for immunofluorescence with antibodies against CHD7 (1 : 7500; Cell Signaling Technology, Beverly, MA, USA), Ki67 (1 : 200; Vector Laboratories, Burlingame, CA, USA), TH (1 : 150; Pel-Freeze, Rogers, AR, USA), cleaved caspase 3 (Casp3; 1 : 800, Cell Signaling Technology), CALB (1 : 5000; Sigma), CALR (1 : 250; EMD Millipore, Billerica, MA, USA), GFAP (1 : 500; Sigma), O4 (1 : 800; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA), Tuj1 (1 : 400; Covance, Princeton, NJ, USA), DCX (1 : 4000; a gift from Jack M. Parent) and SOX2 (1 : 250; EMD Millipore). All secondary antibodies were used at 1 : 200 with Alexa Fluor 488, Alexa Fluor 555 or biotinylated secondary antibodies (Vector Laboratories) conjugated with streptavidin–horseradish peroxidase and Alexa Fluor 488 Tyramide Signal Amplification or Alexa Fluor 555 Tyramide Signal Amplification (Life Technologies, Carlsbad, CA, USA). Nuclear staining was performed with 4′,6-diamidino-2-phenylindole (DAPI, 1 : 100; Life Technologies). Images were produced by single channel fluorescence microscopy on a Leica upright DMRB microscope and processed in Photoshop CS5.1.
OB interneuron quantification
Seven-week-old Chd7+/+ and Chd7Gt/+ littermate mice (n = 3 per genotype) were processed for immunofluorescence with antibodies against TH, CALR and CALB. Images were captured by single channel fluorescence microscopy on a Leica upright DMRB microscope and processed in Adobe Photoshop CS5.1. For quantification of OB interneurons, 25 images using the 10× objective lens were taken of nine OB sections per genotype and analyzed using the cell counter function of Image J (National Institutes of Health, Bethesda, MD, USA) by sampling 10 squares (40 µm × 40 µm) in the GCL and GL.
FI measurements
Tamoxifen-treated 7-week-old mice (n = 3 per genotype) were processed for immunofluorescence with antibodies against GFAP and DCX. Images were captured at the same exposure for each antibody by single channel fluorescence microscopy on a Leica upright DMRB microscope. FI was measured on 25–45 images using the 20× objective lens from nine OB, RMS and SVZ sections per sample, then processed in ImageJ by using the freehand selection and measurement tools. Experimental fluorescence intensities were reported as FI units per μm2.
SVZ proliferation and apoptosis studies
Seven-week-old and P1 mice (n = 3 per genotype) were processed for immunofluorescence with antibody against Ki67 and Casp3. Nuclei were stained with DAPI. Images were captured by single channel fluorescence microscopy on a Leica upright DMRB microscope and processed in Adobe Photoshop CS5.1. For quantification of Ki67+ cells, 36 images using the 20× objective lens were taken of nine SVZ sections per sample, and the number of Ki67+ cells and total number of cells present in the SVZ were counted using the cell counter function of Image J. For quantification of Casp3+ cells, 27 SVZ sections per genotype were scored for the total number of Casp3+ cells per section.
Cortex and corpus callosum measurements
Seven-week-old control, Nestin Cond Het and Nestin Cond KO mice (n = 3 per genotype) were perfusion fixed, and brains were sectioned and processed for H&E staining as above. Cortical thickness measurements were made on 27 SVZ sections per genotype by drawing a line perpendicular to, and extending from, the lateral ventricle to the outer edge of the Ctx. For CC thickness, measurements were made on 27 SVZ sections per genotype by drawing a line at the midline transversely through the CC. Both measurements were performed using the Leica Applications Suite—Advanced Fluorescence software (Leica Camera AG) on 10× images.
Neurosphere cultures
Seven-week-old adult and P1 mice (n = 3 per genotype) were euthanized, whole brain was dissected and coronal sections were cut between the pre-optic area and caudal OB. The lateral wall of the lateral ventricle was microdissected, dissociated in trypsin/EDTA (Life Technologies) and DNase1, filtered through a 0.45-µm mesh (BD Biosciences, East Rutherford, NJ, USA) and counted on a hemacytometer. For 1°NS formation assays, 1000 cells were plated on ultra-low-binding six-well plates (Corning, Corning, NY, USA) under self-renewal conditions (media with epidermal growth factor [EGF] and FGF), incubated at 37°C with 6.5% CO2 for 8 days then measured and imaged using an Olympus IX81 motorized inverted microscope (Olympus, Tokyo, Japan). For 1°NS differentiation, single neurospheres were plated under adherent conditions with FGF and without EGF [24-well tissue culture-treated plates (Corning) with poly-d-lysine and human fibronectin (Biomedical Technologies, Stoughton, MA, USA)], then incubated at 37°C with 6.5% CO2 for 12 days. 2°NSs were generated by dissociating single 1°NSs and re-plating them under self-renewing conditions. Measurement, counting, imaging and differentiation of 2°NSs were performed as described for 1°NSs. Neurosphere diameter (n = 150 per genotype) was measured using the arbitrary line tool in the Olympus cellSens Dimension software package. Neurosphere frequency was determined by counting the total number of neurospheres >50 µm in diameter in each well of a six-well plate and dividing by the total number of cells plated per well. Statistical significance was determined by unpaired Student's t-test.
Neurosphere differentiation, staining and scoring
1°NS and 2°NSs were differentiated as described above, then treated with 1 : 800 anti-O4 antibody for 1 h and fixed with ice cold 95% ethanol/5% glacial acetic acid at −20°C for 20 min. Fixed neurospheres were blocked for 1 h at room temperature with light rocking with blocking buffer [0.05% octylphenoxypolyethoxyethanol (NP-40, Sigma), 2 mg/ml bovine serum albumin (Sigma) in phosphate-buffered saline]. Alexa Fluor 647-conjugated goat anti-mouse IgM (1 : 500, Life Technologies) diluted in blocking buffer was added to each well and incubated at room temperature for 1 h with light rocking. Each well was washed three times with blocking buffer, then a cocktail of 1 : 500 anti-Tuj1 and 1 : 200 anti-GFAP primary antibodies was added to each well and incubated at room temperature for 1 h with light rocking. Each well was washed three times with blocking buffer, then incubated in a cocktail of 1 : 500 Alexa Fluor 555 goat anti-mouse IgG2a and 1 : 500 Alexa Fluor 488 goat anti-mouse IgG1 at room temperature for 1 h with light rocking. Each well was washed three times and nuclei were stained with 1 : 1000 DAPI for 15 min. Stained colonies were imaged with an Olympus IX81 motorized inverted microscope and scored for the presence or absence of cell type-specific immunofluorescence (n = 36 neurospheres per genotype). Statistical significance was determined by unpaired Student's t-test.
RA and citral treatment of neurosphere cultures
Neurosphere assays were performed as above on control, Nestin-ERT2 Cond Het and Nestin-ERT2 Cond KO P1 mice. 2°NSs (n = 3 per genotype) were plated in ultra-low-binding six-well plates in self-renewal medium or medium supplemented with 0.1 μM RA (Sigma), 1 μM RA, 1 μM citral (Sigma) or 10 μM citral (n = 18 wells per genotype). For differentiation, untreated 2°NSs from each genotype were plated in 24-well plates in differentiation medium or medium supplemented with 1 μM RA, 1 μM citral or 10 μM citral (n = 18 neurospheres per condition), and scored for differentiation as above.
Embryonic RA and citral treatment and inner ear paint fills
Timed pregnancies were established between Chd7Gt/+ males and females with the morning of the plug scored as E0.5. At E7.5, pregnant females were treated with either RA (Sigma; in corn oil at 5 mg/kg by oral gavage), or citral (Sigma; in corn oil at 3 μM/g body weight by intraperitoneal injection). At E14.5, Chd7+/+ (n = 12 embryos; 24 ears) and Chd7Gt/+ (n = 18 embryos; 36 ears) embryos were dissected and processed for inner ear paint filling. Briefly, embryos were washed in PBS, fixed in Bodian's fixative, cleared in methyl salicylate, heads were bisected and ears were injected with a solution with 3% Wite-Out (BIC, Shelton, CT), diluted in methyl salicylate (9).
Gene expression analysis
Subventricular zones from littermate control and Ubc Cond KO P1 mice (n = 4 per genotype) were microdissected, and RNA was isolated using the RNAqueous-Micro RNA Isolation Kit (Ambion, Austin, TX, USA). cDNA was generated using the Superscript First-Strand cDNA Synthesis system with random hexamers (Life Technologies). Relative gene expression levels were assayed using TaqMan Gene Expression Master Mix and TaqMan probes (Applied Biosystems, Foster City, CA, USA) for Gapdh, Ccnd1, Ccnd2, Cdk4, Cdkn1a, Cdkn1b, Rarb, Rxrg, Neurod1 and Sox2 or SYBR Green Master Mix (Applied Biosystems) for 45S pre-rRNA (35). Each sample was run in triplicate using an Applied Biosystems StepOne Plus Real-Time qPCR System. The gene expression level of Gapdh was used as an internal positive control. The difference in threshold cycle (CT) between the assayed gene and Gapdh for any given sample was defined as the change in threshold cycle (ΔCT). The difference in ΔCT between two samples was defined as ΔΔCT, which represents a relative difference in expression of the assayed gene. The fold change of the assayed gene relative to Gapdh was defined as 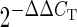 . Unpaired Student's t-test was performed on Gapdh-normalized ΔCT values for each sample to determine statistical significance.
. Unpaired Student's t-test was performed on Gapdh-normalized ΔCT values for each sample to determine statistical significance.
P19 neurosphere cultures
P19 embryonal carcinoma cells were passaged in minimum essential medium alpha (MEMα) (Life Technologies) + 10% fetal bovine serum (FBS) (Life Technologies) + 1× penicillin/streptomycin (Life Technologies) and gently treated with 0.25% trypsin. Cells were plated on 0.7% agarose in bacterial culture plates in MEMα + 7.5% calf serum + 2.5% FBS + 1 μM RA + 1× penicillin/streptomycin. Neurospheres were confirmed to form 2 days after plating by bright field microscopy, and medium was changed 2 and 4 days after initial plating. Six days after plating, neurospheres were collected by gentle centrifugation, then fixed for 20 min with formaldehyde and processed for ChIP.
Chromatin immunoprecipitation and quantitative real-time PCR
ChIP with anti-CHD7 antibody (Cell Signaling) and normal rabbit IgG (EMD Millipore) was performed using the EZ ChIP Chromatin Immunoprecipitation Kit (EMD Millipore), following the manufacturer's instructions on crosslinked P19 neurospheres. Conserved and non-conserved regions within 10 kb of the transcription start site for each of Rarb, Rxrg and Neurod1 were identified using the 60 vertebrate PhastCons track in the UCSC Genome Browser (build GRCm38/mm10) (52). A conserved region was defined by a series of adjacent logarithm of odds score hits conserved in ≥80% of the vertebrates. Primers were designed against nine conserved regions and one non-conserved region for each gene and assayed for a single melting point by qRT-PCR melt curve analysis using SYBR Green reagents (Applied Biosystems). Primer pairs that demonstrated non-specific amplification were removed prior to further analysis. Nine replicates of qRT-PCR were performed on the input, IgG and anti-CHD7 samples for each primer pair. Percent input was calculated for both the IgG and anti-Chd7 samples across each primer pair.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (T32 DC00011 and F31 DC 013227-01 to J.A.M., F31 DC010955-01 to W.S.L., R01 CA160356 and R01HD056369 to P.C.S., P30-DC05188 to Y.R. and D.M.M., and R01 DC009410 to Y.R. and D.M.M.), University of Michigan Rackham Graduate School and Department of Biological Chemistry grants to J.A.M., CHARGE Syndrome Foundation grants to E.A.H. and P.C.S., Hearing Health Foundation grant to E.A.H., the Berte and Alan Hirschfield Foundation and the R. Jamison and Betty Williams Professorship to Y.R., and University of Michigan Donita B. Sullivan MD Research Professorship Funds to D.M.M.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Jack M. Parent at the University of Michigan for providing the anti-DCX antibody and for critically reviewing the manuscript. W.S.L. is currently at the Department of Developmental Neurobiology, St Jude's Children's Research Hospital, Memphis, TN 38105, USA. E.A.H. is currently at the Centre for Neuroregeneration, University of Edinburgh, Edinburgh, Scotland, UK.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Pagon R.A., Graham J.M., Jr, Zonana J., Yong S.L. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr. 1981;99:223–227. doi: 10.1016/s0022-3476(81)80454-4. [DOI] [PubMed] [Google Scholar]
- 2.Issekutz K.A., Graham J.M., Jr, Prasad C., Smith I.M., Blake K.D. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am. J. Med. Genet. A. 2005;133A:309–317. doi: 10.1002/ajmg.a.30560. [DOI] [PubMed] [Google Scholar]
- 3.Pinto G., Abadie V., Mesnage R., Blustajn J., Cabrol S., Amiel J., Hertz-Pannier L., Bertrand A.M., Lyonnet S., Rappaport R., et al. CHARGE Syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J. Clin. Endocrinol. Metab. 2005;90:5621–5626. doi: 10.1210/jc.2004-2474. [DOI] [PubMed] [Google Scholar]
- 4.Layman W.S., McEwen D.P., Beyer L.A., Lalani S.R., Fernbach S.D., Oh E., Swaroop A., Hegg C.C., Raphael Y., Martens J.R., et al. Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum. Mol. Genet. 2009;18:1909–1923. doi: 10.1093/hmg/ddp112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergman J.E., Janssen N., Hoefsloot L.H., Jongmans M.C., Hofstra R.M., van Ravenswaaij-Arts C.M. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J. Med. Genet. 2011;48:334–342. doi: 10.1136/jmg.2010.087106. [DOI] [PubMed] [Google Scholar]
- 6.Legendre M., Gonzales M., Goudefroye G., Bilan F., Parisot P., Perez M.J., Bonniere M., Bessieres B., Martinovic J., Delezoide A.L., et al. Antenatal spectrum of CHARGE syndrome in 40 fetuses with CHD7 mutations. J. Med. Genet. 2012;49:698–707. doi: 10.1136/jmedgenet-2012-100926. [DOI] [PubMed] [Google Scholar]
- 7.Vissers L.E., van Ravenswaaij C.M., Admiraal R., Hurst J.A., de Vries B.B., Janssen I.M., van der Vliet W.A., Huys E.H., de Jong P.J., Hamel B.C., et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 8.Janssen N., Bergman J.E., Swertz M.A., Tranebjaerg L., Lodahl M., Schoots J., Hofstra R.M., van Ravenswaaij-Arts C.M., Hoefsloot L.H. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum. Mutat. 2012;33:1149–1160. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
- 9.Hurd E.A., Capers P.L., Blauwkamp M.N., Adams M.E., Raphael Y., Poucher H.K., Martin D.M. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm. Genome. 2007;18:94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 10.Hurd E.A., Poucher H.K., Cheng K., Raphael Y., Martin D.M. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development. 2010;137:3139–3150. doi: 10.1242/dev.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurd E.A., Adams M.E., Layman W.S., Swiderski D.L., Beyer L.A., Halsey K.E., Benson J.M., Gong T.W., Dolan D.F., Raphael Y., et al. Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear. Res. 2011;282:184–195. doi: 10.1016/j.heares.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurd E.A., Micucci J.A., Reamer E.N., Martin D.M. Delayed fusion and altered gene expression contribute to semicircular canal defects in Chd7 deficient mice. Mech. Dev. 2012;129:308–323. doi: 10.1016/j.mod.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergman J.E., Bosman E.A., van Ravenswaaij-Arts C.M., Steel K.P. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. Eur. J. Hum. Genet. 2010;18:171–177. doi: 10.1038/ejhg.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosman E.A., Penn A.C., Ambrose J.C., Kettleborough R., Stemple D.L., Steel K.P. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 2005;14:3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- 15.Lledo P.M., Merkle F.T., Alvarez-Buylla A. Origin and function of olfactory bulb interneuron diversity. Trends Neurosci. 2008;31:392–400. doi: 10.1016/j.tins.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luskin M.B. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11:173–189. doi: 10.1016/0896-6273(93)90281-u. [DOI] [PubMed] [Google Scholar]
- 17.Wichterle H., Turnbull D.H., Nery S., Fishell G., Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- 18.Anchan R.M., Drake D.P., Haines C.F., Gerwe E.A., LaMantia A.S. Disruption of local retinoid-mediated gene expression accompanies abnormal development in the mammalian olfactory pathway. J. Comp. Neurol. 1997;379:171–184. [PubMed] [Google Scholar]
- 19.Thompson Haskell G., Maynard T.M., Shatzmiller R.A., Lamantia A.S. Retinoic acid signaling at sites of plasticity in the mature central nervous system. J. Comp. Neurol. 2002;452:228–241. doi: 10.1002/cne.10369. [DOI] [PubMed] [Google Scholar]
- 20.Romand R., Krezel W., Beraneck M., Cammas L., Fraulob V., Messaddeq N., Kessler P., Hashino E., Dolle P. Retinoic acid deficiency impairs the vestibular function. J. Neurosci. 2013;33:5856–5866. doi: 10.1523/JNEUROSCI.4618-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang T.W., Zhang H., Parent J.M. Retinoic acid regulates postnatal neurogenesis in the murine subventricular zone-olfactory bulb pathway. Development. 2005;132:2721–2732. doi: 10.1242/dev.01867. [DOI] [PubMed] [Google Scholar]
- 22.Haskell G.T., LaMantia A.S. Retinoic acid signaling identifies a distinct precursor population in the developing and adult forebrain. J. Neurosci. 2005;25:7636–7647. doi: 10.1523/JNEUROSCI.0485-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mark M., Ghyselinck N.B., Chambon P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009;7:e002. doi: 10.1621/nrs.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng W., Khan M.A., Bellvis P., Zhu Z., Bernhardt O., Herold-Mende C., Liu H.K. The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell. 2013;13:62–72. doi: 10.1016/j.stem.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V.P., Velez M., Bhandoola A., Brown E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sclafani A.M., Skidmore J.M., Ramaprakash H., Trumpp A., Gage P.J., Martin D.M. Nestin-Cre mediated deletion of Pitx2 in the mouse. Genesis. 2006;44:336–344. doi: 10.1002/dvg.20220. [DOI] [PubMed] [Google Scholar]
- 27.Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P.C., Bock R., Klein R., Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 28.Marin O., Rubenstein J.L. A long, remarkable journey: tangential migration in the telencephalon. Nat. Rev. Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- 29.Pastrana E., Silva-Vargas V., Doetsch F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. 2011;8:486–498. doi: 10.1016/j.stem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds B.A., Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez-Buylla A., Garcia-Verdugo J.M., Tramontin A.D. A unified hypothesis on the lineage of neural stem cells. Nat. Rev. Neurosci. 2001;2:287–293. doi: 10.1038/35067582. [DOI] [PubMed] [Google Scholar]
- 32.Ables J.L., Decarolis N.A., Johnson M.A., Rivera P.D., Gao Z., Cooper D.C., Radtke F., Hsieh J., Eisch A.J. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J. Neurosci. 2010;30:10484–10492. doi: 10.1523/JNEUROSCI.4721-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson S.A., Eisenstat D.D., Shi L., Rubenstein J.L. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- 34.Beukelaers P., Vandenbosch R., Caron N., Nguyen L., Moonen G., Malgrange B. Cycling or not cycling: cell cycle regulatory molecules and adult neurogenesis. Cell Mol. Life Sci. 2012;69:1493–1503. doi: 10.1007/s00018-011-0880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zentner G.E., Hurd E.A., Schnetz M.P., Handoko L., Wang C., Wang Z., Wei C., Tesar P.J., Hatzoglou M., Martin D.M., et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum. Mol. Genet. 2010;19:3491–3501. doi: 10.1093/hmg/ddq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LaMantia A.S., Colbert M.C., Linney E. Retinoic acid induction and regional differentiation prefigure olfactory pathway formation in the mammalian forebrain. Neuron. 1993;10:1035–1048. doi: 10.1016/0896-6273(93)90052-s. [DOI] [PubMed] [Google Scholar]
- 37.Kikonyogo A., Abriola D.P., Dryjanski M., Pietruszko R. Mechanism of inhibition of aldehyde dehydrogenase by citral, a retinoid antagonist. Eur. J. Biochem. 1999;262:704–712. doi: 10.1046/j.1432-1327.1999.00415.x. [DOI] [PubMed] [Google Scholar]
- 38.Layman W.S., Hurd E.A., Martin D.M. Reproductive dysfunction and decreased GnRH neurogenesis in a mouse model of CHARGE syndrome. Hum. Mol. Genet. 2011;20:3138–3150. doi: 10.1093/hmg/ddr216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouazoune K., Kingston R.E. Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc. Natl. Acad. Sci. USA. 2012;109:19238–19243. doi: 10.1073/pnas.1213825109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rochette-Egly C., Germain P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs) Nucl. Recept. Signal. 2009;7:e005. doi: 10.1621/nrs.07005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roybon L., Deierborg T., Brundin P., Li J.Y. Involvement of Ngn2, Tbr and NeuroD proteins during postnatal olfactory bulb neurogenesis. Eur. J. Neurosci. 2009;29:232–243. doi: 10.1111/j.1460-9568.2008.06595.x. [DOI] [PubMed] [Google Scholar]
- 42.Engelen E., Akinci U., Bryne J.C., Hou J., Gontan C., Moen M., Szumska D., Kockx C., van Ijcken W., Dekkers D.H., et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat. Genet. 2011;43:607–611. doi: 10.1038/ng.825. [DOI] [PubMed] [Google Scholar]
- 43.MacPherson P.A., Jones S., Pawson P.A., Marshall K.C., McBurney M.W. P19 cells differentiate into glutamatergic and glutamate-responsive neurons in vitro. Neuroscience. 1997;80:487–499. doi: 10.1016/s0306-4522(97)00102-4. [DOI] [PubMed] [Google Scholar]
- 44.McBurney M.W., Reuhl K.R., Ally A.I., Nasipuri S., Bell J.C., Craig J. Differentiation and maturation of embryonal carcinoma-derived neurons in cell culture. J. Neurosci. 1988;8:1063–1073. doi: 10.1523/JNEUROSCI.08-03-01063.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takada I., Mihara M., Suzawa M., Ohtake F., Kobayashi S., Igarashi M., Youn M.Y., Takeyama K., Nakamura T., Mezaki Y., et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-gamma transactivation. Nat. Cell. Biol. 2007;9:1273–1285. doi: 10.1038/ncb1647. [DOI] [PubMed] [Google Scholar]
- 46.Carter C.S., Vogel T.W., Zhang Q., Seo S., Swiderski R.E., Moninger T.O., Cassell M.D., Thedens D.R., Keppler-Noreuil K.M., Nopoulos P., et al. Abnormal development of NG2+PDGFR-alpha+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat. Med. 2012;18:1797–1804. doi: 10.1038/nm.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferri A.L., Cavallaro M., Braida D., Di Cristofano A., Canta A., Vezzani A., Ottolenghi S., Pandolfi P.P., Sala M., DeBiasi S., et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- 48.O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jongmans M.C., Admiraal R.J., van der Donk K.P., Vissers L.E., Baas A.F., Kapusta L., van Hagen J.M., Donnai D., de Ravel T.J., Veltman J.A., et al. CHARGE Syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2006;43:306–314. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanlaville D., Etchevers H.C., Gonzales M., Martinovic J., Clement-Ziza M., Delezoide A.L., Aubry M.C., Pelet A., Chemouny S., Cruaud C., et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J. Med. Genet. 2006;43:211–217. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanlaville D., Verloes A. CHARGE Syndrome: an update. Eur. J. Hum. Genet. 2007;15:389–399. doi: 10.1038/sj.ejhg.5201778. [DOI] [PubMed] [Google Scholar]
- 52.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.