

Abstract
Nearly all proteins entering the lumen of the endoplasmic reticulum (ER) become glycosylated en route to a cellular organelle, the plasma membrane, or the extracellular space. Many glycans can be attached to proteins, but the most common are the N-linked glycans (oligosaccharides). These chains are added very soon after a protein enters the ER, but they undergo extensive remodeling (processing), especially in the Golgi. Processing changes the sensitivity of the N-glycan to enzymes that cleave entire sugar chains or individual monosaccharides, which also changes the migration of the protein on SDS gels. These changes can be used to indicate when a protein has passed a particular subcellular location. This unit details some of the methods used to track a protein as it trafficks from the ER to the Golgi toward its final location.
Keywords: ER/Golgi, oligosaccharide, glycan, N-glycosylation, glycosidase, intracellular trafficking
INTRODUCTION
Carbohydrate chain modifications are often used to monitor glycoprotein movement through the secretory pathway. This is because stepwise sugar-chain processing is unidirectional and generally corresponds to the forward or anterograde movement of proteins. This unit offers a group of techniques that will help analyze the general structure of carbohydrate chains on a protein and, therefore, glycan-processing mileposts. The minimum requirements are that the protein can be labeled metabolically (UNIT 10.18) and immunoprecipitated (UNIT 10.16) and clearly seen on a gel or blot (UNIT 10.8). The sugar chains themselves are not analyzed, but their presence and structure are inferred from gel mobility differences after one or more enzymatic digestions. This approach is most often used in combination with [35S]Met pulse-chase metabolic labeling protocols, but can be applied to any suitably labeled protein (e.g., biotinylated or 125I-labeled). As the glycans mature, they become either sensitive or resistant to highly specific glycosidases. Some of these enzymes cleave intact glycans from the protein—e.g., endo H, endo F2, endo F3, peptide:N-glycosidase F (PNGase F), endo D, and O-glycosidase. Others strip only terminal sugars (e.g., sialidase) or degrade a selected portion of the chain (e.g., endo-β-galactosidase). The techniques can be adapted to count the number of N-linked glycans on a protein. One unusual protease, O-sialoglycoprotease, degrades only proteins containing tight clusters of O-linked sialylated sugar chains. These techniques work best on average size proteins (<100 kDa) that contain a few percent carbohydrate by weight, where a gel shift of 1 kDa can be seen. A summary of the enzymes and their applications is shown in Table 17.13A.1.
Table 17.13A.1.
Enzymes Described in This Unit
| Enzyme | Indications and uses | Monitorsa |
|---|---|---|
| Endo D | Transient appearance of highly processed, sensitive forms prior to addition of GlcNAc by GlcNAc transferase I | Cis to medial Golgi |
| Endo F2 | Presence of biantennary chains ± core fucose | Medial Golgi |
| Endo F3 | Presence of core fucosylated biantennary α chains and/or triantennary chains ± core fucosylation | Medial Golgi |
| Endo-β-galactosidase | Presence of poly-N-acetyllactosamines | Trans-Golgi and TGN |
| Endo H | Conversion of high mannose to complex type N-linked chains | Cis to medial Golgi |
| O-Glycosidase | Presence of Galβ1,3GalNAc-α-Thr/Ser O-linked chains | Cis to medial Golgi |
| PNGase F | Presence of N-linked chains; cleaves nearly all N-linked chains; only enzyme that cleaves tetrantennary chains | Medial Golgi |
| Sialidase | Acquisition of sialic acids | Trans-Golgi and TGN |
| O-Sialoglycoprotease | Presence of mucin-like proteins with cluster of sialylated glycans | Trans-Golgi and TGN |
Abbreviation: TGN, trans-Golgi network.
All enzymes except O-sialoglycoprotease (Accurate Chemical and Scientific), endo D (Seikagaku), and endo-β-galactosidase (MP Biomedicals or Seikagaku) are available from Calbiochem or Sigma-Aldrich. Other providers are ProZyme, New England Biolabs, Roche Applied Science, and Takara Biochemical.
This unit provides information on how to measure changes in carbohydrate structure and how these changes relate to protein trafficking. Fortunately, the techniques are independent of mechanistic views, although it should be borne in mind that the organization and distribution of many of these indicator enzymes are cell-type dependent.
The starting material for these protocols is assumed to be [35S]Met-labeled, immunoprecipitated protein bound to ~20 μl of protein A–Sepharose beads (as described in UNIT 10.16). The trace amount of protein is eluted by heating in a small volume of 0.1% SDS, diluted in the appropriate buffer, and then digested with one or more enzymes in a small volume. The digest is analyzed on an appropriate SDS-PAGE system that can detect a 1- to 2-kDa size change. A change in the mobility of the protein after digestion is evidence that the carbohydrate chain was sensitive to the enzyme, and therefore that the protein had encountered a certain enzyme in the processing pathway. Alternatively, the analysis can be done by two-dimensional isoelectric focusing (IEF)/SDS-PAGE or two-dimensional nonequilibrium pH gradient electrophoresis (NEPHGE)/SDS-PAGE (UNIT 10.4) to see the loss of charged sugar residues or of anionic glycans. The same digestions and SDS-PAGE analysis also apply to proteins that are radioiodinated or biotinylated, or to immunoprecipitates derived from subcellular fractions separated on sucrose or Percoll gradients.
It is important to present the glycosylation pathways, as a detailed description of the pathways is needed to appreciate how they will be used in this unit. A single protein can have more than one kind of glycan (N-linked and O-linked), and each individual N-linked chain can mature into a different final form. The same is true for O-linked chains. Each is described below.
THE N-LINKED PATHWAY
The N-linked glycan maturation pathway is most frequently used for tracking protein movement through the Golgi complex. A common feature of all N-linked chains is the core region pentasaccharide shown in Figure 17.13A, which consists of three mannose units and two N-acetylglucosamine units. The mannose units comprise the trimannosyl core, and two of these residues are α-linked to the only β-linked mannose in the molecule. The β-linked mannose is bound to one of the two N-acetylglucosamines. Because they are β1-4 linked to each other, resembling the polysaccharide chitin, this is called a chitobiose disaccharide. Initially, all N-glycosylated proteins begin life when a preformed, lipid-associated glycan is transferred within the lumen of the endoplasmic reticulum (ER) to Asn of proteins having an Asn-X-Thr/Ser sequence. This precursor glycan contains three glucose (Glc), nine mannose (Man), and two N-acetylglucosamine (GlcNAc) sugar residues, and has the structure shown in Figure 17.13A. There are several ways to depict this structure. The short-hand symbol method is the most convenient, but be sure to note the linkages of the individual sugars, as they are important. The α and β symbols denote the anomeric configuration of the sugar, and the number indicates which hydroxyl group of the next sugar is involved in the glycosidic linkage. In all cases, the anomeric position is 1, except in sialic acid where it is 2. The symbols chosen for the sugar residues are those used by Essentials of Glycobiology, Second Edition, Cold Spring Harbor Laboratory Press (Varki et al., 2008; http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=glyco2) and the Consortium for Functional Glycomics (http://www.functionalglycomics.org).
The details of the pathway are presented in Figures 17.13A.2 and 17.13A.3, along with the sensitivity to each endoglycosidase or glycoamidase. The figures show the steps between high-mannose and hybrid types (Fig. 17.13A.2) and complex types (Fig. 17.13A.3). The three Glc residues [filled (blue) circles] are removed from properly folded proteins within the ER by two different glycan-processing α-glucosidases. The first α1-2Glc is cleaved by α-glucosidase I, and the next two α1-3Glc residues by α-glucosidase II (Fig. 17.13A.2, step 1). An ER-associated α-mannosidase removes one Man residue [shaded (green) circle; Fig. 17.13A.2, step 2]. The protein then moves on to the first step in Golgi-localized processing—the removal of the three remaining α1-2 Man units by Golgi α-mannosidase I to produce Man5GlcNAc2 (Fig. 17.13A.3, steps 3 and 4). Many proteins have only high-mannose-type glycans with five to nine Man residues, and no further processing occurs. Alternatively, one to five GlcNAc residues [filled (blue) squares] can be added to the trimannosyl core, and these are usually extended with galactose [Gal; open (yellow) circles] and sialic acid [Sia; shaded (purple) diamonds] residues. These extensions, called antennae, are the hallmarks of complex-type glycans. The transformation of the precursor sugar chain into various high-mannose or complex types is called glycan processing (Kornfeld and Kornfeld, 1985).
Figure 17.13A.2.
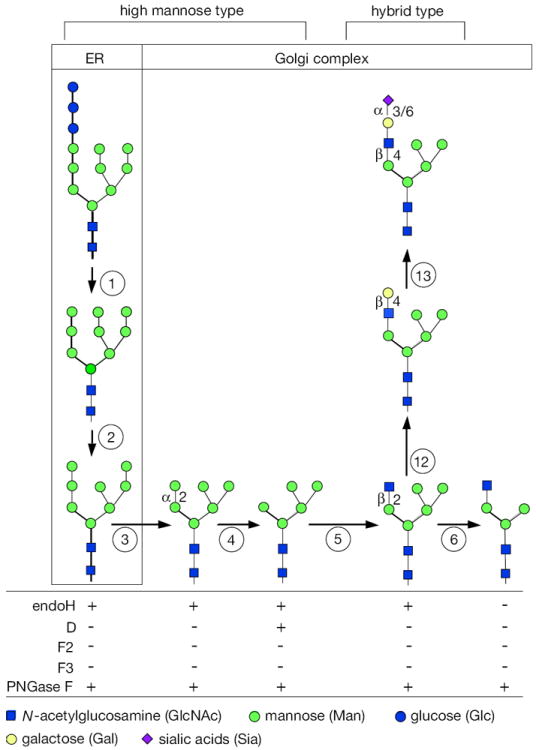
N-linked glycan maturation pathway for high-mannose and hybrid types, and sensitivities to various enzymes. Brackets (top) show the structures designated as high-mannose and hybrid chains. The boxes indicate ER or Golgi localization. The pathway begins with the precursor glycan (see Fig. 17.13A.1). Each successive numbered step in circles represents a glycosidase or glycosyl transferase that generates a new sugar chain with different sensitivities to the various endoglycosidases or PNGase F. (1) precursor glycan is trimmed by α-glucosidases I and II, removing three Glc. (2) ER mannosidase removes one Man. (3) α-Mannosidase I in Golgi complex removes two Man to make Man6GlcNAc2, with a single remaining α1-2Man. (4) The final α1-2Man is removed by a Golgi complex α-mannosidase I. (5) GlcNAc transferase I adds GlcNAc to Man5GlcNAc2. (6) α-Mannosidase II or α-mannosidase IIx (MX) removes the α1-3 and α1-6Man units to make GlcNAc1Man3GlcNAc2. Sensitivity to various enzymes (bottom) changes when moving from left to right, but remains the same within vertical columns. NOTE: This continued maturation to form complex chains is shown in Figure 17.13A.3. Additionally, these figures are not comprehensive; many glycosylation steps have not been included, but they do not affect the sensitivities to the enzymes listed. For the color version of the figure go to http://www.currentprotocols.com/protocol/mb1713a.
Figure 17.13A.3.
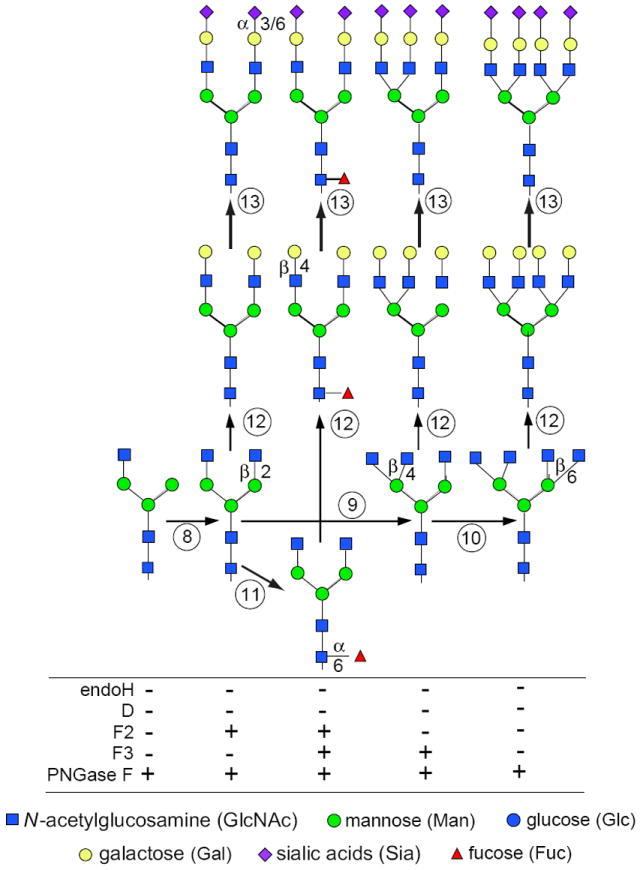
N-linked glycan maturation pathway for complex types, and sensitivities to various enzymes (see Fig. 17.13A.2 for additional details). (8) GlcNAc transferase II adds a second GlcNAc to initiate a biantennary chain. (9) GlcNAc transferase IV adds a third GlcNAc to initiate a triantennary chain. (10) GlcNAc transferase V adds a fourth GlcNAc to initiate a tetraantennary chain. (11) Fucosyltransferase adds α1-6Fuc to the core region of complex chains. (12) β1-4Gal is added to available GlcNAc residues of hybrid and complex chains. (13) α2-3 or α2-6Sia is added to Gal residues of hybrid and complex chains. For the color version of the figure go to http://www.currentprotocols.com/protocol/mb1713a.
Man5GlcNAc2 is an important intermediate because it can have several fates. The first is the well-established addition of one GlcNAc residue by GlcNAc transferase I (Fig. 17.13A.2, step 5). This is the first step toward the formation of complex chains. However, simply adding Gal and Sia to the terminal GlcNAc of this glycan forms a hybrid structure (Fig. 17.13A.2, steps 12 and 13), where the left side of the molecule looks like a complex chain having one antenna, and the right side still resembles a high-mannose chain. The GlcNAc1Man5GlcNAc2 structure is the required substrate for α-mannosidase II, which removes the two terminal Man units from the upper branch of the chain (i.e., the α1-3Man and α1-6 Man units; Fig. 17.13A.2, step 6). This enzyme only works after the addition of the first GlcNAc.
Man5GlcNAc2 is a substrate for α-mannosidase IIx (Chui et al., 1997), an isozyme of α-mannosidase II. Previously α-Man IIx was thought to have a different specificity than α-Man II (Akama et al., 2006).
GlcNAc transferase II now adds a second GlcNAc to the α1-6-linked Man (Fig. 17.13A.3, step 8). This molecule can also have several fates. First, fucose (Fuc) can be added to the GlcNAc residue linked to the Asn of the protein (Fig. 17.13A.3, step 11). Second, one to three more GlcNAc residues can be added to the core mannose residues to initiate tri- and tetraantennary chains (Fig. 17.13A.3, steps 9 and 10), and even pentaantennary chains (not shown). GlcNAc additions are considered to occur in the medial Golgi regions. Each GlcNAc-based branch can be individually modified, but they are usually extended by one Gal (Fig. 17.13A.3, step 12) and terminated by a Sia (Fig. 17.13A.3, step 13). Both of these sugars are usually thought to be added in trans-Golgi cisternae or in the trans-Golgi network (TGN). Sometimes selected antennae are also fucosylated in the TGN. One or more terminal Gal residues can be extended by variable-length poly-N-acetyllactosamines (Galβ4-GlcNAc repeats) capped by a Sia. GlcNAc and Gal can be sulfated as a late, perhaps even final, step of processing. These extensions/modifications are thought to occur in the late Golgi complex and TGN, but their order and compartmental segregation are not well understood. Other modifications of N-linked sugar chains are known, but there are fewer tools available to analyze their biosynthetic localization.
THE O-LINKED PATHWAY
For practical purposes, only a portion of the O-linked pathway—i.e., the addition of the first few sugars—will be presented. However, it is very important to remember that some of the same outer chain structures such as Sia, poly-N-acetyllactosamines, and Fuc residues are common to both N- and O-linked glycans.
α-N-Acetylgalactosamine [α-GalNAc; open (yellow) square] is the lead-off sugar for the O-linked pathway (Fig. 17.13A.4; also see Fig. 17.13A.1 for symbols). It is added to Ser/Thr residues that occur in the proper configuration, generating a broad variety of acceptor sequences. These sequences often cluster as repeats within mucin-like domains. GalNAc is added in the earliest parts of the Golgi complex, not cotranslationally. GalNAc can be further extended by at least six different sugars. The most common is the addition of a β1-3Gal (Fig. 17.13A.4, step 1), forming a disaccharide that is one of the few O-linked chains that can be diagnosed by enzymatic digestions. This disaccharide is often capped by a Sia (Fig. 17.13A.4, step 2). Additional sugars such as Sia (Fig. 17.13A.4, step 3) or GlcNAc followed by Gal (Fig. 17.13A.4, steps 4 and 5) can be added. Structural analysis can be done by sequential exoglycosidase digestion, but given the complexity and heterogeneity of the sugar chains, such analysis is not a very useful indicator for tracking protein movement through the Golgi complex. Many O-linked chains have terminal Sia residues and, when tightly clustered on Ser/Thr residues, these chains promote proteolysis by O-sialoglycoprotease regardless of the structure of the underlying sugar chain.
Figure 17.13A.4.
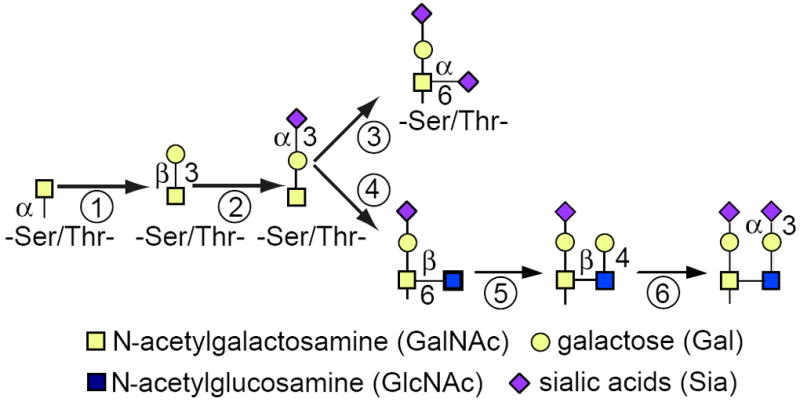
A small portion of the O-GalNAc pathway. The first step of the O-linked pathway occurs in the early Golgi complex with the addition of α-GalNAc. There are at least six other sugars that can be added at this point in this complex pathway. Often β1-3Gal is added (1), quickly followed by α2-3Sia (2). The presence of these structures can be detected with a combination of O-glycosidase and sialidase. Additional sugars can be added as shown. α2-6Sia (3) or β1-6GlcNAc (4) followed by β1-4Gal (5) and α2-3Sia (6) on Gal. Each of these sugars must be removed before O-glycosidase can cleave the disaccharide. For the color version of the figure go to http://www.currentprotocols.com/protocol/mb1713a.
Figure 17.13A.1.
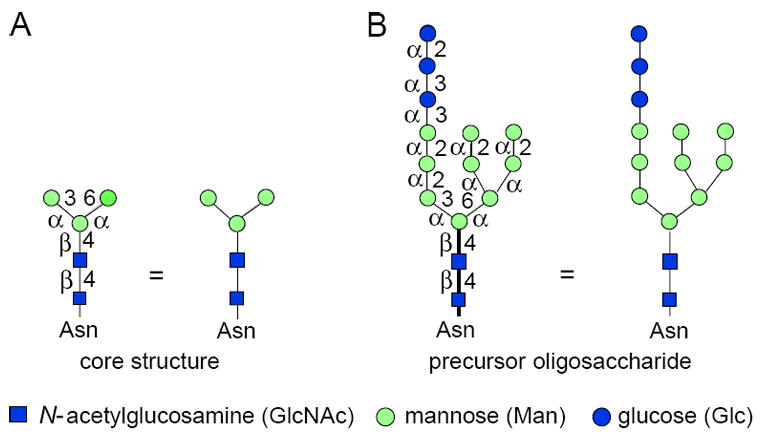
Symbol structures for the core region and precursor of N-linked sugar chains. Each sugar is given a symbol and abbreviation at the bottom of the figure. Each one except sialic acid uses its anomeric carbon (C-1) for linking to other sugars. Sialic acid uses C-2 for glycosidic linkage to other sugars. Glycosidases and glycosyltransferases are anomeric specific and distinguish α or β configurations of each sugar. The core structure (A) is common to all N-linked chains and is composed of three Man and two GlcNAc residues. The α or β configuration of each sugar is indicated, and the OH group to which that sugar is linked is shown on the bar linking the two symbols. Thus, GlcNAcβ1-4GlcNAcβ is represented by two filled squares with β and 4 between them. When a structure is first presented, it will have full display such as that on the left side; if it is repeated, only the symbols will be used, as shown immediately to the right. The precursor glycan (B) for all N-linked chains is synthesized in the ER and transferred cotranslationally to the peptide containing an available Asn-X-Thr/Ser sequon. For the color version of the figure go to http://www.currentprotocols.com/protocol/mb1713a.
Another type of O-linked glycosylation is the addition of glycosaminoglycan (GAG) chains to form proteoglycans. This occurs by a different pathway than the α-GalNAc linkage. Instead, the chains begin by addition of a β-xylose (Xyl) residue to Ser and are then elongated by two Gal residues and a glucuronic acid (GlcA) residue. This core structure can be further elongated by the addition of GlcAβ1-3GalNAcβ disaccharides to form the backbone of chondroitin/dermatan sulfate chains, or by GlcAβ1-3GlcNAcα to form the backbone of heparan sulfate chains. Biosynthesis and movement of these proteins have also been followed through the Golgi complex. Initiation begins in late ER/early Golgi complex, and the core tetrasaccharide is probably finished within the medial Golgi, but the addition of chondroitin chains appears to be confined to the TGN. In addition to the well-known O-linked GAG chains, there is clear evidence for the existence of a class of N-linked GAG chains.
ENDOGLYCOSIDASE H DIGESTION
Endoglycosidase H (endo H) cleaves N-linked glycans between the two N-acetylglucosamine (GlcNAc) residues (Fig. 17.13A.5) in the core region of the glycan chain (Fig. 17.13A.1) on high-mannose and hybrid, but not complex, glycans. In this protocol, a fully denatured protein is digested with endo H to obtain complete release of sensitive glycans.
Figure 17.13A.5.
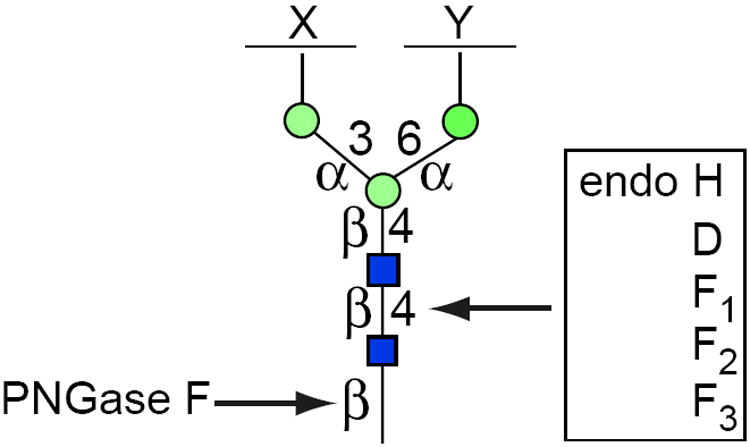
PNGase F and endoglycosidase-sensitive bonds in the core of N-linked glycans. PNGase F is a glycoamidase that severs the bond between GlcNAc and Asn, liberating the entire sugar chain and converting Asn into Asp. The endoglycosidases (H, D, and Fs) cleave the bond between the two GlcNAc residues in the core region, leaving one GlcNAc still bound to the protein. The differential specificity of the endoglyosidases is based on the structure of the sugar chain in a fully denatured protein. Incomplete denaturation may not expose all sensitive linkages. X and Y are unspecified sugar residues. For the color version of the figure go to http://www.currentprotocols.com/protocol/mb1713a.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.5 M sodium citrate, pH 5.5
1% (w/v) phenylmethylsulfonyl fluoride (PMSF) in isopropanol
0.5 U/ml endoglycosidase H (endo H; natural or recombinant)
10 × SDS sample buffer (UNIT 10.2A)
Water baths, 30° to 37°C and 90°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 10.3A)
-
Add 20 to 30 μl of 0.1 M 2-ME/0.1% SDS to immunoprecipitate in a microcentrifuge tube, mix well, and heat denature 3 to 5 min at 90°C.
Use the larger amount of reagent (≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Protein solubilization in nonionic detergents such as Triton X-100 or Nonidet P-40 is not always sufficient to completely expose all susceptible cleavage sites. Only strong denaturation with SDS exposes all sites for maximum cleavage.
Cool and microcentrifuge for 1 sec at 1000 × g to collect condensed droplets in the bottom of the tube.
Place 10-μl aliquots of solubilized, denatured protein (supernatant) in each of two clean microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
6 μl 0.5 M sodium citrate, pH 5.5
20 μl H2O
2 μl 1% PMSF (in isopropanol)
1 μl 0.5 U/ml endo H (enzyme digest only; substitute with water in control).
The PMSF prevents proteolysis. Nonionic detergent is not required to prevent inactivation of endo H as long as high-purity SDS is used.
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
Incubate overnight at 30°C to 37°C.
Immediately prior to electrophoresis, inactivate endo H by adding 4 μl of 10× SDS sample buffer and heating 5 min at 90°C.
-
Analyze protein by one-dimensional SDS-PAGE (UNIT 10.2) and autoradiography (APPENDIX 3A).
The presence of high mannose and/or hybrid N-linked oligsaccharide chains will be evidenced by increased mobility of the digested proteins on SDS-PAGE.
-
BASIC PROTOCOL 2
ENDOGLYCOSIDASE D DIGESTION
Like endo H, endo D also cleaves between the two GlcNAc residues in the core of the N-linked sugar chains (Fig. 17.13A.5). However, its narrow substrate specificity makes it useful for detecting the transient appearance of just a few early processing intermediates. It requires that the 2 position of the α1-3-linked core Man be unsubstituted. This intermediate sometimes arises after more extensive processing, but prior to addition of the first GlcNAc or action of α-mannosidase II. Cells with a defect in GlcNAc I transferase (e.g., Lec 1 CHO cells) do not add the first GlcNAc residue (Fig. 17.13A.2, step 5), and N-linked glycans will remain sensitive to endo D because they cannot modify the α1-3Man residue.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.5 M NaH2PO4, pH 6.5
10% (w/v) Triton X-100 or Nonidet P-40 (NP-40)
0.5 U/ml endoglycosidase D (endo D)
10 × SDS sample buffer (UNIT 10.2A)
Water baths, 37° and 90°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS by heating for 3 to 5 min at 90°C.
Use the larger amount of reagent (≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Cool and microcentrifuge at 1000 × g for 1 sec to collect condensed droplets in the bottom of the tube.
Transfer 10-μl aliquots of supernatant into two microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
2 μl 10% Triton X-100 or NP-40 (20-fold excess over SDS)
2 μl 0.5 M NaH2PO4, pH 6.5
5 μl H2O
1 μl 1 IU/ml endo D (enzyme digest only; substitute with water in control).
The 20-fold excess of nonionic detergent is essential to prevent inactivation of endo D by SDS.
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
Incubate overnight at 37°C.
Immediately prior to electrophoresis, inactivate by adding 2 μl of 10× SDS sample buffer and heating for 5 min at 90°C to 95°C.
-
Analyze protein by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
Endo D sensitivity is detected by increased electrophoretic mobility of the digested proteins on SDS-PAGE.
-
BASIC PROTOCOL 3
ENDOGLYCOSIDASE F2 DIGESTION
Endo F2, like endo H and endo D, cleaves between the two GlcNAc residues in the chitobiose core (Fig. 17.13A.5). It preferentially releases biantennary complex-type glycans from glycoproteins, but does not cleave tri- or tetraantennary chains.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.5 M sodium acetate, pH 4.5 (APPENDIX 2)
10% (w/v) Triton X-100 or Nonidet P-40 (NP-40)
0.1 M 1,10-phenanthroline in methanol
200 mU/ml endoglycosidase F2 (endo F2)
4 × SDS sample buffer (UNIT 10.2A)
Water baths, 30° to 37°C and 90°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS by heating for 3 to 5 min at 90°C.
Use the larger amount of reagent (≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Cool and microcentrifuge at 1000 × g for 1 sec to collect condensed droplets in the bottom of the tube.
Transfer 10-μl aliquots of supernatant into two microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
15 μl 0.5 M sodium acetate, pH 4.5
3 μl 0.1 M 1,10-phenanthroline in methanol
2 μl 10% Triton X-100 or NP-40 (20-fold excess over SDS)
1 μl 200 mU/ml endo F2 (enzyme digest only; substitute 0.5 M sodium acetate in control).
A 10- to 20-fold excess of nonionic detergent is required to stabilize the enzyme.
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
-
Incubate the mixture overnight at 30° to 37°C.
Some inactivation of the enzyme occurs at 37°C, even with nonionic detergent present; however, if the enzyme is present in sufficient excess, incubation can generally be carried out successfully at 37°C.
Immediately before electrophoresis, inactivate by adding 8 μl of 4× SDS sample buffer and heating for 5 min at 90°C to 95°C.
-
Analyze the protein by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
Sensitivity to endo F2 is detected by increased electrophoretic mobility on SDS-PAGE.
-
BASIC PROTOCOL 4
ENDOGLYCOSIDASE F3 DIGESTION
Endoglycosidase F3 (endo F3) is another endoglycosidase with a narrow substrate range and, therefore, high specificity: it cleaves triantennary chains, but not high-mannose, hybrid, nonfucosylated biantennary or tetraantennary chains. A core-fucosylated biantennary chain is the only other demonstrated substrate. When both endo F3 and endo F2 digestions are done in parallel on a sample, it can provide evidence for chain branching and core fucosylation. The approach is essentially the same as for the other endoglycosidases.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.5 M sodium acetate, pH 4.5 (APPENDIX 2)
10% (w/v) Triton X-100 or NP-40
0.1 U/ml endoglycosidase F3 (endo F3)
10× SDS sample buffer (UNIT 10.2A)
Water baths, 37° and 90°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS and heat denature 3 to 5 min at 90°C.
Use the larger amount of reagent (≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Cool and microcentrifuge for 1 sec at 1000 × g to collect condensed droplets at the bottom of the tube.
Transfer 10-μl aliquots of supernatant into two microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
2 μl 10% Triton X-100 or NP-40 (20-fold excess over SDS)
4 μl 0.5 M sodium acetate, pH 4.5
5 μl H2O
1 μl 0.1 U/ml endo F3 (enzyme digest only; substitute with water in control).
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
Incubate overnight at 37°C.
Immediately prior to electrophoresis, inactivate by adding 2 μl of 10× SDS sample buffer and heating for 5 min at 90°C to 95°C.
-
Analyze by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
Sensitivity to endo F3 is detected by increased mobility on SDS-PAGE.
-
BASIC PROTOCOL 5
PEPTIDE: N-GLYCOSIDASE F DIGESTION
PNGase F is a glycoamidase that cleaves the bond between the Asn residue of the protein and the GlcNAc residue that joins the carbohydrate to the protein (Fig. 17.13A.5). Because it liberates nearly all known N-linked glycans from glycoproteins, it is the preferred enzyme for complete removal of N-linked chains. It is the only enzyme that releases tetra- and pentaantennary chains. The glycoprotein sample must be denatured and digested with PNGase F to remove N-linked glycans completely.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.5 M Tris·Cl, pH 8.6 as determined at 37°C (APPENDIX 10.16)
10% (w/v) Triton X-100 or Nonidet P-40 (NP-40)
200 to 250 mU/ml peptide:N-glycosidase F (PNGase F)
10× SDS sample buffer (UNIT 10.2A)
Water baths, 30° to 37°C and 90°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS by heating 3 to 5 min at 90°C.
Use the larger amount of reagent ≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Cool and microcentrifuge for 1 sec at 1000 × g to collect condensed droplets in the bottom of the tube.
Transfer 10-μl aliquots of supernatant into two microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
3 μl 0.5 M Tris·Cl, pH 8.6
5 μl H2O
2 μl 10% NP-40 or Triton X-100
-
5 μl 200 to 250 mU/ml PNGase F (enzyme digest only; substitute with 0.5 M Tris·Cl in control).
Sodium phosphate or HEPES buffer, pH 7.0, can be used instead of Tris·Cl. Avoid potassium buffers because these may cause precipitation of a potassium SDS salt. Use of a nonionic detergent is essential, because SDS inactivates PNGase F. A 10-fold weight excess of any of the above nonionic detergents over the amount of SDS will stabilize the enzyme.
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
Incubate overnight at 30° to 37°C.
Immediately prior to electrophoresis, inactivate the enzyme by adding 2.5 μl of 10× SDS sample buffer and heating 3 to 5 min at 90°C.
-
Analyze the protein by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
The presence of N-linked glycan chains will be evidenced by increased electrophoretic mobility on SDS-PAGE.
-
SUPPORT PROTOCOL
ESTIMATING THE NUMBER OF N-LINKED GLYCANS ON A GLYCOPROTEIN
One widely used application of endo H or PNGase F digestion is estimation of the number of N-linked glycans on a given glycoprotein. This is done by creating a ladder of partially digested molecules that each differ by only one N-linked sugar chain. The number of separate bands in a one-dimensional polyacrylamide gel (less one for the totally deglycosylated protein) provides an estimate of the number of N-linked chains. The conditions used to generate partially deglycosylated protein must be determined for each protein studied, because the sensitivity of each chain may be different, even when all of them are completely exposed by denaturation. For this protocol, either the incubation time or the amount of enzyme can be varied to determine the best conditions to produce a ladder of partial digests. Usually five or six points are enough to provide a reasonable estimate (Fig. 17.13A.6). Of course, it is important to use enough enzyme to obtain complete deglycosylation. This is best done by monitoring the effects of endo H or PNGase F on newly synthesized [35S]Met pulse-labeled protein just after synthesis, but before any N-linked glycans processing has occurred. Pulse labeling of protein for 10 min with [35S]Met followed by digestion is the best way to be sure that all chains are removed.
Figure 17.13A.6.
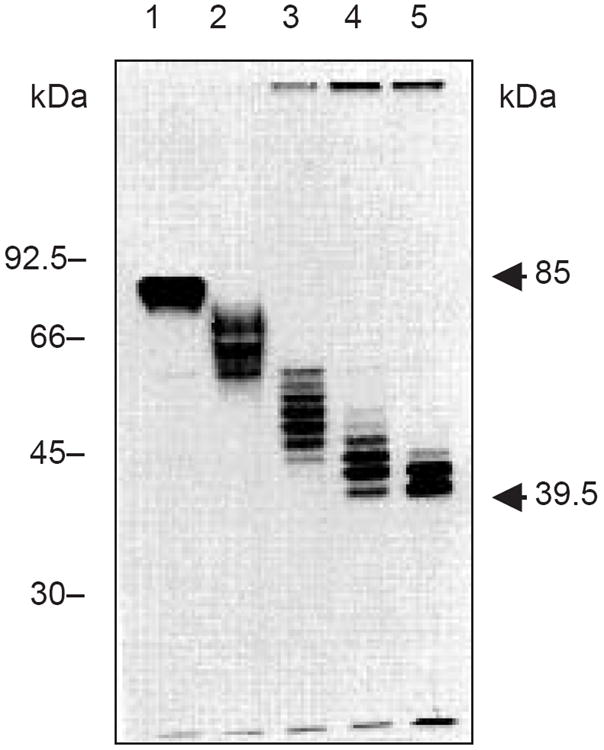
Data from the estimation of the number of glycosylation sites on lysosome-associated membrane protein 1 (LAMP-1; Viitala et al., 1988). LAMP-1 contains eighteen potential N-linked sites. Graded digestion with increasing amounts of PNGase F was used to generate this ladder of glycoforms. Each band contains at least one less N-linked chain than the band above it. An average N-linked carbohydrate chain has an apparent mass of ~1.5 to 3 kDa. Lysosomal membrane glycoprotein was immunoprecipitated from [35S]Met-labeled cells and the sample was digested with PNGase F for 0 min (lane 1), 5 min (lane 2), 20 min (lane 3), 45 min (lane 4), and 24 hr (lane 5). Figure courtesy of Dr. Minoru Fukuda.
-
Add 0.1 M 2-ME/0.1% SDS solution to the total volume of immunoprecipitated protein required and heat denature by incubating 3 to 5 min at 90°C.
Each digestion reaction requires 20 μl of immunoprecipitate. Thus, 120 to 140 μl is sufficient for one control plus five or six digests.
Cool and centrifuge for 1 sec at 1000 × g to collect condensed droplets at the bottom of the tube.
Aliquot 10 μl supernatant to the number of microcentrifuge tubes required to cover the concentration range (e.g., 0.01 to 1 mU/ml PNGase F) or incubation times (e.g., 5 to 60 min) plus one for an undigested control.
Add remaining reagents as specified for endo H (see Basic Protocol 1, step 4) or PNGase F (see Basic Protocol 5, step 4), adjusting the enzyme concentration as desired.
-
Incubate at 30°C for the desired length of time.
High enzyme concentration (10 mU/ml) and prolonged incubation (16 hr) must be among the conditions included, in order to ensure that there is a data point for maximum deglycosylation.
For varying enzyme concentrations, incubate for the same amount of time, but the duration of incubation should be shorter than what would give complete digestion because the goal is to obtain increasing extent of incomplete cleavage.
After the desired incubation time, inactivate enzyme by adding 0.1 volume of 10× SDS sample buffer and heating 5 min at 90° to 95°C.
-
Analyze the sample from each concentration/time point, including undigested sample, by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
Most newly formed N-linked chains will have a molecular weight in the range of 1500 to 2200, and loss of one chain is sufficient to change the migration of a protein. This procedure has been used to count up to eighteen N-linked sites on one molecule. A sample result is shown in Figure 17.13A.6.
Basic Protocol 6
SIALIDASE (NEURAMINIDASE) DIGESTION
Sialic acids are the terminal sugars on many N- and O-linked glycans. The great majority are released with broad-specificity sialidases (neuraminidases) such as that from Arthrobacter ureafaciens. Because sialic acids are charged, their loss usually changes the mobility on one-dimensional SDS polyacrylamide gels, but it will always change the mobility on a two-dimensional gel. Since one-dimensional analysis is easier, it can be tried first.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
10% (w/v) Triton X-100 or Nonidet P-40 (NP-40)
0.5 M sodium acetate, pH 5.0 (APPENDIX 2)
1 IU/ml neuraminidase from Arthrobacter ureafaciens
10× SDS sample buffer (UNIT 10.2A)
Water baths, 37° and 90°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A), for IEF/SDS-PAGE or NEPHGE/SDS-PAGE (UNIT 10.3), and for autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS by heating 3 to 5 min at 90°C.
Denaturation is less important here, because the sialic acids are exposed at the ends of the sugar chains. In most instances, the denaturation step can probably be omitted and the digestion done while the protein is still bound to the beads.
Cool and microcentrifuge for 1 sec at 1000 × g to collect condensed droplets in the bottom of the tube.
Transfer 10-μl aliquots of supernatant to two clean microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
2 μl 10% Triton X-100 or NP-40 (20-fold excess over SDS)
4 μl 0.5 M sodium acetate, pH 5.0
5 μl H2O
1 μl 1 IU/ml neuraminidase (enzyme digest only; substitute with water for control).
This amount of neuraminidase should be in great excess. Addition of nonionic detergent is not needed if the digestion is done while the protein was still bound to the beads.
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
-
Incubate overnight at 37°C.
This time can be shortened to 2 hr, if necessary, but longer incubations are better.
-
Immediately prior to electrophoresis, inactivate the enzyme by adding 2 μl of 10× SDS sample buffer and heating 3 to 5 min at 90°C.
If the protein will be analyzed by IEF or NEPHGE, addition of sample buffer is replaced by lysis buffer used for these techniques.
-
Analyze the protein using either the appropriate one-dimentional SDS-PAGE system (UNIT 10.2A) or a two-dimentional IEF/SDS-PAGE or NEPHGE/SDS-PAGE system (UNITS 10.3 & 10.4), and detect by autoradiography (APPENDIX 3A).
Removal of sialic acids usually results in a decrease in apparent molecular weight on one-dimensional gel analysis, or an increase in the isoelectric point of the protein analyzed by two-dimentional gel analysis.
-
BASIC PROTOCOL 7
ENDO-Β-GALACTOSIDASE DIGESTION
The endo-β-galactosidase from Escherichia freundii degrades poly-N-acetyllactosamine chains (Galβ1-4GlcNAcβ1-3)n found on both N- and O-linked glycans. The variable length of these repeating units usually causes the protein to run as a broad band or a smear on the gel. Although not all linkages are equally cleaved by this enzyme (see Fig. 17.13A.7), sensitive proteins that often run as broad bands or smears on gels—e.g., lysosome-associated membrane protein 1 (LAMP-1)—produce both sharper bands and lower molecular weight species after digestion.
Figure 17.13A.7.
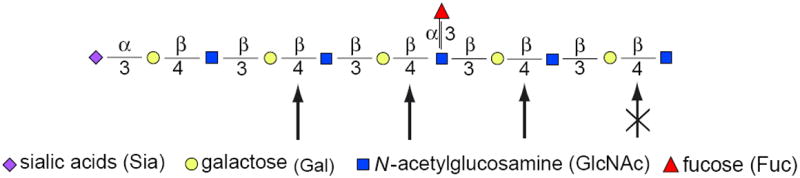
Endo-β-galactosidase-sensitive linkages in poly-N-acetyllactosamines. Linear, unsubstituted poly-N-acetyllactosamine units (GlcNAcβ1-3Galβ1-4) are sensitive to digestion with endo-β-galactosidase, while substitutions—such as sulfate esters (S) or branches starting with GlcNAc (not shown)—completely block digestion. Substitution of neighboring GlcNAc with Fuc or sulfate esters slows the rate, but does not block cleavage. Sensitive sites are shown with bold arrows, slowly hydrolyzed sites with a lighter arrow, and resistant bonds are struck out. Various substitutions are possible, leading to broad bands on gels. This will create variable sensitivities, but even partial sensitivity should give a sharper, more defined band. For the color version of the figure go to http://www.currentprotocols.com/protocol/mb1713a.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.5 M sodium acetate buffer, pH 5.8 (APPENDIX 2)
10% (w/v) Triton X-100 or Nonidet P-40 (NP-40)
100 mU/ml endo-β-galactosidase
10× SDS sample buffer (UNIT 10.2A)
Water baths, 37° and 95°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS by heating 3 to 5 min at 95°C.
Use the larger amount of reagent (≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Cool and microcentrifuge 1 sec at 1000 × g to collect condensed droplets in the bottom of the tube.
Transfer 10-μl aliquots of supernatant into two microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
2 μl 10% Triton X-100 or NP-40 (20-fold excess over SDS)
4 μl 0.5 M sodium acetate, pH 5.8
5 μl H2O
1 μl 100 mU/ml endo-β-galactosidase (enzyme digest only; substitute with water in control).
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
Incubate overnight at 37°C.
Immediately prior to electrophoresis, inactivate by adding 3 μl of 10× SDS sample buffer and heating for 5 min at 90°C.
-
Analyze protein by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
If the protein has poly-N-acetyllactosamine chains, its mobility should increase after digestion.
-
BASIC PROTOCOL 8
ENDO-α-N-ACETYLGALACTOSAMINIDASE DIGESTION
This enzyme (also known as O-glycosidase or O-glycanase) has limited utility because it is highly specific for cleaving only one O-linked disaccharide, Galβ1-3GalNAcα-Ser/Thr. Adding any more sugars, including sialic acid, renders the molecule resistant to cleavage and requires removal of each residue before the enzyme will work. Prior sialidase digestion is sometimes used (see Basic Protocol 6), and this can be done while the protein is still bound to the immunoprecipitation beads.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.5 M sodium citrate phosphate buffer, pH 6.0, containing 500 μg/ml BSA (complete buffer supplied with enzyme)
10% (w/v) Triton X-100 or Nonidet P-40 (NP-40)
300 mU/ml endo-α-N-acetylgalactosaminidase (use according to manufacturer’s directions)
10× SDS sample buffer (UNIT 10.2A)
Water bath, 37° and 95°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS by heating 3 to 5 min at 95°C.
Use the larger amount of reagent (≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Cool and microcentrifuge 1 sec at 1000 × g to collect condensed droplets in the bottom of the tube.
Transfer 10-μl aliquots of supernatant into two microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
2 μl 10% Triton X-100 or NP-40 (20-fold excess over SDS)
4 μl 0.5 M sodium citrate phosphate buffer, pH 6.0, with 500 μg/ml BSA
3 μl H2O
1 μl 300 mU/ml endo-α-N-acetylgalactosidase (enzyme digest only; substitute with water in control).
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
ncubate overnight at 37°C.
Immediately prior to electrophoresis, inactivate by adding 2 μl of 10× SDS sample buffer and heating for 5 min at 90°C.
-
Analyze protein by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
If the protein contains the disaccharide unit, the mobility of the protein should increase. Presence of only a single unit (mol wt. ~400 Da) may be difficult to detect unless a high-resolution gel is used.
-
BASIC PROTOCOL 9
O-SIALOGLYCOPROTEASE DIGESTION
Digestion with O-sialoglycoprotease requires that the substrate have a tight cluster of sialylated O-linked glycans. Proteins with a single O-linked chain or a few widely spaced chains will not be cleaved. This property makes the enzyme a valuable diagnostic tool.
Materials
Immunoprecipitated protein of interest (UNIT 10.16)
0.1 M 2-mercaptoethanol (2-ME)/0.1% (w/v) SDS (ultrapure electrophoresis grade; prepare fresh)
0.4 M HEPES buffer, pH 7.4
10% (w/v) Triton X-100 or Nonidet P-40 (NP-40)
2.4 mg/ml O-sialoglycoprotease (O-sialoglycoprotein endoglycoprotease; reconstituted according to manufacturer’s directions)
10× SDS sample buffer (UNIT 10.2A)
Water baths, 37° and 95°C
-
Additional reagents and equipment for SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A)
-
Denature immunoprecipitated protein in 20 to 30 μl of 0.1 M 2-ME/0.1% SDS by heating 3 to 5 min at 95°C.
Use the larger amount of reagent (≥30 μl) for more complete recovery. This treatment may also release some (unlabeled) antibody molecules.
Cool and microcentrifuge 1 sec at 1000 × g to collect condensed droplets in the bottom of the tube.
Transfer 10-μl aliquots of supernatant into two microcentrifuge tubes, one for a control (no enzyme) and the other to digest (plus enzyme).
-
Add in the following order, mixing after each addition:
2 μl 10% Triton X-100 or NP-40 (20-fold excess over SDS)
4 μl 0.4 M HEPES buffer, pH 7.4
5 μl H2O
2 μl 2.4 mg/ml O-sialoglycoprotease (enzyme digest only; substitute with water in control).
O-Sialoglycoprotein endopeptidase is a partially purified enzyme, and the specific activity is relatively low. A quantity of 1.0 μg of this enzyme preparation will cleave 5 μg of sensitive substrate per hour at 37°C. Human glycophorin A can serve as a positive control.
The amount of water can be varied. Other solutions (or an increased amount of sample) may be substituted for water if needed, but potassium buffers should be avoided because they precipitate SDS as a potassium salt. The reaction volume can be scaled up proportionally if required, but in nearly all cases the enzyme will be in vast excess.
Incubate overnight at 37°C.
Immediately prior to electrophoresis, inactivate by adding 2.5 μl of 10× SDS sample buffer and heating for 5 min at 90°C.
-
Analyze protein by one-dimensional SDS-PAGE (UNIT 10.2A) and autoradiography (APPENDIX 3A).
If the digestion was successful, the target protein will be undetectable or may be cleaved into small fragments.
-
COMMENTARY
Background Information
The results of digestion of a hypothetical protein with two N-linked carbohydrate chains as it moves through the ER and Golgi complex with various enzymes are shown in Figure 17.13A.8. At 0 min, both N-linked chains are high-mannose type. They have lost their Glc residues and one Man residue in the ER. Both are sensitive to endo H and PNGase F digestion, yielding a protein with only two remaining GlcNAc residues in the case of endo H digestion, and no carbohydrate at all in the case of PNGase F digestion. These sugar chains are resistant to the other enzyme digestions.
Figure 17.13A.8.
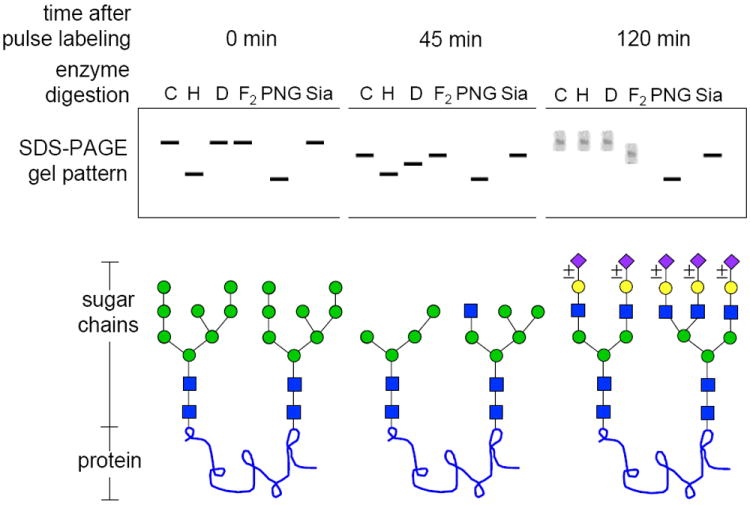
Schematic diagram showing results that could be obtained for a hypothetical protein with two N-linked glycosylation sites as it moves through the Golgi complex. Assume that the protein has been biosynthetically labeled with an amino acid precursor (such as [35S]Met) for 10 min and chased in the absence of label for 45 min and 120 min. The protein is then precipitated with a specific antibody. At each time point, equal amounts of the sample are analyzed by fluorography after one-dimensional SDS-PAGE, either without any digestion (control; C) or following digestion with endo H (H), endo D (D), endo F2 (F2), PNGase F (PNG), or sialidase (Sia). Glycan structures consistent with the banding patterns are shown below the schematic gel pattern. For the color version of the figure go to http://www.currentprotocols.com/protocol/mb1713a.
At 45 min, the protein is in the medial Golgi complex and both sugar chains have been processed by Golgi α-mannosidase I. However, one of the chains (left) has been partially processed by Golgi α-mannosidase IIx to an endo H–resistant/endo D–sensitive chain. The other chain (right) has received a GlcNAc residue from GlcNAc transferase I, but has not encountered Golgi α-mannosidase II. This chain is still endo H sensitive and endo D resistant. Both chains are sensitive to PNGase F, but neither is sensitive to endo F2 or sialidase digestion.
At 120 min, both chains have fully matured to complex-type chains. One is a sialylated biantennary chain and the other is a sialylated triantennary chain. Note that each sialic acid is marked ±, indicating that not all molecules are fully sialylated, accounting for the broader bands. Neither sugar chain is sensitive to endo H or endo D digestion. The biantennary chain (left) is sensitive to endo F2 cleavage, leaving one GlcNAc residue on the protein, but the triantennary chain (right) is not sensitive to this enzyme. Both chains are cleaved by PNGase F. All sialic acids are removed by sialidase to produce a sharp band, but the underlying sugar chains remain.
Endoglycosidase H
Endo H from Streptomyces plicatus cleaves the bond between the two GlcNAc residues in the core of N-linked glycans. One GlcNAc residue remains attached to the protein or peptide and the remainder of the chain is released as an intact unit (Tarentino et al., 1989). The glycan structures cleaved by endo H are shown schematically in Figure 17.13A.2. Substrates for endo H include all high-mannose glycans and certain hybrid types, but not bi-, tri-, tetra- or pentaantennary (complex) chains. Endo H also cleaves glycans that have α1-6 fucose residues bound to the reducing (protein to carbohydrate linkage) GlcNAc residue (Tarentino et al., 1989).
Endo H sensitivity is the most common way to trace the movement of newly synthesized glycoproteins from the endoplasmic reticulum (ER) into the Golgi complex. Proteins remain sensitive to endo H while they are in the ER and in early regions of the Golgi complex; they become endo H–resistant after they are processed by enzymes located in the medial Golgi complex. Endo H cleaves the N-linked glycans from proteins as long as they have not lost the α1-3Man residue cleaved by Golgi mannosidase II or mannosidase IIx (Fig. 17.13A.2). After removal of that mannose, the glycan becomes endo H resistant. Endo H is the best enzyme for identifying high-mannose and hybrid chains and their general transition toward complex chains.
Endoglycosidase D
The narrow substrate specificity of endo D makes it useful for detecting a very restricted set of processing intermediates, particularly for distinguishing α-mannosidase II from α-mannosidase IIx processing. When the results of endo D digestion are combined with the results of an endo H digestion of the same sample, it can help determine which of the two alternate processing pathways is being used. The key requirement for endo D cleavage is an unsubstituted 2 position on the α1-3Man that forms the trimannosyl core (Fig. 17.13A.1). This window of endo D sensitivity occurs only after removal of the α1-2Man residue by α-mannosidase I and before the addition of a GlcNAc residue to this same position by GlcNAc transferase I (Beckers et al., 1987; Davidson and Balch, 1993). The immediate precursor of both pathways is Man6GlcNAc2, which is endo D resistant/endo H sensitive. The better-known α-mannosidase II pathway involves the removal of the last α1-2Man residue by α-mannosidase I (making the glycan endo D sensitive/endo H sensitive), and then the addition of GlcNAc via GlcNAc transferase I (making it endo D resistant/endo H sensitive). α-Mannosidase II removes two of the remaining five Man residues, specifically the α1-3Man and α1-6Man that form the upper branch. Once this occurs, this endo D–resistant molecule also becomes endo H resistant. This structure is also a substrate for GlcNAc transferase II on the way to forming a biantennary chain.
Mutant cell line CHO Lec1 lacks GlcNAc transferase I activity and cannot synthesize either complex or hybrid chains. These cells can be used to measure the kinetics of acquiring endo D sensitivity (Beckers et al., 1987). This cell line can be obtained through ATCC (CRL-1735). The chains become permanently endo D sensitive, because they lack GlcNAc transferase I and cannot be converted back to an endo D–resistant form. These chains will also remain endo H sensitive, because α-mannosidase II requires GlcNAc transferase I. However, rarely, chains are more extensively processed, and become endo H resistant. Again, the combination of endo D and endo H digestions can reveal which pathway was used.
CHO Lec1 cells are also useful for tracking the movement of a protein from the ER into the earliest Golgi compartment where α-mannosidase I is located. Acquisition of endo D sensitivity requires the action of this enzyme. The advantage of using Lec1 cells is that the proteins remain permanently endo D sensitive and there is no risk of kinetically missing that small window of sensitivity before the sugar chain might become endo D resistant once again.
Endoglycosidase F and peptide N-glycosidase F
Elder and Alexander (Elder and Alexander, 1982; Alexander and Elder, 1989) made a landmark discovery when they identified an enzyme in culture filtrates of the bacterium Flavobacterium meningosepticum that cleaved N-linked glycans from glycoproteins. This preparation had a broad substrate specificity. The endoglycosidase activity in this preparation (endo F) was actually due to a set of enzymes, each with a more restricted substrate range. Like endo H, the endo F enzymes cleave the sugar chains between the two core GlcNAc residues (Fig. 17.13A.5). Endo F was originally thought to be a single enzyme, but it is now known that each of the three enzymes has a distinct specificity (Plummer and Tarentino, 1991). The specificity of endo F1 is very similar to that of endo H, while endo F2 prefers biantennary chains, and endo F3 will cleave core fucosylated biantennary and triantennary, but not tetraantennary, chains (Fig. 17.13A.2 and Fig. 17.13A.3).
Plummer et al. (1984) carefully analyzed Flavobacterium filtrates and found that the very broad substrate range was actually due to a glycoamidase activity rather than an endoglycosidase activity. The glycoamidase releases the entire carbohydrate chain from the protein by cleaving the Asp-GlcNAc bond (Fig. 17.13A.5). The enzyme is called by various names, including peptide:N-glycosidase F (PNGase F), glycopeptidase F, and N-glycanase, but the proper name is peptide N-4(N-acetyl-β-glucosaminyl)asparagine amidase F. PNGase F has the broadest specificity, and it releases most of the N-linked glycans from proteins.
Endo F2 and endo F3
Endo F2 prefers biantennary chains over high-mannose chains by ~20 fold. Thus, endo H and PNGase F are better choices for broadly distinguishing high-mannose from complex chains as described above in the endo H protocol (Tarentino and Plummer, 1994).
Many proteins have core-fucosylated N-linked glycans, and the addition of fucose can be used as an additional trafficking marker. Endo F3 will hydrolyze triantennary chains, but endo F2 will not. Endo F2 hydrolyzes biantennary chains; however, endo F3 will also hydrolyze core-fucosylated biantennary chains only a bit more slowly than it does triantennary chains. Thus, if all the chains on a protein are sensitive to endo F2 (biantennary) and to endo F3, this is evidence for the presence of a core fucose on those chains.
The enhancement of endo F3 activity on biantennary chains with a core α1-6Fuc points out that some specificities are really a matter of relative rates of cleavage. If both endo F2 (biantennary) and endo F3 (triantennary and biantennary with core fucose) cleave the protein, they may be acting on different chains on the same molecule. If there is only a single chain, repeat the experiment under the same conditions using 10- to 20-fold dilutions of each enzyme. If both still cleave the chain about equally, it is evidence for core fucosylation of a biantennary chain.
PNGase F
PNGase F has the broadest specificity of all the enzymes that cleave N-linked glycans. It is indifferent to all extended structures on the chains, such as sulfate, phosphate, poly-N-acetyllactosamines, polysialic acids, and even the occasional glycosaminoglycan chain. Most of the modifications in the Man3GlcNAc2 core region also make no difference in chain cleavage. The only glycan structural feature that confers PNGase F resistance is the presence of an α1-3Fuc on the GlcNAc bound to Asn (Tretter et al., 1991). This modification is commonly found in plants and in some insect glycoproteins, but it is rare in most mammalian cell lines. However, caution is warranted, as there is evidence that some mammalian cells do have the critical α1-3Fuc transferase, and some studies show that a majority of N-linked chains of bovine lung are actually PNGase F resistant! It is not known how common this resistance may be. It is thus important to document N-glycosylation with proteins still in the ER (see Support Protocol) before they might be processed to a PNGase F–resistant form.
Sialidase
Sialidases are also called neuraminidases because the most common form of sialic acid is N-acetyl neuraminic acid. The sialic acids are a family with over forty different members, but fortunately the very great majority of them can be removed from the glycans by the broad-spectrum sialidase from Arthrobacter ureafaciens (AUS). It can even digest polysialic acids, a rare modification found on only a few proteins such as neuronal cell adhesion molecule (NCAM). Sialidases with selected specificities from other sources are available but would not usually be needed. AUS has an optimum pH of 5.0, with ~30% of maximum activity at pH 7.
Because sialic acids are charged, they affect gel mobility of proteins more than would be expected from their nominal molecular weight. The magnitude of the gel shift depends on the number of residues. It is difficult to estimate their number by sialidase digestion, but the mobility change is usually sufficient if there are several sialic acid residues. On the other hand, if a protein has only one sialic acid, its presence could be missed using standard one-dimensional SDS-PAGE. To be certain of the effects of sialidase, the sample can be analyzed by a two-dimensional system, using IEF or NEPHGE in the first dimension (UNITS 10.3 & 10.4). The loss of even a single sialic acid will be evident because it changes the isoelectric point.
AUS will remove sialic acids from both N- and O-linked chains, so the type of chain carrying them must be determined independently using PNGase F or possibly O-glycosidase in combination with sialidase. A protein will generally be partially or completely resistant to O-glycosidase because the required disaccharide, Galβ1-3GalNAc, is usually extended and often sialylated. Until the sialic acid is removed, it will be resistant.
The presence of sialic acid (sialidase sensitivity) is often used as an indication of the transport of a protein into the trans-Golgi network (TGN). This may be true in general, but it is important to remember that the distribution of Golgi enzymes is cell type dependent. For instance, α-mannosidases I and II, which are typically considered cis/medial Golgi enzymes, are strongly expressed on the brush border of enterocytes—hardly a Golgi compartment. There are other similar examples of various distributions of sialyl transferase. Moreover, there are different sialyl transferases and each may have its own unique distribution. Although one should be cautious, it is probably safe to place sialyl transferase in the late Golgi compartment rather than an early one.
Endo-α-N-acetylgalactosaminidase
This enzyme from Diplococcus pneumoniae also goes by various names, including O-glycosidase and O-glycanase. This enzyme has a narrow substrate range and cleaves only Galβ1-3GalNAcα-Ser/Thr. These are only the first two sugars added in the diverse O-linked pathway that can produce glycans with a dozen or more sugar units. A portion of the pathway is shown in Figure 17.13A.4. Fortunately, many, but far from all, O-linked chains have the simple trisaccharide structure and would be sensitive to cleavage after removing the Sia. Thus, sequential individual digestions or mixed digestions can be used. As both Gal and GalNAc (and probably Sia) are added in the early Golgi complex, sensitivity to the enzyme shows that the protein carries O-linked chains, but matching enzyme sensitivity and a Golgi compartment to further chain extension is difficult. Combining a battery of exoglycosidases (sialidase, α-fucosidase, α-N-acetylgalactosaminidase, and β-hexosaminidase) with endo-α-N-acetylgalactosaminidase will probably remove most O-linked sugar chains, except sulfated ones. The bottom line is that it is easy to use the enzyme in combination with sialidase to show that a protein has simple O-linked chains, but it is difficult to conclude much more concerning either the structure of the sugar chain or intracellular trafficking.
Endo-β-galactosidase
Escherichia freundii endo-β-galactosidase is one of several enzymes that specifically degrade poly-N-acetyllactosamines by cleaving linear chains of GlcNAcβ1-3Galβ1-4 repeats at the Galβ1-4 linkage. Any substitution on the galactose itself blocks cleavage; however, modifications of the neighboring sugars can slow hydrolysis (Fig. 17.13A.7). For instance, fucosylation and/or sulfation of nearby GlcNAc slows cleavage, but chain branching or sulfation at Gal block it. Even with these potential complexities, digestion with endo-β-galactosidase will sharpen a broad band even if it does not cleave every linkage. The repeating GlcNAcβ1-3Galβ1-4 units can be found on both N- and O-linked chains, so sensitivity to PNGase F digestion can potentially distinguish the location. Lysosome-associated membrane protein 1 (LAMP-1) has poly-N-acetyllactosamine repeats on N-linked chains. Remember that glycosylation is not template driven, so glycans often exist as a continuum of different structures on individual proteins. For example, heavily sulfated poly-N-acetyllactosamine repeats are also known as keratan sulfate and are degraded by keratanases.
O-Sialoglycoprotease
O-Sialoglycoprotease (also called O-glycoprotease or O-sialoglycoprotein endopeptidase; Mellors and Lo, 1995) is a neutral metalloprotease produced by Pasteurella haemolytica. This enzyme requires clusters of sialylated glycans on Ser or Thr residues (Norgard et al., 1993). Having a single sialylated O-linked sugar chain on a protein will not lead to degradation, nor will having a nonsialylated sugar chain. Therefore, this enzyme can be used in a typical pulse-chase experiment to indicate whether there are tightly grouped O-linked chains and when they are sialylated. Adding the initial α-GalNAc in the early Golgi complex is insufficient to cause proteolysis; sialylation is specifically required, and this may occur in a later Golgi compartment. Proteolysis can generate smaller fragments of a target protein that are still visible on gels, or the fragments may be so small that they are not even seen on the gels. It depends on the protein. Many leukocyte antigens such as the P-selectin ligand, or others such as CD43, CD44, CD45, and CD34 found on hematopoietic stem cells, are all substrates. As sialic acids are found on both N- and O-linked chains (in clusters and not), sequential digestions using PNGase F, sialidase, and O-sialoglycoprotease endopeptidase in different orders can reveal different kinds of sialylated glycans. If used as part of a pulse-chase protocol, they can reveal different kinetic subcompartments. Not all sialyl transferases are necessarily within the same Golgi compartment.
Critical Parameters
For nearly all of the digestions, complete denaturation of the protein can be important, as maximum deglycosylation occurs only when the sugar chains of glycoproteins are completely exposed. This is not important for sialidase digestions since sialic acids are exposed. Usually endo H digestions will work without full denaturation, but the unprocessed high-mannose chains remain unprocessed because they are often less exposed to the processing enzymes. This may or may not be true of the target protein, but it is better to be safe and denature it completely before digestion. Heating the immunoprecipitate with 0.1 M 2-mercaptoethanol/0.1% SDS is the best way to release the protein in a denatured state. Assuming that the immunoprecipitate contains <10 μg of protein, 30 μl of 0.1% SDS still provides a three-fold excess over the protein. Adding SDS presents another problem: free SDS may denature the digesting enzyme before it has a chance to finish its job. For most enzymes, be sure that the free SDS concentration is <0.01%. The best way to do this is to add a 10- to 20-fold weight excess of nonionic detergent with a low critical micellar concentration (e.g., Triton X-100 or NP-40). These detergents will form mixed micelles with the free SDS and keep it from denaturing the added enzymes.
The amount of enzyme and the incubation time recommended in the protocols are in excess and should be sufficient to cleave any of the sensitive linkages. The incubation times can be shortened, if necessary, but it is better to keep the enzyme concentration as indicated.
Many of the digestions (e.g., sialidase, O-sialoglycoprotease) can be adapted for use on membrane preparations or on live cell surfaces by simply omitting the ionic and nonionic detergents and decreasing the incubation time. The problem is that some linkages may not be exposed and/or sensitive to the digestion. Thus, the usefulness of this approach needs to be determined on a case-by-case basis.
Enzyme sources and availability
All enzymes except O-sialoglycoprotease (Accurate Chemical and Scientific), endo D (Calbiochem), and endo-β-galactosidase (MP Biomedicals or Seikagaku) are available from Calbiochem or Sigma-Aldrich. Other providers are ProZyme, New England Biolabs, Roche Applied Science, and Takara Biochemical.
Endo H
Endo H has a broad pH optimum between 5.5 and 6.5, and phosphate or citrate/phosphate buffers can be used in place of citrate. Endo H is very stable to proteases, freezing and thawing, and prolonged incubations. No additives are required for storage of the enzyme. At concentrations below 5 to 10 μg/ml (200 to 400 mU/ml), endo H will bind to glass, so it should be stored in plastic vials (e.g., screw-cap microcentrifuge tubes).
Endo D
Endo D has a broad pH optimum of 4 to 6.5. One unit of enzyme activity will cleave 1 μmole of a Man5GlcNAc3 glycopeptide per min at 37°C. After reconstitution, the enzyme is stable for 6 months at –20°C. It should be frozen in small aliquots to avoid the need for freezing and thawing.
Endo F
Commercial endo F preparations are mixtures of endo F1, F2, and F3. Endo F preparations should not be used for routine deglycosylation or to draw conclusions about the structure of the released glycans unless the specificity is clearly defined.
Endo F2
Endo F2 has a broad pH optimum of 4 to 6 and retains >50% of its activity at pH 7. The enzyme is sensitive to SDS, but adding nonionic detergents prevents denaturation of the enzyme by SDS. Although the enzyme is stable at 4°C for months, it can be frozen in aliquots at −70°C as long as repeated freeze/thaw cycles are avoided. The 1,10-phenanthroline can be used to inhibit a trace of a zinc metalloprotease that may be present.
PNGase F
The pH optimum for PNGase F is 8.6, but 80% of full activity occurs between 7.5 to 9.5 with a range of buffers including phosphate, ammonium bicarbonate, Tris·Cl, and HEPES. Borate buffers inhibit the enzyme. Commercial PNGase F is available from natural or recombinant sources (two main suppliers, see Enzyme sources And Availability, above) and is stable for 6 months at 4°C, or indefinitely at −70°C. However, it should be stored in small aliquots and repeated freeze/thaw cycles should be avoided. PNGase F will bind to glass and plastic surfaces and should not be stored in dilute solutions (<0.1 mU/ml). All of the unit activities of commercial preparations are based on cleavage of dansylated glycopeptides; they are expressed in nmoles/min, which are actually mU, not true International Units (1 International Unit = 1 μmole/min). SDS inactivates PNGase F, but adding a ten-fold weight excess of nonionic detergents protects the enzyme (Tarentino et al., 1989; Tarentino and Plummer, 1994).
Sialidase
Recombinant AUS is available as a liquid (see Enzyme sources and availability). The native enzyme can be purchased as a lyophilized powder and should be reconstituted in water at 1 to 10 mU/μl, according to the manufacturer’s directions. The native enzyme is stable for 1 year at 4°C if unopened. After reconstitution, it should be used within 7 days and stored at 4°C. The recombinant enzyme is stable for 1 year at 4°C. Treatment with sialidase is also used in assays of protein transport to the cell surface.
Endo-α-N-acetylgalactosaminidase (O-glycosidase)
Endo-α-N-acetylgalactosaminidase has a pH optimum of 6.0 and has 50% activity at 5.5 to 7.0. The thiol inhibitor parachloromercuric benzoate (PCMB; 1 mM) inactivates the enzyme, and 1 mM EDTA inhibits it (63%), as do Mn2+ and Zn2+ (50%). Chloride also inhibits the enzyme, so HCl-containing buffers should be avoided. The enzyme will have full access to the sugar chains only after denaturating the protein with SDS, but the excess SDS needs to be removed by forming mixed micelles with nonionic detergents. The enzyme is stable at 4°C and at −20°C, but freeze/thaw cycles should be avoided.
Endo-β-galactosidase
This enzyme is free of contaminating endo- and exoglycosidases. It has a pH optimum of 5.8 and can be stored at −20°C, for up to 2 years. Dissolved enzyme is only stable for 1 month at −20°C. EDTA, Ca2+, Mn2+, and Mg2+ do not affect stability or activity, but PCMB inactivates it.
O-Sialoglycoprotease
The partially purified enzyme is supplied by Accurate Chemical and Scientific as a lyophilyzed powder containing nonsubstrate bovine serum proteins and HEPES buffer. The enzyme should be reconstituted according to the manufacturer’s instructions, divided into aliquots appropriate for a single use, and stored at −20°C. Freeze/thaw cycles inactivate the enzyme, and it is inhibited by EDTA or 1,10-phenanthroline. It is possible to check the activity with a positive control of glycophorin A, which is available through Sigma-Aldrich.
Troubleshooting
Most of the procedures should work as described, but there is a chance that the enzyme is inactive because of a variety of factors such as age, poor storage, or excess SDS. To check activity, it is worthwhile to run a positive control digestion using the same solutions including SDS and nonionic detergents as for the samples. Since the positive controls are simply glycoproteins that are visualized by Coomassie or silver staining, this requires running a separate gel for staining. This should not be required on a routine basis if the enzymes are used and stored as directed. The most likely culprit in failed digestions is using SDS solutions that are too old or too impure.
Anticipated Results
If the digestions are effective, the labeled band will usually show increased mobility on the gel. In rare instances, digestions can actually decrease mobility. The amount of change will depend on the contribution of that component to the overall mass of the protein. As mentioned before, a gel system that allows visualization of a 1-kDa change should be used. Proteins that are >100 kDa may cause problems for fine resolution. Here are a few numbers to keep in mind.
The smallest N-linked chain (Man3GlcNAc2) will have a mass of ~0.9 kDa.
Two sialic acids on a single N-linked biantennary chain will have a mass of ~0.6 kDa, but their loss may appear larger. If they occur on clustered O-linked chains (sialoglycoprotease sensitive), the apparent size difference will be even larger.
Most poly-N-acetyllactosamines are three or more repeats, and therefore their mass would be −1 kDa. The protein will probably run as a heterogeneous smear or broad band before digestion.
A single O-glycosidase-sensitive disaccharide (0.4 kDa) may be below detection limits.
Time Considerations
All digestions can be done overnight for convenience, but the amount of enzymes should be sufficient for complete digestion in less time. The gels are run the next day, but the time needed for the developsent of autoradiograms will depend on the strength of the signal. A low-abundance protein labeled for 10 to 30 min with [35S]Met may give a weak signal and require long exposures (e.g., 2 weeks). Trafficking of abundant glycoproteins such as viral coat proteins require only short exposure times (e.g., a few hours).
Literature Cited
- Akama TO, Nakagawa H, Wong NK, Sutton-Smith M, Dell A, Morris HR, Nakayama J, Nishimura S, Pai A, Moremen KW, Marth JD, Fukuda MN. Essential and mutually compensatory roles of α-mannosidase II and a-mannosidase IIx in N-glycan processing in vivo in mice. Proc Nati Acad Sei U S A. 2006;13(103):8983–8988. doi: 10.1073/pnas.0603248103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander S, Elder JH. Endoglycosi-dases from Flavobacterium meningosepticum: Application to biological problems. Methods Enzymol. 1989;179:505–518. doi: 10.1016/0076-6879(89)79151-5. [DOI] [PubMed] [Google Scholar]
- Beckers CJM, Keller DS, Balch WE. Semi-intact cells permeable to macro-molecules: Use in reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex. Cell. 1987;50:523–534. doi: 10.1016/0092-8674(87)90025-0. Describes the use of Lee 1 CHO cells and endo D to study processing. [DOI] [PubMed] [Google Scholar]
- Chui D, Oh-Eda M, Liao YE, Panneerselvam K, Lai A, Marek KW, Freeze HH, Moremen KW, Fukuda MN, Marth JD. Alpha-mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell. 1997;90:157–167. doi: 10.1016/s0092-8674(00)80322-0. Demonstrates the importance of α-mannosidase IIx. [DOI] [PubMed] [Google Scholar]
- Davidson HW, Balch WE. Differential inhibition of multiple vesicular transport steps between the endoplasmic reticulum and trans Golgi network. J Biol Chetn. 1993;268:4216–4226. [PubMed] [Google Scholar]
- Elder JH, Alexander S. Endo-β-iV-acetylglucosaminidase F: Endoglycosidase from Flavobacterium meningosepticum that cleaves both high mannose and complex glyco-proteins. Proc Nati Acad Sei U S A. 1982;79:4540–4544. doi: 10.1073/pnas.79.15.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. Landmark review of processing. [DOI] [PubMed] [Google Scholar]
- Mellors A, Lo RY. O-Sialoglycopro-tease from Pasteurella haemolytica. Methods Enzymol. 1995;248:728–740. doi: 10.1016/0076-6879(95)48049-8. [DOI] [PubMed] [Google Scholar]
- Norgard KE, Moore KL, Diaz S, Stults NL, Ushiyama S, McEver RR, Cummings RD, Varki A. Characterization of a specific ligand for P-selectin on myeloid cells. A minor glycoprotein with sialylated O-linked oligosaccharides. J Biol Chem. 1993;268:12764–12774. [PubMed] [Google Scholar]
- Plummer TH, Jr, Tarentino AL. Purification of the oligosaccharide-cleaving enzymes of Flavobacterium meningosepticum. Glycobi-ology. 1991;1:257–263. doi: 10.1093/glycob/1.3.257. [DOI] [PubMed] [Google Scholar]
- Plummer TH, Jr, Elder JH, Alexander S, Phelan AW, Tarentino AL. Demonstration of peptide: iV-glycosidase F activity in endo-β-N-acetylglucosaminidase F preparations. J Biol Chem. 1984;259:10700–10704. [PubMed] [Google Scholar]
- Tarentino AL, Plummer TH., Jr Enzymatic deglycosylation of asparagine-linked glycans: Purification, properties, and specificity of oligosaccharide-cleaving enzymes from Flavobacterium meningosepticum. Methods Enzymol. 1994;230:44–57. doi: 10.1016/0076-6879(94)30006-2. Best review of the use of these enzymes. [DOI] [PubMed] [Google Scholar]
- Tarentino AL, Trimble RB, Plummer TH., Jr Enzymatic approaches for studying the structure, synthesis, and processing of glycopro-teins. Methods Cell Biol. 1989;32:111–139. doi: 10.1016/s0091-679x(08)61169-3. [DOI] [PubMed] [Google Scholar]
- Tretter V, Altmann F, März L. Peptide-N4-(W-acetyl-β-glucosaminyl) asparagine anudase F cannot release glycans with fucose attached α1 → 3 to the asparagine-linked N-acetylglucosamine residue. Eur J Biochem. 1991;199:647–652. doi: 10.1111/j.1432-1033.1991.tb16166.x. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Esko JD, Freeze HH, Stanley R, Bertozzi CR, Hart GW, Etzler ME. Essentials of Gly-cobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor; New York: 2008. http://www.ncbi.nlm.nih.gov/bookshelflbr.fcgi?book=glycol. [Google Scholar]
- Viitala J, Carlsson SR, Siebert RD, Fukuda M. Molecular cloning of cDNAs encoding lamp A, a human lysosomal membrane glycoprotein with apparent Mr approximately equal to 120,000. Proc Nati Acad Sei U S A. 1988;85:3743–3747. doi: 10.1073/pnas.85.11.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]