

Abstract
The medical significance of N-glycosylation is underlined by a group of inherited human disorders called Congenital Disorders of Glycosylation (CDG). One key step in the biosynthesis of the Glc3Man9Glc-NAc2-PP-dolichol precursor, essential for N-glycosylation, is the translocation of Man5GlcNAc2-PP-dolichol across the endoplasmic reticulum membrane. This step is facilitated by the RFT1 protein. Recently, the first RFT1-deficient CDG (RFT1-CDG) patient was identified and presented a severe N-glycosylation disorder. In the present study, we describe three novel CDG patients with an RFT1 deficiency. The first patient was homozygous for the earlier reported RFT1 missense mutation (c.199C4T; p.R67C), whereas the two other patients were homozygous for the missense mutation c.454A4G (p.K152E) and c.892G4A (p.E298 K), respectively. The pathogenic character of the novel mutations was illustrated by the accumulation of Man5GlcNAc2-PP-dolichol and by reduced recombinant DNase 1 secretion. Both the glycosylation pattern and recombinant DNase 1 secretion could be normalized by expression of normal RFT1 cDNA in the patients’ fibroblasts. The clinical phenotype of these patients comprised typical CDG symptoms in addition to sensorineural deafness, rarely reported in CDG patients. The identification of additional RFT1-deficient patients allowed to delineate the main clinical picture of RFT1-CDG and confirmed the crucial role of RFT1 in Man5GlcNAc2-PPdolichol translocation.
Keywords: glycosylation, CDG, RFT1, dolichol
Introduction
N-glycosylation is a posttranslational modification of proteins found in eukaryotic and prokaryotic organisms [Weerapana and Imperiali, 2006]. In eukaryotes, protein N-linked glycans are involved in many essential biological processes including the immune response, intracellular targeting, cell–cell recognition, protein folding, and protein stability [Varki, 1993].
The eukaryotic N-glycosylation pathway starts with the assembly of the Glc3Man9GlcNAc2 oligosaccharide precursor on a dolichol-PP carrier in a well-ordered process known as the dolichol cycle [Burda and Aebi, 1999]. This dolichol cycle starts with the elongation of dolichol-P to Man5GlcNAc2-PP-dolichol on the cytosolic face of the endoplasmic reticulum (ER) membrane, while the elongation of Man5GlcNAc2-PP-dolichol to the complete Glc3Man9GlcNAc2-PP-dolichol occurs in the ER lumen. For this reason, the Man5GlcNAc2-PP-dolichol intermediate has to be translocated across the ER membrane. In yeast, the ER membrane protein Rft1 was shown to facilitate the translocation of Man5GlcNAc2-PP-dolichol to the ER lumen in a bidirectional and ATP-independent manner [Helenius et al., 2002]. Once the fully assembled oligosaccharide precursor is synthesized, Glc3Man9GlcNAc2 is transferred onto selected asparagine residues of polypeptide chains by the oligosaccharyltransferase (OST) complex [Burda and Aebi, 1999].
Congenital disorders of glycosylation (CDG) are a group of inherited human disorders characterized by deficient protein glycosylation. Up to now, 14 different CDG types deficient in protein N-glycosylation site occupancy have been identified. The analysis of the serum sialotransferrin pattern is the most widely used method to screen for N-glycosylation disorders, which can be classified into two subgroups: defects of oligosaccharide precursor assembly and transfer to proteins (formerly known as CDG-I) and defects of N-glycoprotein processing (formerly known as CDG-II) [Eklund and Freeze, 2006; Freeze, 2007; Jaeken and Matthijs, 2007; Leroy, 2006].
Recently, the first RFT1-deficient patient (MIM] 612015) was identified [Haeuptle et al., 2008]. Haeuptle and coworkers described a young girl presenting with marked psychomotor retardation, hypotonia, seizures, hepatomegaly, and coagulopathy. This patient was homozygous for the missense mutation c.199C4T (p.R67C) in the RFT1 gene (MIM] 611908) and accumulated Man5GlcNAc2-PP-dolichol as shown by lipid-linked oligosaccharides (LLO) analysis. However, no Man5GlcNAc2 was transferred onto glycoproteins, which pointed to a deficient translocation of the accumulated LLO across the ER membrane.
In the present study, we describe three additional RFT1deficient patients including two novel pathogenic point mutations. The identification of these additional patients allowed us to refine the clinical phenotype characteristic for RFT1 deficiency, designated as RFT1-CDG according to the suggested novel nomenclature [Jaeken et al., 2008].
Materials and Methods
Cell Culture
Primary skin fibroblasts from healthy controls and patients were cultured at 371C under 5% CO2 in DMEM/F12 (Life Technologies, Washington, DC) supplemented with 10% fetal bovine serum (Clone III, HyClone, Logan, UT).
Research on patients’ cells was prospectively reviewed and approved by the Ethics Committee of the University of Leuven.
Mutation Analysis
Total RNA was isolated from 2 ×107 fibroblasts using the TRIzol LS reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The human RFT1 cDNA was prepared and the protein-coding region was amplified by PCR as described before [Haeuptle et al., 2008]. The PCR products were sequenced (Synergene Biotech, Zurich, Switzerland) after removal of the unincorporated nucleotides with QIAquick columns (Qiagen, Chatsworth, CA). Carrier analysis in the parents and healthy siblings was performed on DNA extracted from blood. Primers were designed to amplify exons 3, 4, and 9 of the RFT1 gene, based on the genomic sequence (NM_052859.2). Primer sequences are available on request. These exons were amplified using standard PCR conditions, subsequently sequenced with the Big Dye Terminator Ready reaction cycle sequencing kit V3.1 (Applera, Norwalk, CT) and analyzed on an ABI3100 Avant (Applera). The numbering of the nucleotide changes is based on the cDNA sequence (NM_052859.2) with 11 corresponding to the A of the ATG translation initiation codon in the reference sequence (www.hgvs.org/mutnomen).
Metabolic Radiolabeling
Fibroblasts (8 ×106 per labeling) were grown overnight in a 175 cm2 tissue culture flask. After 24 hr, cells were preincubated in 0.5 mM glucose for 45 min and then pulse radiolabeled for 1 hr with 150 mCi of 2-[3H]-mannose (16 Ci/mmol, Amersham Biosciences, Buckinghamshire, England). After metabolic labeling, the cells were scraped with 1.1 ml MeOH/H2O (8:3) followed by the addition of 1.2 ml CHCl3. Sequential extraction of oligosaccharide material was performed as previously described [Cacan and Verbert, 1997].
Analysis of Oligosaccharide Material
Glycoprotein fractions obtained at the end of the sequential extraction were digested overnight at room temperature with trypsin (1 mg/ml; Sigma, St. Louis, MO) in 0.1 M ammonium bicarbonate buffer, pH 7.9. The resulting glycopeptides were treated with 0.5 U PNGase F (Roche, Indianapolis, IN) in 50 mM phosphate buffer, pH 7.2 for 4 hr to release the oligosaccharides from the peptides. The oligosaccharides were desalted on Bio-Gel P2 columns and eluted with 5% acetic acid. LLO fractions obtained after sequential extraction were subjected to mild acid treatment (0.1 M HCl in THF) for 2 hr at 501C. Purification of the released oligosaccharides was performed as described above.
The oligosaccharides were separated by HPLC on an amino derivated Asahipak NH2P-50 column (250 mm x4.6 mm; Asahi, Malden, MA) applying a gradient of acetonitrile/H2O ranging from 70:30 to 50:50 over 90 min at a flow rate of 1 ml/min. Oligosaccharides were identified on the basis of their retention times compared to standard glycans [Foulquier et al., 2002]. Elution of the radiolabeled oligosaccharides was monitored by continuous b-counting with a flo-one b detector (Packard).
Complementation of the LLO profile
Lentiviruses containing RFT1 and EGFP cDNAs were prepared as previously described [Haeuptle et al., 2008]. Briefly, 3 ×106 HEK293 T cells were transfected with 20 mg pLenti6-hRFT1 or pLenti6-EGFP and 36 mg of the packing plasmid mix (Invitrogen) by calcium–phosphate precipitation. Two days after transfection, the supernatants were collected and used to transduce the patient fibroblasts. After selection with 5 mg/ml blasticidin (Invitrogen), the cells were metabolically labelled with 2-[3 H]-mannose and the extracted LLO were analyzed by HPLC [Zufferey et al., 1995].
Complementation of DNase 1 Secretion
Bovine DNase I cDNA was subcloned into the pSVK3 vector as previously reported [Nishikawa et al., 1997]. Patients’ fibroblasts were transduced with an adenovirus expressing bovine DNase I [Eklund et al., 2005; Fujita et al., 2008]. Two days posttransduction, the cells were washed twice with PBS and incubated at 371C with 1 ml DMEM without methionine/cysteine, containing 10% dialyzed fetal bovine serum, 10 mM NH4Cl, and 0.2 mCi [35S]-Met/Cys labelling mixture. After a labeling time of 4 hr, the culture medium was harvested and DNase 1 was immunoprecipitated as previously described [Nishikawa and Mizuno, 2001]. The immunoprecipitated samples were subjected to SDS-PAGE using a 13% acrylamide gel. After electrophoresis, the gels were rinsed with a mixture of 7% acetic acid and 10% methanol for 15 min and soaked in Amplify solution (Amersham Pharmacia Biotech) for 15 min. The gels were dried, autoradiography was carried out, and the intensity of the bands corresponding to DNase I was quantified by scanning densitometery (using Quantity One software PDI). Band intensity was normalized against protein levels.
Results
Biochemical and Molecular Diagnosis
Three patients with a type 1 pattern of serum sialotransferrins were further investigated. Subsequently, phosphomannomutase (PMM2-CDG) and phosphomannose isomerase (PMI-CDG) deficiencies were excluded on the basis of enzymatic activity measurements in the patients’ fibroblasts (data not shown).
To identify a defect in the assembly of the oligosaccharide precursor within the dolichol cycle, structural analysis of the LLO was performed by HPLC after metabolic labeling. In healthy control fibroblasts, the LLO profile is characterized by the fully assembled oligosaccharide precursor Glc3Man9GlcNAc2-PPdolichol. In contrast, an accumulation of Man5GlcNAc2-PPdolichol in combination with only a minor amount of complete LLO was detected in all three patients (Fig. 1A–D). Additionally, protein-linked glycan structures were analyzed. In both healthy control and patients, Man8GlcNAc2,Man9GlcNAc2,and Glc1Man9 GlcNAc2 structures were obtained (Fig. 1E–H). Notably, no Man5GlcNAc2 structure could be detected on the patients’ glycoproteins.
Figure 1.
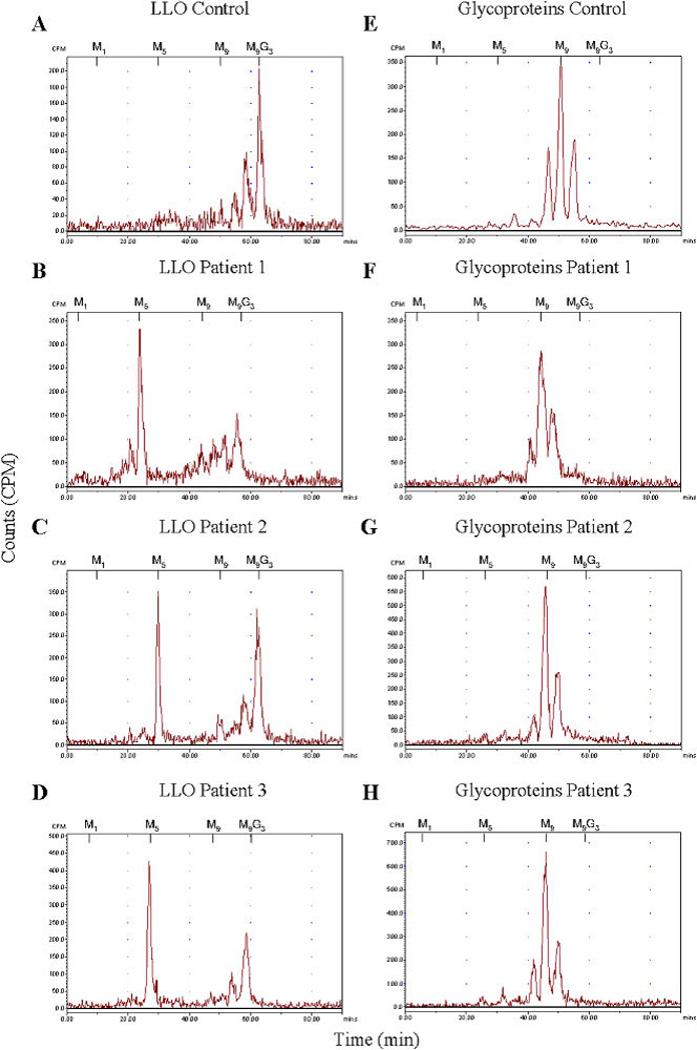
A–D: HPLC analysis of the LLO in control and patients’ fibroblasts revealing the accumulation of Man5GlcNAc2-PP-dolichol in the three patients. E–H: Protein N-linked oligosaccharides of control and patients were separated by HPLC, demonstrating that no aberrant glycan structures were detected in the patients. The retention times of the standard oligosaccharides Glc3Man9GlcNAc2-PP-dolichol (G3M9) and Man1-9 GlcNAc2-PP-dolichol (M1–9) are marked above the HPLC profiles.
The accumulation of Man5GlcNAc2-PP-dolichol on LLO combined with normal protein-linked glycans is typical for a defect in the translocation of Man5GlcNAc2-PP-dolichol to the ER lumen and was recently reported in a RFT1-deficient patient (MIM] 612015) [Haeuptle et al., 2008]. To this end, mutation analysis of the RFT1 cDNA was performed in the present patients and all three carried RFT1 mutations. The first patient was homozygous for the earlier reported missense mutation (c.199C4T; p.R67C) [Haeuptle et al., 2008], whereas the parents were heterozygous carriers of this mutation (data reviewed but not shown). Two novel point mutations were identified in two other patients. Sequencing of the second patient’s RFT1 cDNA revealed an A-to-G transition at nucleotide position 454, leading to the conversion of a lysine into a glutamic acid at position 152 on the protein level (data reviewed but not shown). Both parents as well as a healthy sibling were heterozygous for this mutation. In model eukaryotic organisms, the lysine at position 152 is mostly conserved and a conversion of a lysine into a glutamic acid was not observed (data not shown). The third patient was homozygous for a G-to-A transition at nucleotide position 892, causing a glutamic acid to lysine change at position 298 (data reviewed but not shown). The mutated glutamic residue is strictly conserved among eukaryotes (data not shown). No material of the parents and siblings was available for carrier analysis.
In addition, all converted amino acids were found in hydrophilic domains of the RFT1 protein predicted to be oriented to the ER lumen (TMpred [Hofmann and Stoffel, 1993]; TMHMM [Krogh et al., 2001]) (Fig. 2). As previously reported, the p.R67C mutation is located in the first luminal loop of the RFT1 protein [Haeuptle et al., 2008]. The p.K152E mutation was found in the second luminally oriented 25 amino acid long hydrophilic stretch and the p.E298K mutation was positioned in the largest luminal loop, bearing a potential N-glycosylation site at position p.N227 and ranging over 130 amino acids (Fig. 2).
Figure 2.
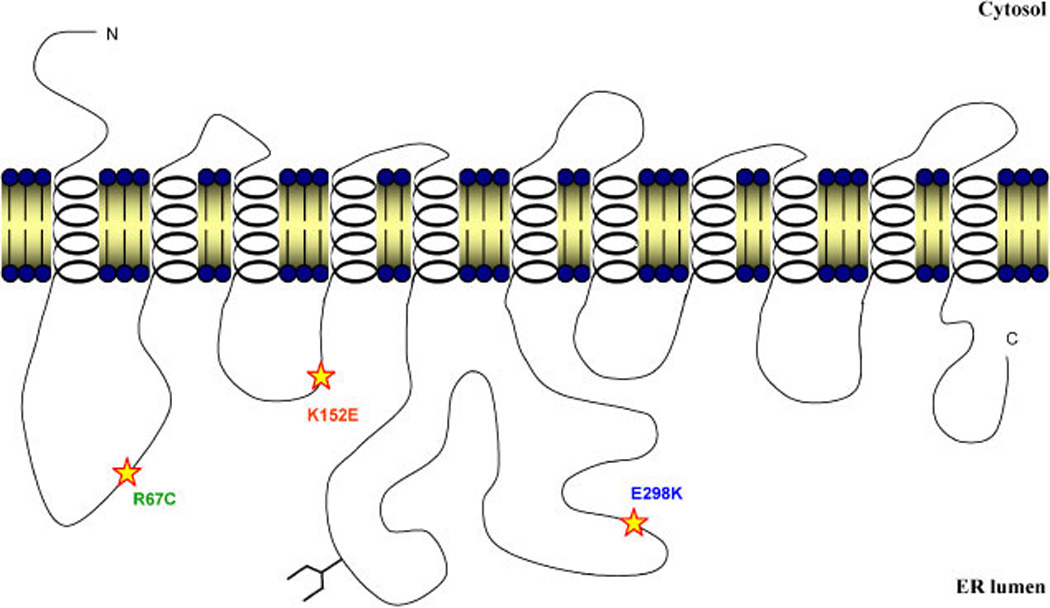
Schematic representation of the identified missense mutations in the predicted topology of the RFT1 protein. All three mutations were found in the predicted luminal loops of the protein. The orientation of the model was supported by the favoured position of the 11 transmembrane domains and the localization of a potential N-glycosylation site (p.N227) (TMpred [Hofmann and Stoffel, 1993]; TMHMM [Krogh et al., 2001]). [Color figure can be viewed online at www.interscience.wiley.com.]
Complementation of the LLO Profile
To demonstrate the pathogenicity of the two new missense mutations, wild-type RFT1 cDNA was transduced into patients’ fibroblasts using a lentiviral construct to complement the Man5GlcNAc2-PP-dolichol accumulation. Healthy control and patients’ fibroblasts were thus infected with recombinant lentiviruses expressing either the wild-type RFT1 cDNA or EGFP as a negative control. Compared to EGFP expression in the patients’ fibroblasts, analysis after RFT1 expression revealed a normalization of the LLO profile characterized by decreased levels of Man5GlcNAc2-PP-dolichol and increased levels of the complete Glc3Man9GlcNAc2-PP-dolichol (Fig. 3). EGFP expression in the patients’ fibroblasts did not affect the LLO profile, as expected.
Figure 3.
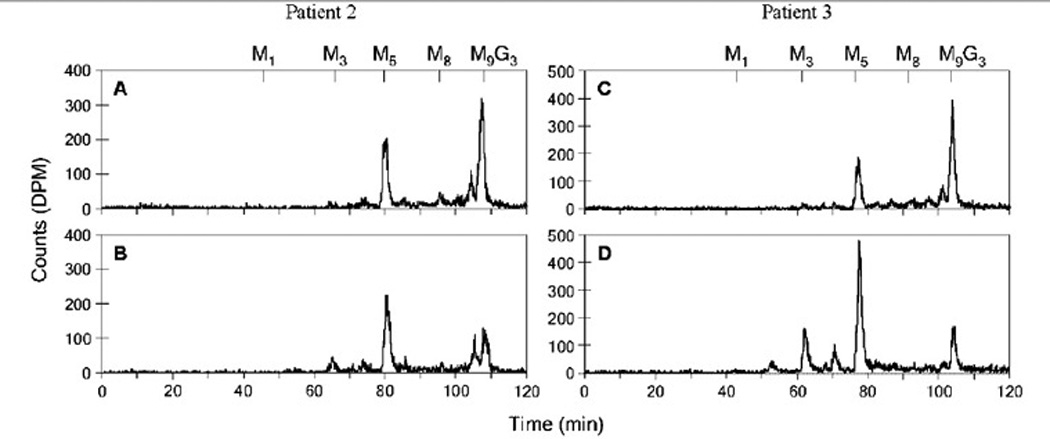
Complementation of the Man5GlcNAc2-PP-dolichol accumulation. Fibroblasts of patient 2 (A) and patient 3 (C) expressing wild-type RFT1 cDNA, leading to a decreased accumulation of Man5GlcNAc2-PP-dolichol. Lentiviral mediated expression of EGFP in the fibroblasts of patient 2 (B) and patient 3 (D) showed no effect. The retention times of the standard oligosaccharides Glc3Man9GlcNAc2-PP-dolichol (G3M9) and Man1-9GlcNAc2-PP-dolichol (M1-9) are marked above the HPLC profiles.
Complementation of DNase 1 Secretion
In another alternative method to prove the pathogenicity of the two new mutations, secretion of recombinant bovine DNase 1 was investigated in all three RFT1-deficient patients. A modified version of bovine DNase 1 has a single potential N-glycosylation site (p.N106) and is secreted when expressed in human fibroblasts [Nishikawa and Mizuno, 2001]. However, upon expression in the fibroblasts of CDG patients, DNase 1 secretion was strongly reduced [Eklund et al., 2005; Fujita et al., 2008]. As shown in Figure 4 (lanes 3, 5, and 7), DNase 1 secretion was significantly reduced in all three RFT1-deficient patients. Transduction of the patients’ fibroblasts with lentiviruses coding for wild-type RFT1 restored the levels of DNase 1 secretion (Fig. 4, lanes 4, 6, and 8), thus demonstrating the pathogenicity of the new mutations.
Figure 4.
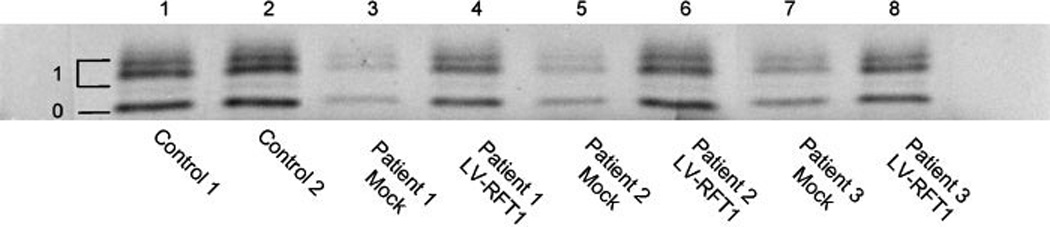
Rescue of DNase 1 secretion in the patients’ fibroblasts. Secretion of DNase 1 is reduced in the fibroblasts of the three RFT1deficient patients (lanes 3, 5, and 7). Expression of wild-type RFT1 cDNA (lanes 4, 6, and 8) restored the secretion of DNase 1 to a level equal to controls (lanes 1 and 2). Symbols: o 5 nonglycosylated DNase 1; 1 5 singly glycosylated DNase 1.
Clinical Phenotype
To address the main clinical characteristics of RFT1 deficiency, the clinical phenotypes of the three different patients were compared. Patient 1 was the first child of healthy, unrelated North Americans of Scottish–English origin and presented with respiratory insufficiency, severe generalized epilepsy with intractable seizures, infantile spasms, microcephaly, failure to thrive, hypotonia, sensorineural deafness, and decreased visual acuity. In addition, this patient showed severe mental retardation, with virtually no development, and had normal liver function. Dysmorphic features included micrognathia, short neck, small nose, drooping eyelids, valgus feet, and adducted thumbs. This patient also had severe feeding problems requiring gastrostomy and died at the age of 8 months. Brain magnetic resonance imaging (MRI) at early age did not reveal cerebral or cerebellar atrophy. However, at autopsy, weight and gyral/sulcal pattern consistent with a degree of cerebral atrophy was reported (Table 1).
Table 1.
Clinical Phenotype of the Three New RFT1-Deficient Patients Compared to the Phenotype of the Originally Reported Case (KS) [Clayton and Grunewald, 2009; Haeuptle et al., 2008; Imtiaz et al., 2000]
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | ||
|---|---|---|---|---|---|
| Gender | female | male | male | female | |
| Origin | Scottish | Italian | Algerian | English | |
| Vital status | died 8 m | alive 5.5 y | alive 2.2 y | died 4.3 y | |
| Consanguinity | − | + | + | − | |
| Feeding problems | + | + | + | + | |
| Failure to thrive | + | + | + | + | |
| Severe mental retardation | + | + | + | + | |
| Microcephaly | + | + | − | na | |
| Hypotonia | + | + | + | + | |
| Epilepsy | + | + | + | + | |
| Decreased visual acuity | + | + | + | + | |
| Sensorineural deafness | + | + | + | + | |
| Myoclonic jerks | + | + | + | + | |
| Respiratory insufficiency | + | − | + | na | |
| Pulmonary infections | − | + | + | + | |
| Coagulopathy | na | + | na | + | |
| Dysmorphic features: | micrognathia | + | − | + | na |
| short neck | + | − | + | na | |
| valgus feet | + | + | + | na | |
| adducted thumbs | + | na | + | − | |
| kyphoscoliosis | na | + | + | ||
| inverted nipples | − | + | + | + | |
| Brain MRI: | cerebral atrophy | −* | + | − | + |
| cerebellar atrophy | − | − | − | + |
na = not available
Cerebral atrophy reported at autopsy
Patient 2 was the second son of healthy, consanguineous Italian parents. During the first year of life, he presented with severe developmental delay, microcephaly, nystagmus, sensorineural deafness, relapsing aspiration pneumonia, a generalized hypotonia, and inverted nipples. Additionally, this patient was frequently hospitalized because of drug-resistant epilepsy and feeding problems requiring gastrostomy. At the age of 4 years, computer tomography scan of the brain revealed a stroke-like episode affecting the left frontal lobe, and the following year he started to suffer from recurrent deep venous thrombosis of the left leg. Clinical examination at the age of 5 years showed a severe mentally retarded child with microcephaly, spastic tetraparesis, and kyphoscoliosis. Serial brain MRI examinations showed progressive cortical and subcortical atrophy with no cerebellar involvement. Liver function has always been normal (Table 1).
The third patient was the ninth child of healthy, consanguineous parents from Algerian origin. This family has two children presenting with the autosomal recessive disorder Hemophagocytic lymphohistiocytosis (MIM] 267700). One child died at the age of 3 months, while the other is still alive due to bone marrow transplantation at the age of 4 months. In addition, one son died at the age of 4 days and presented with similar characteristics as patient 3. Patient 3 presented with respiratory insufficiency, hypotonia, body spasm, failure to thrive, epilepsy, bilateral glaucoma, and sensorineural deafness. Dysmophic features included slightly inverted nipples, infiltrated ears, short neck, retrognathism, glossoptosis, adducted thumbs, and valgus feet. Brain MRI did not show cerebral or cerebellar atrophy. This patient also had feeding problems and chronic infections of the respiratory tract. In addition, he showed severe mental retardation with the absence of visual contact. Liver function was normal (Table 1).
Discussion
Based on the analysis of LLO and protein-linked glycan profiles in fibroblasts, three potential RFT1-deficient patients were identified. All three patients showed an accumulation of the LLO Man5GlcNAc2-PP-dolichol, whereas no Man5GlcNAc2 was detected on glycoproteins. Mutation analysis of the RFT1 gene revealed that one patient was homozygous for the earlier reported missense mutation (c.199C4T, p.R67C). This is the second report of the p.R67C mutation in a patient of British origin [Imtiaz et al., 2000], and there are no indications that both patients are related. This could thus point to a founder effect of the p.R67C mutation. The other patients were homozygous for the new missense mutations c.454A4G (p.K152E) and c.892G4A (p.E298 K). The pathogenic character of these novel mutations was demonstrated by the complementation of the abnormal LLO profile and reduced DNase 1 secretion upon expression of wild-type RFT1 in the patients’ fibroblasts.
All three RFT1 mutations identified so far are located in one of the hydrophilic loops predicted to be within the ER lumen. It can be assumed that these regions are of major importance for the translocation of Man5GlcNAc2-PP-dolichol in the ER lumen or for the maintenance of Man5GlcNAc2-PP-dolichol on the luminal side. Further structural analysis will be required to confirm the predicted orientation of the RFT1 protein. Determination of the occupancy of the putative N-glycosylation site at position p.N227 (Fig. 2) would certainly contribute to establish the topology of the RFT1 protein.
In yeast, the Rft1 protein was genetically identified as a protein mediating the translocation of LLO across the ER membrane [Hselenius et al., 2002]. However, recent evidence suggests that the RFT1 protein would not be the flippase enzyme itself, but would play a critical accessory role in translocating Man5GlcNAc2PP-dolichol to the ER lumen [Frank et al., 2008; Sanyal et al., 2008]. Anyhow, the identification of three additional RFT1-deficient patients clearly underscores the major importance of RFT1 in this translocation event.
Finally, the identification of three additional patients allowed us to refine the clinical phenotype characteristic for RFT1 deficiency (MIM] 612015). All four known RFT1-deficient patients showed very similar characteristics including severe mental retardation, hypotonia, epilepsy, myoclonic jerks, decreased visual acuity, sensorineural deafness, and feeding problems (Table 1). In comparison to other CDG defects, RFT1 deficiency is thus mainly a neurological disorder. Strikingly, sensorineural deafness was found in all four RFT1-deficient patients [Imtiaz et al., 2000], and might represent a characteristic clinical feature of RFT1-CDG, because deafness has been reported in only a few other CDG patients [Hutchesson et al., 1995; Imtiaz et al., 2000; Kranz et al., 2007].
Acknowledgments
We thank Dr. MC. Nassogne for providing the clinical data of patient 3. We also thank Dr. N. Seta for the initial molecular and biochemical workup of patient 2. Additionally, we are thankful to L. Keldermans (Leuven) and C. Maag (Zürich) for their help with the project. This work was supported by the European Commission (Sixth Framework Programme, contract LSHM-CT.2005-512131 to EUROGLYCANET; http://www.euroglycanet.org); Marie Curie (Marie Curie European Reintegration Grant to F.F.); the “Ministère de la Recherche et de l’Enseignement supérieur” (“Allocation de Recherche” to W.V.); the Research Foundation (FWO) Flanders (Grant G.0553.08 to G.M.) and the National Institute of Digestive and Kidney Diseases (Grant DKR0155615 to H.H.F.), and the Rocket Williams Fund.
References
- Burda P, Aebi M. The dolichol pathway of N-linked glycosylation. Biochim Biophys Acta. 1999;1426:239–257. doi: 10.1016/s0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- Cacan R, Verbert A. Glycosyltransferases of the phosphodolichol pathways. In: Verbert A, editor. Methods on glycoconjugates: a laboratory manual. Chur, Switzerland: Harwood Academic Publishers; 1997. pp. 191–199. [Google Scholar]
- Clayton P, Grunewald S. Comprehensive description of the phenotype of the first case of congenital disorder of glycosylation due to RFT1 deficiency (CDG In) J Inherit Metab Dis. 2009 doi: 10.1007/s10545-009-1108-x. (in press). [DOI] [PubMed] [Google Scholar]
- Eklund EA, Freeze HH. The congenital disorders of glycosylation: a multifaceted group of syndromes. NeuroRX. 2006;3:254–263. doi: 10.1016/j.nurx.2006.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund EA, Merbouh N, Ichikawa M, Nishikawa A, Clima JM, Dorman JA, Norberg T, Freeze HH. Hydrophobic Man-1-P derivatives correct abnormal glycosylation in Type I congenital disorder of glycosylation fibroblasts. Glycobiology. 2005;15:1084–1093. doi: 10.1093/glycob/cwj006. [DOI] [PubMed] [Google Scholar]
- Foulquier F, Harduin-Lepers A, Duvet S, Marchal I, Mir AM, Delannoy P, Chirat F, Cacan R. The unfolded protein response in a dolichyl phosphate mannose-deficient Chinese hamster ovary cell line points out the key role of a demannosylation step in the quality-control mechanism of N-glycoproteins. Biochem J. 2002;362:491–498. doi: 10.1042/0264-6021:3620491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CG, Sanyal S, Rush JS, Waechter CJ, Menon AK. Does Rft1 flip an N-glycan lipid precursor? Nature. 2008;454:E3–E4. doi: 10.1038/nature07165. [DOI] [PubMed] [Google Scholar]
- Freeze HH. Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med. 2007;7:389–396. doi: 10.2174/156652407780831548. [DOI] [PubMed] [Google Scholar]
- Fujita N, Tamura A, Higashidani A, Tonozuka T, Freeze HH, Nishikawa A. The relative contribution of mannose salvage pathways to glycosylation in PMI-deficient mouse embryonic fibroblast cells. FEBS J. 2008;275:788–798. doi: 10.1111/j.1742-4658.2008.06246.x. [DOI] [PubMed] [Google Scholar]
- Haeuptle MA, Pujol FM, Neupert C, Winchester B, Kastaniotis AJ, Aebi M, Hennet T. Human RFT1 deficiency leads to a disorder of N-linked glycosylation. Am J Hum Genet. 2008;82:600–606. doi: 10.1016/j.ajhg.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius J, Ng DTW, Marolda CL, Walter P, Valvano MA, Aebi M. Translocation of lipid-linked oligosaccharides across the ER membrane requires Rft1 protein. Nature. 2002;415:447–450. doi: 10.1038/415447a. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W. TMbase—a database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler. 1993;374:166. [Google Scholar]
- Hutchesson ACJ, Gray RGF, Spencer DA, Keir G. Carbohydrate deficient glycoprotein syndrome; multiple abnormalities and diagnostic delay. Arch Dis Child. 1995;72:445–446. doi: 10.1136/adc.72.5.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiaz F, Worthington V, Champion M, Beesley C, Charlwood J, Clayton P, Keir G, Mian N, Winchester B. Genotypes and phenotypes of patients in the UK with carbohydrate-deficient glycoprotein syndrome type 1. J Inherit Metab Dis. 2000;23:162–174. doi: 10.1023/a:1005669900330. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Hennet T, Freeze HH, Matthijs G. On the nomenclature of congenital disorders of glycosylation (CDG) J Inherit Metab Dis. 2008;31:669–672. doi: 10.1007/s10545-008-0983-x. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genom Hum Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- Kranz C, Basinger AA, Güc-savas-Calikoglu M, Sun L, Powell CM, Henderson FW, Aylsworth AS, Freeze HH. Expanding spectrum of congenital disorder of glycosylation Ig (CDG-Ig): sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am J Med Genet Part A. 2007;143A:1371–1378. doi: 10.1002/ajmg.a.31791. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Leroy JG. Congenital disorders of N-glycosylation including diseases associated with O-as well as N-glycosylation defects. Pediatr Res. 2006;60:643–656. doi: 10.1203/01.pdr.0000246802.57692.ea. [DOI] [PubMed] [Google Scholar]
- Nishikawa A, Gregory W, Frenz J, Cacia J, Kornfeld S. The phosphorylation of bovine DNase I Asn-linked oligosaccharides is dependent on specific lysine and arginine residues. J Biol Chem. 1997;272:19408–19412. doi: 10.1074/jbc.272.31.19408. [DOI] [PubMed] [Google Scholar]
- Nishikawa A, Mizuno S. The efficiency of N-linked glycosylation of bovine DNase I depends on the Asn-Xaa-Ser/Thr sequence and the tissue of origin. Biochem J. 2001;355:245–248. doi: 10.1042/0264-6021:3550245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S, Frank CG, Menon AK. Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry. 2008;47:7937–7946. doi: 10.1021/bi800723n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E, Imperiali B. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology. 2006;16:91R–101R. doi: 10.1093/glycob/cwj099. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Knauer R, Burda P, Stagljar I, te Heesen S, Lehle L, Aebi M. STT3, a highly conserved protein required for yeast oligosaccharyl transferase activity in vivo. EMBO J. 1995;14:4949–4960. doi: 10.1002/j.1460-2075.1995.tb00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]