

Abstract
The cooperative binding of N2 by late transition metals and main-group metals is a promising strategy for N-N bond weakening and activation. We report the use of activated Rieke magnesium for reduction of iron and cobalt complexes supported by bulky β-diketiminate ligands. Binding of N2 is accompanied by assembly of a linear M-NN-Mg-NN-M (M = Co, Fe) core with N-N bonds that are weakened, as judged by infrared spectroscopy. Both the cobalt and iron complexes require THF solvent, because of Mg-THF binding. The cobalt complex can be isolated as a pure solid, but the iron complex is stable only in solution. These results demonstrate the correlation between the binding mode and N-N weakening in heterobimetallic N2 complexes.
Keywords: Iron, Cobalt, Magnesium, Dinitrogen, Backbonding
Introduction
The catalytic reduction of N2 to NH3 via the Haber-Bosch process is one of the most important industrial reactions performed worldwide, as NH3 is a precursor for many nitrogen containing compounds including fertilizers required for food production.[1,2] A number of heterogeneous zerovalent metal catalysts (Fe, Ru, Os, U, and Co-Mo) are capable of catalyzing the Haber-Bosch process, but an iron catalyst with K+ additives is the most widely used due to its low cost.[1,3] Coordination chemists have studied the binding of N2 to iron complexes to understand the potential mechanisms in more detail.[4,5] Despite these efforts, there have been few reports where the N-N bond is substantially weakened (as judged by the N-N bond length) in an iron N2 complex,[5] and even fewer where the N-N bond has been cleaved in well-characterized products.[6,7]
Interestingly, each published example of complete N-N bond cleavage at iron involved an N2 complex with cooperative binding between Fe and an alkali metal cation. Splitting of N2 to two nitrides in a β-diketiminate supported system gave a product in which one of the two nitrides had close interactions with two K+ ions.[6] The stepwise cleavage of N2 in a tris(phosphino)borane system involved an intermediate in which bound N2 had a side-on interaction with a Na+ ion.[7] These results motivate the more thorough investigation of the involvement of s-block metal cations in N-N bond weakening and cleavage.
Our studies have primarily involved low-coordinate end-on bridging N2 complexes with an MNNM core (M = Fe, Co) supported by bulky β-diketiminate ligands that led to a three-coordinate trigonal-planar geometry at the metal.[8] Neutral, formally iron(I) compounds were found to have weakened N-N bonds (1.18 – 1.19 Å; 1778–1810 cm−1), with significantly longer N-N bonds than in the analogous cobalt compound (1.14 Å) due to better π-backbonding from the higher-energy d orbitals of iron. These compounds were reduced by elemental Na or by potassium/graphite (Scheme 1) to give tetrametallic N2 complexes, M′2[M-NN-M] (M′ = Na, K; M = Fe, Co), with alkali metal cations coordinated to the aryl rings of the ligand and to the N2 ligand. The N-N bond was further weakened in both the formally iron(0) (1.21–1.24 Å; 1583 – 1625 cm−1) and cobalt(0) (1.21–1.22 Å; 1598 cm−1) complexes. Complexes with Na+ were similar to those with K+. In contrast to the formally univalent complexes, the weakening of the N-N bond in zerovalent cobalt complexes was nearly identical to that in zerovalent iron complexes.
Scheme 1.
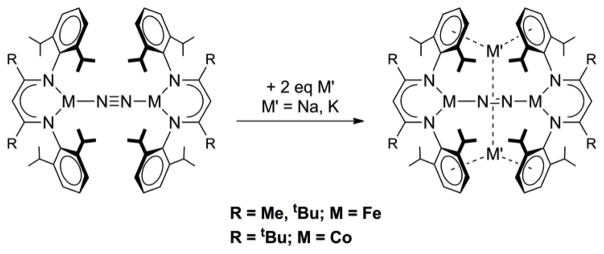
Previously reported reduction of β-diketiminate-supported iron and cobalt dinitrogen complexes to the formal zerovalent state. Note that the alkali metals have side-on interactions with the bound N2.
Here, we show that the use of Mg[9] as a reductant gives a different type of heterobimetallic product in iron and cobalt complexes of nitrogen. This represents a rare example of a single series of transition metal-N2 complexes in which the binding of Na+, K+, and Mg2+ can be directly compared. In addition to the preparation of the new compounds, we report here a combination of crystallographic, spectroscopic, analytical, and reactivity studies on the heterobimetallic complexes.
Results
Formation of a Cobalt-N2 Complex Containing Mg
The reduction of LtBuCoCl with 3 molar equivalents of Rieke magnesium (Mg*)[9] in THF under an argon atmosphere yielded the previously reported Co(I) complex, LtBuCo(THF).[10] This known complex was identified by 1H NMR spectroscopy, and by its distinct dark green color in THF. However, when the reaction was performed under an N2 atmosphere, the initial formation of LtBuCo(THF) was followed by transformation to a dark red mixture over 90 min (Scheme 2). The dark red product 1 was poorly soluble in THF and Et2O and was insoluble in nonpolar solvents, so the mixture was filtered to give a mixture of dark red powder and the remaining Mg*. Because of the low solubility of 1, extraction, filtration and crystallization gave an isolated yield of only 6%. However, since the crude sample of 1 + Mg* had similar spectroscopic properties to the isolated solid, it is likely that the conversion to 1 is significantly higher.
Scheme 2.

Reduction of LtBuCoCl with Rieke magnesium (Mg*) gave {LtBuCo(μ-N2)}2Mg(THF)4 (1).
Compound 1 could be crystallized. Elemental analysis of the crystals showed an amount of N indicating the presence of one N2 per cobalt, and the CHN analysis was consistent with the formulation {LtBuCo(μ-N2)}2Mg(THF)4. A single crystal of 1, from a THF solution layered with Et2O at −45 °C, gave weak diffraction data that were only suitable for confirming the connectivity of 1 (R ~ 20%). The refined structure displayed a central Mg(THF)42+ dication bound to two complex anions of the formally cobalt(0) fragment LtBuCoN2−, and was fully consistent with the determined microanalytical results. In this structure, the Mg2+ had an octahedral geometry with the LtBuCoN2− anions trans to one another.
The infrared spectrum of 1 in dilute THF revealed an intense band at 1878 cm−1, which is assigned as the N-N stretching vibration (νNN). The frequency of the νNN band agreed with those obtained from solid 1 (1868 cm−1) and the solid mixture of 1 and Mg* (1867 cm−1). Compound 1 was also prepared under an atmosphere of isotopically labeled 15N2 to yield 1-15N2, which gave shifts in the νNN bands for 1-15N2 in THF (1816 cm−1) and the solid mixture of 1-15N2 and Mg* (1805 cm−1). The observation of N-N stretching vibrations in the IR spectrum is consistent with the {LtBuCo(μ-N2)}2Mg core, and inconsistent with a symmetric complex (in an MNNM core, the N-N stretching motion is totally symmetric and therefore forbidden in the infrared spectrum).
Analogous Fe-Mg-N2 Complexes are Metastable
The reduction of a bright red solution of the iron(II) complex LtBuFeCl in THF with 3–5 molar equivalents of Mg* under a nitrogen atmosphere gave a dark green mixture (2), which after filtration exhibited a νNN band at 1808 cm−1. Likewise, the reduction of a yellow THF solution of [LMeFe(μ-Br)]2 with 3–5 molar equivalents of Mg* produced a dark green mixture (3), which after filtration exhibited a νNN band at 1818 cm−1. Solutions of 2 or 3 in THF decomposed within 12 h at room temperature and within 3 days at −45 °C. THF was the only suitable solvent for 2 and 3: use of other solvents (pentane, Et2O, diglyme, and mixtures of these) for the reactions with Mg* gave the previously characterized bridging N2 complexes LtBuFeNNFeLtBu and LMeFeNNFeLMe,[8] even in the presence of excess Mg*. Also, addition of Et2O or nonpolar solvents to THF solutions of 2 or 3 resulted in decomposition to solutions containing predominantly LtBuFeNNFeLtBu or LMeFeNNFeLMe. Therefore, we were able to characterize 2 and 3 only in THF solution.
Infrared spectroscopy of the THF solutions of 2 and 3 showed the presence of intense νNN bands at 1808 cm−1 and 1818 cm−1, respectively, which shifted to 1748 cm−1 and 1758 cm−1 with the use of 15N2. These data clearly establish the presence of bound N2 in the major products, and the IR activity again rules out structures with N2 at the center. These data are most consistent with a Fe-N2-Mg-N2-Fe core similar to the one in the cobalt analogue above; additional data supporting the presence of two N2 units per molecule will be presented below.
57Fe Mössbauer spectroscopy was used to learn more about the composition of the iron-containing species in the solutions. Frozen THF solutions (80 K) of 2 and 3 gave zero-field spectra with one predominant doublet accounting for 60–65% of the intensity. This doublet had an isomer shift δ = 0.64 mm/s (2) or 0.61 mm/s (3), and quadrupole splitting |ΔEQ| = 2.39 mm/s (2) or 2.46 mm/s (3). The similarity in the major component of each spectrum supports the idea that these are the analogous compounds 2 and 3 as proposed. These isomer shifts are similar to that for LMeFeNNFeLMe (δ = 0.58 mm/s),11 another compound with low-valent iron having backbonding to N2. However, since these isomer shifts are within the isomer shift ranges for both high-spin iron(II) and iron(I)12,13 (and no high-spin iron(0) complexes have been studied by Mössbauer spectroscopy), it is difficult to use these data to specify the oxidation state.
Though the above measurements support the idea that 2 and 3 have structures analogous to 1, we desired additional solution experiments testing the presence of two N2 ligands in 2 and 3. Since the compounds could not be isolated, CHN analysis was unsuitable, and different strategies were used. For compound 3, the reaction with Et3SiCl (see below) releases the bound N2, and thus the quantity of N2 released is a useful test for the amount of N2 in 3. Monitoring the pressure change upon addition of Et3SiCl to a solution of 3 showed the production of 0.68 ± 0.03 mol of N2 per Fe, in two independent experiments with 5 or 9 equiv of Et3SiCl added per Fe. Taking into account the Mössbauer evidence indicating that the major component is roughly 60% of the solution, these manometry experiments indicate a 1:1 Fe-N2 stoichiometry in 3, consistent with the proposed formulation {LMeFe(μ-N2)}2Mg(THF)4.
For compound 2, attempted manometry experiments were less reproducible. Therefore, we designed an alternative test based on the Fe:N2 stoichiometry in the putative Fe-N2-Mg-N2-Fe product (2:2 Fe:N2), which has more N2 per iron than the well-characterized complex LtBuFeNNFeLtBu (2:1 Fe:N2).[8] Because of the different stoichiometry, formation of a Fe-N2-Mg-N2-Fe compound from the FeNNFe compound should require additionalN2 (see the bottom of Scheme 3). Reduction of LtBuFeNNFeLtBu in THF with a slight excess of Mg* (4 equivalents) under an N2 atmosphere yielded 2 after 90 min, as confirmed by solution IR spectroscopy. However, LtBuFeNNFeLtBu did not react with Mg* under an Ar atmosphere. Therefore, the presence of N2 is required for the formation of 2 from LtBuFeNNFeLtBu. Thus, the compiled evidence and analogies to the cobalt system are most consistent with assignment of compounds 2 and 3 as {LtBuFe(μ-N2)}2Mg(THF)4 and {LMeFe(μ-N2)}2Mg(THF)4 (Scheme 3).
Scheme 3.

Reduction of Fe(II) halide complexes with Mg* gave metastable products spectroscopically consistent with the formulations {LtBuFe(μ-N2)}2Mg(THF)4 (2) and {LMeFe(μ-N2)}2Mg(THF)4 (3).
Addition of Electrophiles to Fe-Mg-N2 Complexes
The iron complex with less steric hindrance, 3, was selected for a survey of reactivity with acids. Compound 3 was generated in situ and was treated with 50 equiv of HCl or H2SO4, and the production of NH3 and N2H4 from 3 was determined colorimetrically. In five trials with HCl the yield of ammonia was 1.8 ± 0.6%, and no N2H4 was formed. The yield of NH3 did not change when HCl addition to 3 was conducted at −96 °C as opposed to room temperature. Use of 50 equiv of H2SO4 at room temperature gave similar results, with 1.5% yield of NH4+ and 1.3% yield of N2H4. The amount of excess Mg* present (2, 3, 4 or 5 equiv per Fe) had no effect on the yield of NH3. Only a trace amount of NH3 (less than 1% yield) and no N2H4 was observed when 1 and 2 were treated with HCl under similar conditions.
The addition of methyl tosylate (MeOTs) and chlorotriethylsilane (Et3SiCl) to 1, 2, or 3 led to redox chemistry at iron, rather than functionalization of the N2 ligand. For example, the addition of three equivalents of MeOTs to 2 gave a mixture of the iron(II) compounds LtBuFeCl and LtBuFeCH314 as judged by 1H NMR spectroscopy. The addition of two equivalents of Et3SiCl to 2 gave a mixture in which the predominant component by 1H NMR spectroscopy was LtBuFeCl. Analogous mixtures were observed from the reactions of MeOTs and Et3SiCl with 1 and 3.
Discussion
Transition metal-N2 complexes supported by β-diketiminate ligands are known where the N2 ligand binds to the metal in the terminal,[15] end-on bridging (μ-η1:η1-N2),[8, 16, 17] and side-on bridging (μ-η2:η2-N2) modes.[18] However, the first row late transition metal (Fe, Co, Ni) complexes of β-diketiminates invariably have the N2 ligand bridging in the end-on/end-on bridging mode MNNM.[8,17] In the complexes most relevant to this study, reduction of neutral (formally univalent) N2 complexes with alkali metals (Na, K) led to formally zerovalent N2 complexes where the N2 ligand remained bound to the metal centers in an end-on bridging mode.[8,17] The alkali metal cations interacted with the aryl groups of the β-diketiminate ligands and with the N2 ligand, as indicated in Scheme 1 above. Attempts to remove the inner-sphere cations resulted in decomposition of the Fe and Co complexes,[8] while the Ni complexes were stable with a single K+ ion in the singly-reduced form.[17] Thus, all previous complexes have cations in the core to give an overall neutral complex. In the work reported here, we used activated Mg0 as a reducing agent to learn whether a similar interaction between a dication and partially reduced N2 would be effective, and what geometry might be attained.
Here, we found that Rieke magnesium (Mg*) is capable of reducing the cores of the bridging N2 complexes to formally Fe(0) and Co(0) oxidation levels. Interestingly, the Mg products have a different interaction between the cation and the anionic N2 complex than the side-on interactions previously observed in the alkali metal complexes. The Mg2+ cation is inner sphere, but it is bound end-on in contrast with the side-on binding of Na+ and K+. This Mg2+ cation has THF coordination to complete its octahedral coordination sphere, explaining why the complexes are accessible only in THF solvent. The Mg2+ ions are each bound to two trans-disposed LMN2− (M = Co, Fe) anions. Trimetallic N2 complexes exhibiting this bonding orientation are uncommon, but have been observed in {Co(μ-N2)}2Mg, {Co(μ-N2)}2Mg2, and {Fe(μ-N2)}2Mg systems, as well as a {Mo(μ-N2)}2Mg system.[19,20]
The first report of a structurally characterized trimetallic N2 complex of the formulation {Co(μ-N2)}2Mg was {(PMe3)3Co(μ-N2)}2Mg(THF)4, a Co(-I) complex that was synthesized via magnesium reduction of CoCl2/PMe3 in THF.[19a] The X-ray structure of the complex revealed an elongated N-N bond length of 1.18 Å, suggesting significant N-N activation. The originally reported νNN of 2058 cm−1 in THF is most likely in error, corresponding instead to the cobalt(I) complex (PMe3)3CoH(N2), to which the cobalt(-I) species decomposes in the presence of moisture.[19b] The analogues {(PPh3)3Co(μ-N2)}2Mg(THF)4 and {(PPh2Et)3Co(μ-N2)}2Mg(THF)4 were independently reported with more reasonable νNN values of 1840 cm−1 and 1835 cm−1.[19,21] Meanwhile, the formally Co(0) complex {(PhBPiPr3)Co(μ-N2)}2Mg(THF)4 has a slightly higher νNN of 1863 cm−1,[22] and this value is very similar to 1 (1868 cm−1) which is also formally Co(0).
The only previously reported iron species with an analogous FeNNMgNNFe core, {(PPh3)2EtFe(μ-N2)}2Mg(THF)4, is formally Fe(0) and has νNN = 1830 cm−1.[19c] A related mononuclear iron(0) complex (PhBPiPr3)Fe(μ-N2)MgCl(THF)2 also has a stretching frequency of 1830 cm−1.[22] This stretching frequency is near those of 2 (1808 cm−1) and 3 (1818 cm−1) which are also both formally Fe(0). Thus, the νNN values for complexes 1–3 correlate well with the few reported trimetallic N2 complexes, and this provides further support for their formulation as described. Literature correlations suggest that N-N stretching frequencies in this range correspond to N-N bond distances around 1.15 Å, which may be compared to those in free N2 (1.11 Å; 2331 cm−1).[5,21] Gambarotta has reported PDI-supported Fe-NN-Na complexes with similar weakening of NN bonds (1.15–1.17 Å; 1965–1868 cm−1),[23] and Peters has reported others with the SiP3 ligand set (1.13–1.15 Å; 1967–1891 cm−1).[24,25]
The LMN2− anions in 1–3 enable a comparison of the extent of π-backbonding in complexes with interactions between a single metal and the N2 ligand to other late transition metal β-diketiminate N2 complexes where two transition metal centers synergistically influence the extent of π-backbonding into the N2 ligand. These effects are illustrated in Table 1.
Table 1.
Metrical parameters and N-N stretching frequencies for formally zerovalent N2 complexes of the late first row transition metals supported by β-diketiminate ligands.
| Compound | νNN (cm−1)[a] | N-N (Å) | Reference |
|---|---|---|---|
| 1 | 1878 (1816) | N/A | This work |
| 3 | 1818 (1758) | N/A | This work |
| 2 | 1808 (1748) | N/A | This work |
| K2LtBu2Ni2N2 | 1696 (1642) | 1.185(8) | [17a] |
| K2LMe2Fe2N2 | 1625 | 1.215(6) | [8a] |
| K2LtBu2Co2N2 | 1599 (1545) | 1.220(2) | [8c] |
| Na2LtBu2Co2N2 | 1598 (1542) | 1.211(3) | [8c] |
| K2LtBu2Fe2N2 | 1589 (1536) | 1.241(7) | [8b] |
νNN for 15N2 isotopologues in parentheses.
The N2 ligands in 1–3 are somewhat weakened, as judged by comparing to the N-N stretching frequency of free N2 (2331 cm−1). The iron complexes 2 and 3 are more activated than other zerovalent iron complexes having coordination numbers of four or greater, where the N-N stretching frequencies lie in the range from 1912–2141 cm−1 except for Peters’ BP3 and SiP3 complexes that are more activated (1830–1967 cm−1).5a However, using νNN as a judge, the N-N bonds in 1–3 are not as weakened as in complexes with the MNNM core. The νNN of 1 is nearly 300 cm−1 greater than M2[LtBuCoNNCoLtBu] (M = Na, K), while the νNN of 2 and 3 is more than 200 cm−1 greater than their corresponding iron analogues. Thus, the second iron or cobalt center contributes a substantial amount of backbonding to the N2 ligand in the MNNM core. This is consistent with computations on the mononuclear complexes LFeNN−.[8b]
The protonolysis of iron-N2 complexes typically yields NH3 in less than 10% yield (calculated per N atom of N2) if observed at all.[5,26] The only example of an iron complex where a high yield of NH3 was derived from N2 via protonolysis used an iron complex where the N-N bond had been completely cleaved to nitrides.[6] However, Peters and coworkers have described the alkylation and silylation of Fe-bound N2 ligands.[7,22] Some phosphine-supported iron(I) complexes give hydrazine, in up to 47% yield.[24,26d] The protonolysis of cobalt N2 complexes to give NH3 and/or N2H4 has been reported for cobalt complexes only using the formally Co(-I) oxidation state, and gives poor yields.[19] In this work, we observed low yields (<2%) of ammonia and of hydrazine. These low yields were reproducible and greater than zero, which contrasts with earlier studies on alkali-bound [LFeNNFeL]2− complexes which gave no detectable ammonia or hydrazine.[8b] While it is pleasing to see any ammonia, the low yields in the system studied here are not indicative of a reactive N2 unit. We tentatively attribute the low yields of electrophilic attack to steric protection of the N2 unit, because the backbonding in the ground state is stronger than that in Peters’ complexes that are attacked by acids[24] or silyl groups.[22,25]
Conclusions
We have shown that Rieke magnesium is an effective reductant for iron and cobalt complexes, leading to unusual heterobimetallic N2 complexes. The reduction of LtBuCoCl with Mg* under N2 yielded 1, a N2 complex with Mg2+ cation sandwiched between two LtBuCoN2− anions. The IR data of 1 and 1-15N2 support this formulation, and agree well with the few reported compounds of {LCo(μ-N2)}2Mg(THF)4 species in the literature. The Fe analogues 2 and 3 were synthesized similarly, but are stable only in THF solution, and Mössbauer spectroscopy showed that they are 60–65% pure. Despite these difficulties, solution IR measurements supported the presence of a linear MNNMgNNM core. The iron complexes have slightly more backbonding than the cobalt complexes, as expected from the relative electronegativities.
The most important finding in this work is the significant difference between alkali metals vs. alkaline earth metals in the extent of N-N activation in zerovalent dinitrogen complexes. The Mg2+ species have significantly less activation of the N-N bond, as judged by the N-N stretching frequencies. This disparity can be attributed to the different binding modes: side-on binding of Na+ and K+ to N2 assists greatly in the N-N bond weakening, while end-on binding of Mg2+ does not have as much of an influence. We expect that this trend will be useful in the design of future late-metal systems for N2 activation.
Experimental Section
General Considerations
All manipulations were performed under a nitrogen atmosphere (or argon atmosphere where specified) by Schlenk techniques or in an M. Braun glovebox maintained at or below 1 ppm of O2 and H2O. Glassware was dried at 150 °C overnight, and Celite wasdried overnight at 200 °C under vacuum. Pentane, hexane, benzene, diethyl ether, and toluene were purified by passage through activated alumina and Q5 columns from Glass Contour Co. (Laguna Beach, CA). THF was distilled under N2 from a potassium-benzophenone. All solvents were stored over 3 Å molecular sieves. Benzene-d6 was dried and stored over flame-dried alumina. THF-d8 was vacuum transferred from sodium-benzophenone and was stored over 3 Å molecular sieves. Before use, an aliquot of each solvent was tested with a drop of sodium-benzophenone in THF solution. 15N2 (98% isotopic purity) was obtained from Cambridge Isotope Laboratories, and 15N-labeled compounds were handled under an atmosphere of argon. LtBuFeCl,[14] LtBuCoCl,[27] [LMeFe(μ-Br)]2,[28] LtBuFeNNFeLtBu,[8a] LMeFeNNLMe,[8b] and Rieke-Mg (Mg*)[29] were prepared by published procedures. Methyl tosylate (MeOTs), chlorotriethylsilane (Et3SiCl), and HCl solution in Et2O were purchased and were used as received.
1H NMR data were recorded on a Bruker Avance 500 spectrometer at 500 MHz. All resonances in the 1H NMR spectra are referenced to residual protiated benzene (δ 7.16 ppm) or THF (δ 3.58 or 1.73 ppm). IR data were recorded on either a Shimadzu (FTIR-8400S) or Shimadzu IR Prestige-21 FTIR spectrophotometer using a KBr pellet or an airtight CaF2 solution cell. Mössbauer experiments were performed at 80 K using a SeeCo spectrometer with alternating constant acceleration; isomer shifts are relative to iron metal at 298 K. Elemental analyses were obtained from the CENTC Elemental Analysis Facility at the University of Rochester. Microanalysis samples were weighed with a PerkinElmer Model AD-6 Autobalance and their compositions were determined with a PerkinElemer 2400 Series II Analyzer. Air-sensitive samples were loaded in a VAC Atmospheres glovebox under argon. Manometry experiments with 3 were performed by adding Et3SiCl into a round-bottom flask containing a solution of 3 in THF, and simultaneously monitoring the pressure with a mercury-filled U-tube composed of a 1/16-inch Tygon tube attached to a ruler.
Synthesis of {LtBuCo(μ-N2)}2Mg(THF)4 (1)
Under an atmosphere of N2, LtBuCoCl (404 mg, 0.678 mmol) was dissolved in THF (20 mL) to give a dark olive-green solution that was transferred to a Schlenk flask. Mg* (53.7 mg, 2.21 mmol, 3.3 equiv) was added to the solution to produce a dark green mixture. The mixture was stirred at room temperature and a dark red mixture was observed after 90 min. After 10 h, the dark red mixture was filtered through a fritted funnel to collect the majority of the product as a dark red solid that contained trace amounts of Mg*. The solid mixture was dried under reduced pressure to yield 390 mg of this mixture. The identity of the red solid mixture as the product was confirmed by IR spectroscopy via a KBr pellet where an intense νNN band was observed at 1867 cm−1. The product is poorly soluble in THF and Et2O and is not soluble in nonpolar solvents which prevented product isolation free of Mg* in high yield. The volatile components were removed under reduced pressure from the filtrate to give a dark red residue. As much of the residue as possible was dissolved in Et2O (10 mL) and the mixture was filtered through Celite to remove a small amount of insoluble red material. The dark red solution was concentrated to 3 mL and was stored at −45 °C to give a dark red solid (29 mg, 5.7%). The poor solubility of the product prevented characterization by 1H NMR and other solution techniques. IR (THF): 1878 cm−1. IR (KBr): 3051 (w), 2958 (s), 2903 (m), 2871 (m), 1868 (vs), 1530 (w), 1485 (w), 1463 (w), 1445 (w), 1431 (m), 1396 (s), 1365 (s), 1325 (s), 1254 (w), 1220 (w), 1196 (w), 1156 (m), 1129 (w), 1098 (m), 1057 (w), 1037 (s), 960 (w), 937 (w), 921 (w), 891 (m), 802 (w), 774 (w), 766 (w), 756 (m) cm−1. Anal. Calcd for C86H138N8O4MgCo2: C, 69.31; H, 9.35; N, 7.52. Found: C, 69.00; H, 9.42; N, 7.43.
Generation of {LtBuFe(μ-N2)}2Mg(THF)4 (2)
LtBuFeCl (55 mg, 0.093 mmol) was added to a scintillation vial and was dissolved in THF (6 mL) to give a bright red solution. Mg* (12.6 mg, 0.518 mmol, 5.6 equiv) was added to the solution which immediately produced a dark green mixture. The mixture was stirred for 2 h and was filtered through Celite to give a dark green solution. The solution was concentrated to half the original volume and a solution IR spectrum was taken. An intense νNN band was observed at 1808 cm−1. The product is only observed in THF solution, and it decomposed within 12 h at room temperature in THF.
Generation of {LMeFe(μ-N2)}2Mg(THF)4 (3)
[LMeFe(μ-Br)]2 (75 mg, 0.068 mmol) was added to a scintillation vial and was dissolved in THF (6 mL) to give a yellow solution. Mg* (10.4 mg, 0.428 mmol, 6.4 equiv (3.2 equiv per Fe)) was added to the solution. A violet colored mixture was observed within 10 min of Mg* addition, while a dark green mixture was observed within 90 min. After 4 h, the dark green mixture was filtered through Celite to give a dark green solution. The solution was concentrated to half the original volume and a solution IR spectrum was taken. An intense band was observed at 1818 cm−1. The product was only formed in THF, and it decomposed within 12 h at room temperature in THF.
Example of procedure for NH3 and N2H4 quantification
A sample of {LRM(μ-N2)}2Mg(THF)4 was generated in situ as described above in small Schlenk flask that was sealed with a septum. A solution of HCl in Et2O (Aldrich, 2.0 M, 40–50 equiv) was added via syringe. The reaction was allowed to stir overnight (12 h) at room temperature. The white suspension was pumped down to a powder, dissolved in a phosphate buffer solution (50 mM, pH 7.0) and filtered through Celite into a volumetric flask, rinsing at least three times for quantitative transfer. The indophenol method for ammonia[30] and the hydrazine test[31] were used to quantify these N-containing products.
Supplementary Material
Figure 1.

Mössbauer spectra of compounds 2 and 3. The data (black) and fits (grey) are shown; these fits are composed of 3–4 components and each consist of 60–65% of a major doublet (assigned to 2 and 3) as well as some smaller components (see Supporting Information for details).
Acknowledgments
The authors thank Meghan Rodriguez for assistance with colorimetric ammonia and hydrazine tests. This research was supported by the U.S. National Institutes of Health (GM065313).
Footnotes
Supporting Information: Infrared spectra of 1, 2, and 3; Mössbauer spectra and fits for 2 and 3; crystallographic data for 1.
Supporting information for this article is available on the WWW under http://www.eurjic.org/ or from the author.
References
- 1.Schlögl R. In: Handbook of Heterogeneous Catalysis. 2. Ertl G, Knözinger H, Schüth F, Weitkamp J, editors. Vol. 5. Wiley-VCH; Weinheim, Germany: 2008. pp. 2501–2575. [Google Scholar]
- 2.a) Smil V. Sci Am. 1997;277:76–81. [Google Scholar]; b) Leigh GJ. The World’s Greatest Fix. Oxford; New York: 2006. [Google Scholar]
- 3.Mittasch A. Geschichte der Ammoniaksynthese. Verlag Chemie; Weinheim: 1951. [Google Scholar]
- 4.a) Fryzuk MD, Johnson SA. Coord Chem Rev. 2000:200–202. 379–409. [Google Scholar]; b) MacKay BA, Fryzuk MD. Chem Rev. 2004;104:385–401. doi: 10.1021/cr020610c. [DOI] [PubMed] [Google Scholar]
- 5.a) Hazari N. Chem Soc Rev. 2010;39:4044–4056. doi: 10.1039/b919680n. [DOI] [PubMed] [Google Scholar]; b) Crossland JL, Tyler DR. Coord Chem Rev. 2010;254:1883–1894. [Google Scholar]
- 6.Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780–783. doi: 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moret ME, Peters JC. J Am Chem Soc. 2011;133:18118–18121. doi: 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Smith JM, Lachicotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Rodgers KR, Holland PL. J Am Chem Soc. 2001;123:9222–9223. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]; b) Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J Am Chem Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]; c) Ding K, Pierpont AW, Brennessel WW, Lukat-Rodgers G, Rodgers KR, Cundari TR, Bill E, Holland PL. J Am Chem Soc. 2009;131:9471–9472. doi: 10.1021/ja808783u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rieke RD. Science. 1989;246:1260–1264. doi: 10.1126/science.246.4935.1260. [DOI] [PubMed] [Google Scholar]
- 10.Dugan TR, Sun X-R, Rybak-Akimova EV, Olatunji-Ojo O, Cundari TR, Holland PL. J Am Chem Soc. 2011;133:12418–12421. doi: 10.1021/ja2052914. [DOI] [PubMed] [Google Scholar]
- 11.Stoian SA, Vela J, Smith JM, Sadique AR, Holland PL, Münck E, Bominaar EL. J Am Chem Soc. 2006;128:10181–10192. doi: 10.1021/ja062051n. [DOI] [PubMed] [Google Scholar]
- 12.Dugan TR, Bill E, MacLeod KC, Christian GJ, Cowley RE, Brennessel WW, Ye S, Neese F, Holland PL. J Am Chem Soc. 2012;134:20352–20364. doi: 10.1021/ja305679m. [DOI] [PubMed] [Google Scholar]
- 13.Hendrich MP, Gunderson W, Behan RK, Green MT, Mehn MP, Betley TA, Lu CC, Peters JC. Proc Natl Acad Sci U S A. 2006;103:17107–17112. doi: 10.1073/pnas.0604402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith JM, Lachicotte RJ, Holland PL. Chem Commun. 2001:1542–1543. doi: 10.1039/b103635c. [DOI] [PubMed] [Google Scholar]
- 15.a) Bernskoetter WH, Lobkovsky E, Chirik PJ. Chem Commun. 2004:764–765. doi: 10.1039/b315817a. [DOI] [PubMed] [Google Scholar]; (b) Masuda JD, Stephan DW. Can J Chem. 2005;83:324–327. [Google Scholar]
- 16.Bai G, Wei P, Stephan DW. Organometallics. 2006;25:2649–2655. [Google Scholar]
- 17.a) Pfirrmann S, Limberg C, Herwig C, Stoesser R, Ziemer B. Angew Chem, Int Ed. 2009;48:3357–3361. doi: 10.1002/anie.200805862. [DOI] [PubMed] [Google Scholar]; b) Pfirrmann S, Yao S, Ziemer B, Stosser R, Driess M, Limberg C. Organometallics. 2009;28:6855–6860. [Google Scholar]; c) Horn B, Pfirrmann S, Limberg C, Herwig C, Braun B, Mebs S, Metzinger R. Z Anorg Allg Chem. 2011;637:1169–1174. [Google Scholar]
- 18.Monillas WH, Yap GPA, MacAdams LA, Theopold KH. J Am Chem Soc. 2007;129:8090–8091. doi: 10.1021/ja0725549. [DOI] [PubMed] [Google Scholar]
- 19.a) Hammer R, Klein HF, Schubert U, Frank A, Huttner G. Angew Chem, Int Ed. 1976;15:612–613. [Google Scholar]; b) Miura Y, Yamamoto A. Chem Lett. 1978:937–940. [Google Scholar]; c) Yamamoto A, Miura Y, Ito T, Chen H-L, Iri K, Ozawa F, Miki K, Sei T, Tanaka N, Kasai N. Organometallics. 1983;2:1429–1436. [Google Scholar]; d) Klein HF, König H, Koppert S, Ellrich K, Riede J. Organometallics. 1987;6:1341–1345. [Google Scholar]
- 20.a) O’Donoghue MB, Zanetti NC, Davis WM, Schrock RR. J Am Chem Soc. 1997;119:2753–2754. [Google Scholar]; b) Greco GE, Schrock RR. Inorg Chem. 2001;40:3861–3878. doi: 10.1021/ic001123n. [DOI] [PubMed] [Google Scholar]
- 21.Holland PL. Dalton Trans. 2010;39:5415–5425. doi: 10.1039/c001397h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]
- 23.Scott J, Vidyaratne I, Korobkov I, Gambarotta S, Budzelaar PHM. Inorg Chem. 2008;47:896–911. doi: 10.1021/ic701643d. [DOI] [PubMed] [Google Scholar]
- 24.Mankad NP, Whited MT, Peters JC. Angew Chem, Int Ed. 2007;46:5768–5771. doi: 10.1002/anie.200701188. [DOI] [PubMed] [Google Scholar]
- 25.Lee Y, Mankad NP, Peters JC. Nat Chem. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Leigh GJ, Jimenez-Tenorio M. J Am Chem Soc. 1991;113:5862–5863. [Google Scholar]; b) Hills A, Hughes DL, Jimenez-Tenorio M, Leigh GJ, Rowley AT. J Chem Soc, Dalton Trans. 1993:3041–3049. [Google Scholar]; c) George TA, Rose DJ, Chang Y, Chen Q, Zubieta J. Inorg Chem. 1995;34:1295–1298. [Google Scholar]; d) Hall DA, Leigh GJ. J Chem Soc, Dalton Trans. 1996:3539–3541. [Google Scholar]; e) Gilbertson JD, Szymczak NK, Tyler DR. J Am Chem Soc. 2005;127:10184–10185. doi: 10.1021/ja053030g. [DOI] [PubMed] [Google Scholar]
- 27.Ding K, Dugan TR, Holland PL, Adhikari D, Mindiola DJ. Inorg Synth. 2010;35:43–45. [Google Scholar]
- 28.a) Tonzetich ZJ, Heroguel F, Do LH, Lippard SJ. Inorg Chem. 2011;50:1570–1579. doi: 10.1021/ic102300d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dugan TR. PhD Dissertation. University of Rochester; 2012. [Google Scholar]
- 29.Rieke RD, Li PTJ, Burns TP, Uhm ST. J Org Chem. 1981;46:4323–4324. [Google Scholar]
- 30.Chaney AL, Marbach EP. Clin Chem. 1962;8:130–132. [PubMed] [Google Scholar]
- 31.Watt GW, Chrisp JD. Anal Chem. 1952;24:2006–2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.