

Abstract
An 18-year-old male with a history of Charcot-Marie-Tooth disease (CMT) presented with metastatic Ewing sarcoma to the lungs. He had been followed by several healthcare professionals who ascribed his enlarging 23 cm gluteal mass to his CMT. The patient experienced a significant delay in diagnosis, not uncommon in sarcoma. This case explores the various system and cognitive errors that contributed to this delay.
Keywords: : Ewing sarcoma, Ewing family of tumors, diagnosis, comorbidity, cancer care continuum
The Ewing sarcoma family of tumors (ESFT) is the second most common bone and soft tissue cancer in adolescents and young adults (AYAs) after osteosarcoma,1 yet there is often a delay in confirming the diagnosis. The lack of predisposing factors, non-specific presentation of pain and swelling, and the unique challenges of providing care for AYAs contribute to this problem. We present the case of a young man with a comorbid condition that masked the development of Ewing sarcoma in order to highlight the preventable factors that contributed to the delay in diagnosis.
Case
An 18-year-old man with Charcot-Marie-Tooth disease type 2 (CMT2) presented to the emergency room (ER) in respiratory distress. Diagnosed at age 3, he was wheelchair-bound by age 10 due to progressive limb weakness and scoliosis. CMT2-related phrenic nerve palsy with resultant hypercapnea required him to use nocturnal bilevel positive airway pressure (BPAP) for the past 4 years. In the 2 weeks prior, he noted fever, chest tightness, shortness of breath, and non-productive cough, requiring him to use BPAP continuously. He denied other constitutional symptoms, hemoptysis, recent travel, smoking, or infectious contacts.
The patient had been receiving care from a children's rehabilitation facility and a tertiary care pediatric hospital. At age 18, his medical care was transferred to an adult hospital. However, he had yet to be seen by the adult physicians at the time of presentation. In the preceding 12 months, the patient and his family alerted the pediatric care team to his rapidly worsening scoliosis, along with the development of a left gluteal deformity. He had received follow-up from various team members on a monthly basis, and these changes were attributed to CMT2. His wheelchair was adjusted on multiple occasions to accommodate his changing habitus. A week prior to his ER presentation, his family doctor prescribed cloxacillin for erythema over his left gluteal deformity.
Our ER physical examination revealed oxygen saturation of 95% on BPAP (IPAP max 23 cm H2O, EPAP 6 cm H2O), temperature of 38.8°C, pulse of 136 beats/minute, respiratory rate of 22 breaths/minute, and blood pressure of 115/84 mmHg. He uttered short sentences. Lymphadenopathy and clubbing were absent. Heart sounds were normal, and point of maximal impulse was diffuse. The lower two-thirds of the right lung field were dull to percussion with diminished breath sounds. The abdomen was distended. Leftward scoliosis and a large, firm prominence with overlying erythema of the left gluteal region were noted. There was bilateral pedal edema. Neurologically, he was at his previously documented baseline: distal weakness, muscle wasting and sensory loss in all extremities, and diminished deep tendon reflexes.
Preliminary ER investigations revealed hemoglobin 92 g/L, mean corpuscular volume (MCV) 79.2 fL, leukocytes 12.3×109/L, and platelets 556×109/L. See Figure 1 for chest X-ray. He was admitted to Internal Medicine, and ultrasound-guided thoracentesis removed 1.5 L of serosanguinous exudative fluid with negative cytology. A chest computed tomography (CT) revealed a mass (5.3 cm×5.2 cm) adjacent to the left upper mediastinum, soft tissue pleural nodules, and small bilateral parenchymal nodules (Fig. 2). A pelvic CT revealed a left gluteal mass suspicious for sarcoma (23.5 cm×22.1 cm×19.3 cm) and intra-abdominal lymphadenopathy (Fig. 3). Biopsy of the gluteal mass confirmed a diagnosis of Ewing sarcoma, prompting referral to an oncologist specializing in sarcoma in AYAs. Chemotherapy was initiated as an inpatient 5 days after diagnosis with doxorubicin and cyclophosphamide. Vincristine was omitted given the risk of severe neurotoxicity in patients with CMT.2–5 He received a planned 10 cycles of chemotherapy with doxorubicin plus cyclophosphamide alternating with ifosfamide plus etoposide, followed by radiotherapy to his primary pelvic mass.6,7
FIG. 1.
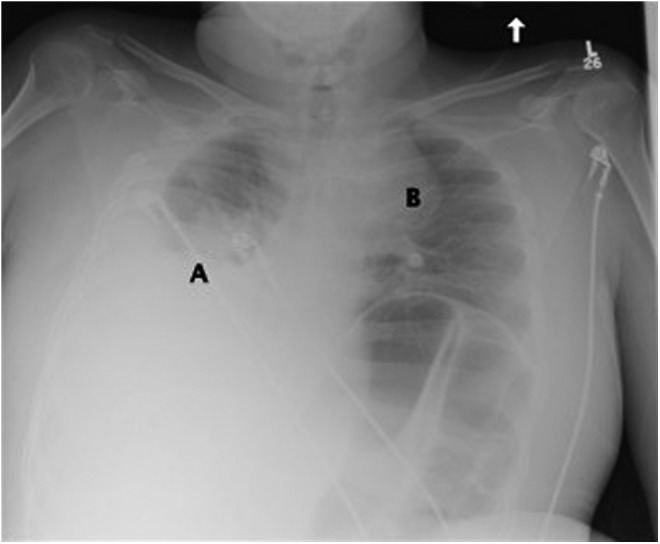
Chest X-ray showing right pleural effusion with associated atelectasis and possible consolidation (A) and a left superior mediastinal mass (B).
FIG. 2.
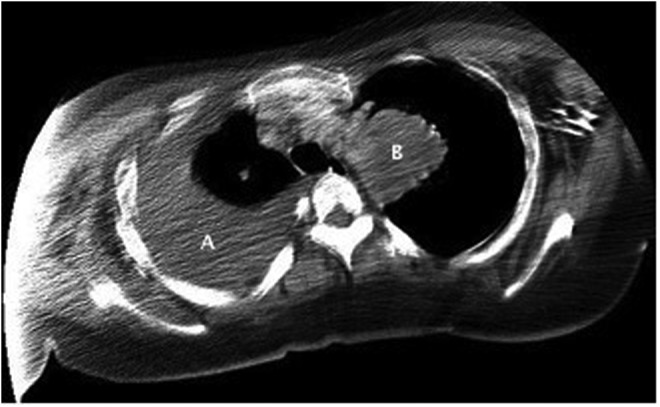
Chest computed tomography showing large right pleural effusion (A) and mass (5.3 cm×5.2 cm) in the upper left hemithorax adjacent to the mediastinum (B).
FIG. 3.

Pelvic computed tomography showing hypodense heterogeneous mass (23.5 cm×22.1 cm×19.3 cm) in left gluteal region (A) extending through the sciatic notch and contiguous with the internal iliac vessels. Retroperitoneal, mesenteric, and upper abdominal lymphadenopathy is present.
Six months after completing radiotherapy, routine surveillance imaging demonstrated progression of his lung disease. He is currently receiving second-line chemotherapy with irinotecan plus temozolomide, and remains alive and well with disease 20 months from his original diagnosis. Throughout his therapy, his CMT symptoms have been stable and he requires BPAP only at night. He is currently taking university courses by correspondence.
Discussion
ESFT can develop in any bone or soft tissue, but is most common in the long bones and pelvis.8 Patients typically complain of weeks or months of localized pain and swelling, often aggravated by exercise and worse at night.9,10 The most important investigation to perform when a young patient presents with persistent pain is a plain radiograph of the bone. This will most often reveal a “permeative” or moth-eaten appearance, associated with periosteal reaction manifested by layers of reactive bone, giving an “onion peel” appearance.11 MRI is subsequently the best modality to assess tumor size and extent.12 Pathologic diagnosis is established via tumor biopsy.13 Since metastases predominantly spread to the lungs, bones, or bone marrow, evaluation can be completed by a thorax CT, radionuclide bone scan, and bone marrow biopsy.
Prognosis is determined by the presence and location of metastases, location and size of the primary tumor, and possibly by patient age. In some case series, older age has been associated with an inferior outcome in patients with ESFT,6,14–16 potentially confounded by the increased prevalence of large pelvic disease and distant metastases at diagnosis.16 A recent randomized trial in patients younger than 30 years of age with ESFT reported a 5-year event-free survival of 71.1% (95% confidence interval [CI]: 67.7–75.0%) and overall survival of 78.6% (95% CI: 74.6–82.1%) when treated with multi-agent chemotherapy and surgery and/or radiotherapy for local control.17 However, outcomes for patients with metastases are considerably worse.6,16
This patient's comorbid condition, CMT2, affected the diagnosis of his malignancy. CMT is the most common inherited neuromuscular disorder, having a prevalence of 1 in 2500 in the United States.18 It represents a heterogeneous group of disorders caused by mutations in more than 40 genes in Schwann cells and neurons that classically manifest with distal sensory and motor deficits and skeletal deformities.19 Our patient possessed the mitofusin-II gene mutation, which causes nearly 100% of CMT2A2 cases.20 It is inherited in an autosomal-dominant fashion. However, it is possible to occur de novo.20 Our patient's parents were not tested, and no family history was available. The typical CMT2A2 phenotype involves early lower extremity involvement, followed by distal upper extremity deficits and more prominent motor than sensory deficits, which were seen in our patient.20 Diagnosis is made with nerve biopsy and genetic testing, and treatment for CMT is supportive, with no currently available disease-modifying therapies. There is no proven association between CMT and cancer, with a paucity of case studies identifying patients with Ewing sarcoma who were retrospectively diagnosed with CMT after treatment with vincristine.21,22
Potential causes for delay in diagnosis
This case highlights several missed opportunities for diagnosis and intervention. The framework for examining medical error was posited by Graber et al. to include no fault error, systemic error, and cognitive error.23
No fault error refers to the masked or unusual presentation of a disease and interference of patient-related factors in diagnosis.23 This case demonstrates both aspects of this inherent form of medical error. A study by Widhe and Widhe found that sarcoma was frequently misdiagnosed as tendonitis, osteitis simplex, osteomyelitis, or sciatica, resulting in a median delay of 19 weeks from presentation to diagnosis in patients younger than 30, but that this delay was decreased to 8 weeks if a radiograph was taken at presentation.8 In this case, the patient's CMT2-related musculoskeletal abnormalities rendered his sarcoma less evident, precluding timely investigation. In addition, the patient minimized his symptoms in order to avoid medical scrutiny, possibly impeding appropriate and timely care. The delay may have been ameliorated had his healthcare providers been able to maintain a high level of suspicion for a rapidly changing condition in the face of his chronic illness, and had the patient been more engaged in his own care, either of which may have led to an earlier X-ray.
Systemic error in this case involved a failure of communication among healthcare providers and the possibility of inadequate training.23 The patient's wheelchair was adjusted monthly by allied health professionals to accommodate the expanding gluteal mass. However, it is not evident from the patient's chart whether these interventions were communicated with other members of the team. It is unclear whether this resulted from a lack of training in the identification of abnormal signs and symptoms in the context of CMT2 or if the perceived responsibility of each provider within the healthcare team was diluted. This could be prevented by a reliable system of team communication and by training healthcare providers to render them knowledgeable and accountable to identify and communicate critical developments.
Cognitive errors refer to incorrect information collection or interpretation, reasoning, and knowledge flaws.23 Cognitive errors in this case include anchoring bias and premature closure. Anchoring bias is the tendency to focus on salient features of the initial presentation and failure to adjust one's perception despite new information emerging later in the clinical process.24 In this case, his primary physician and allied health workers focused on the patient's pre-existing CMT2-related scoliosis when the patient's family expressed concern about the rapid progression of his spinal curvature and left gluteal enlargement. The providers applied the principle of Occam's razor—the simplest and most unifying explanation is most often correct25—in error. The family physician also exhibited premature closure—the acceptance of a diagnosis before it has been fully verified.24 The physician was alerted to the patient's fevers, noted the erythema over the mass, and treated the patient for a presumed infection, but never ordered any further investigations (e.g., pelvic X-ray). Strategies to reduce cognitive errors such as these include establishing awareness of their pervasiveness, developing a routine of considering differential diagnoses for every case, and using handheld aids with algorithms and guidelines.24 Furthermore, regular feedback and accountability for decisions must be in place for practitioners to learn from previous errors.24
Conclusion
This case of an 18-year-old man with CMT2 presenting with metastatic Ewing sarcoma exemplifies the difficulties in diagnosing AYAs with malignancy, particularly in the context of comorbid diseases. While ESFT is the second most common bone tumor in AYAs, it is notoriously misdiagnosed. It is therefore important for physicians to maintain a high degree of suspicion when patients complain of persistent pain or deformity. It is imperative to perform at least a plain radiograph in these patients. This case also highlights the nuances of caring for patients with chronic illnesses like CMT, which is known for having a multitude of manifestations. When a patient's clinical course deviates from the expected trajectory, it is crucial to reevaluate the case and ensure patients receive a thorough evaluation to exclude concomitant illnesses.
Acknowledgments
The authors are thankful to the patient and his family for allowing his case to be reported. The authors would also like to acknowledge Dr. Hans Katzberg for his assistance.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Bleyer A, O'Leary M, Barr R, Ries LAG. (Eds). Cancer epidemiology in older adolescents and young adults 15–29 years of age, including SEER incidence and survival, 1975–2000 (NIH Pub. No 06-5767). Bethesda, MD: National Cancer Institute; 2006 [Google Scholar]
- 2.Chauvenet AR, Shashi V, Selsky C, et al. Vincristine-induced neuropathy as the initial presentation of Charcot-Marie-Tooth disease in acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Pediatr Hematol Oncol. 2003;25(4):316–20 [DOI] [PubMed] [Google Scholar]
- 3.Trobaugh-Lotrario AD, Smith AA, Odom LF. Vincristine neurotoxicity in the presence of hereditary neuropathy. Med Pediatr Oncol. 2003;40(1):39–43 [DOI] [PubMed] [Google Scholar]
- 4.Nakamura T, Hashiguchi A, Suzuki S, et al. Vincristine exacerbates asymptomatic Charcot-Marie-Tooth disease with a novel EGR2 mutation. Neurogenetics. 2012;13(1):77–82 [DOI] [PubMed] [Google Scholar]
- 5.Nishikawa T, Kawakami K, Kumamoto T, et al. Severe neurotoxicities in a case of Charcot-Marie-Tooth disease type 2 caused by vincristine for acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2008;30(7):519–21 [DOI] [PubMed] [Google Scholar]
- 6.Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701 [DOI] [PubMed] [Google Scholar]
- 7.Gupta AA, Pappo A, Saunders N, et al. Clinical outcome of children and adults with localized Ewing sarcoma: impact of chemotherapy dose and timing of local therapy. Cancer. 2010;116(13):3189–94 [DOI] [PubMed] [Google Scholar]
- 8.Jedlicka P. Ewing sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int J Clin Exp Pathol. 2010;3(4):338–47 [PMC free article] [PubMed] [Google Scholar]
- 9.Widhe B, Widhe T. Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg Am. 2000;82(5):667–74 [DOI] [PubMed] [Google Scholar]
- 10.Sneppen O, Hansen LM. Presenting symptoms and treatment delay in osteosarcoma and Ewing's sarcoma. Acta Radiol Oncol. 1984;23(2–3):159–62 [DOI] [PubMed] [Google Scholar]
- 11.Gebhardt MC. Bone tumors in children. Differential characteristics and treatment. Postgrad Med. 1984;76(4):87–96 [DOI] [PubMed] [Google Scholar]
- 12.Panicek DM, Go SD, Healey JH, et al. Soft-tissue sarcoma involving bone or neurovascular structures: MR imaging prognostic factors. Radiology. 1997;205(3):871–5 [DOI] [PubMed] [Google Scholar]
- 13.Meyer JS, Nadel HR, Marina N, et al. Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer. 2008;51(2):163–70 [DOI] [PubMed] [Google Scholar]
- 14.Bacci G, Ferrari S, Bertoni F, et al. Neoadjuvant chemotherapy for peripheral malignant neuroectodermal tumor of bone: recent experience at the Istituto Rizzoli. J Clin Oncol. 2000;18(4):885–92 [DOI] [PubMed] [Google Scholar]
- 15.Craft A, Cotterill S, Malcolm A, et al. Ifosfamide-containing chemotherapy in Ewing's sarcoma: the Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol. 1998;16(11):3628–33 [DOI] [PubMed] [Google Scholar]
- 16.Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108–14 [DOI] [PubMed] [Google Scholar]
- 17.Granowetter L, Womer R, Devidas M, et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol. 2009;27(15):2536–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6(2):98–118 [DOI] [PubMed] [Google Scholar]
- 19.Patzkó A, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep. 2011;11(1):78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Züchner S. Charcot-Marie-Tooth neuropathy type 2A. In: Pagon RA, Adam MP, Bird TD, et al. (Eds). GeneReviews [internet]. Seattle, WA: University of Washington; initial posting February18, 2005; last update August1, 2013. Accessed July29, 2013 from: www.ncbi.nlm.nih.gov/books/NBK1511 [Google Scholar]
- 21.Neumann Y, Toren A, Rechavi G, et al. Vincristine treatment triggering expression of asymptomatic Charcot-Marie-Tooth disease. Med Pediatr Oncol. 1996;26(4):280–3 [DOI] [PubMed] [Google Scholar]
- 22.Igarashi M, Thompson EI, Rivera GK. Vincristine neuropathy in type I and type II Charcot-Marie-Tooth disease (hereditary motor sensory neuropathy). Med Pediatr Oncol. 1995;25(2):113–6 [DOI] [PubMed] [Google Scholar]
- 23.Graber M, Gordon R, Franklin N. Reducing diagnostic errors in medicine: what's the goal? Acad Med. 2002;77(10):981–92 [DOI] [PubMed] [Google Scholar]
- 24.Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78(8):775–80 [DOI] [PubMed] [Google Scholar]
- 25.Wardrop D. Ockham's razor: sharpen or re-sheathe? J R Soc Med. 2008;101(2):50–1 [DOI] [PMC free article] [PubMed] [Google Scholar]