

Abstract
Significance: Dysregulation of cortical and striatal neuronal processing plays a critical role in Huntington's disease (HD), a dominantly inherited condition that includes a progressive deterioration of cognitive and motor control. Growing evidence indicates that ascorbate (AA), an antioxidant vitamin, is released into striatal extracellular fluid when glutamate is cleared after its release from cortical afferents. Both AA release and glutamate uptake are impaired in the striatum of transgenic mouse models of HD owing to a downregulation of glutamate transporter 1 (GLT1), the protein primarily found on astrocytes and responsible for removing most extracellular glutamate. Improved understanding of an AA–glutamate interaction could lead to new therapeutic strategies for HD. Recent Advances: Increased expression of GLT1 following treatment with ceftriaxone, a beta-lactam antibiotic, increases striatal glutamate uptake and AA release and also improves the HD behavioral phenotype. In fact, treatment with AA alone restores striatal extracellular AA to wild-type levels in HD mice and not only improves behavior but also improves the firing pattern of neurons in HD striatum. Critical Issues: Although evidence is growing for an AA-glutamate interaction, several key issues require clarification: the site of action of AA on striatal neurons; the precise role of GLT1 in striatal AA release; and the mechanism by which HD interferes with this role. Future Directions: Further assessment of how the HD mutation alters corticostriatal signaling is an important next step. A critical focus is the role of astrocytes, which express GLT1 and may be the primary source of extracellular AA. Antioxid. Redox Signal. 19, 2115–2128.
Introduction
Well over a century ago, when George Huntington first described the symptoms of the disease that would later bear his name, he made a singular, but enduring contribution to medicine. In one carefully written report, he not only identified the choreic movements and mental impairments that characterize Huntington's disease (HD), but also its hereditary nature, progressive course, and adult onset (56). Other investigators would later characterize the underlying neuropathology. Although HD affects many brain regions, morphological changes are first evident in the striatum and its main source of neuronal input, the cerebral cortex (36, 108, 124). Over the 10–15 year course of HD, neurons in both regions undergo extensive degeneration. Gross examination of advanced HD patients at autopsy shows almost complete atrophy of the striatum and widespread thinning of cortical white matter (45). Dramatic though they are, these morphological changes are not the earliest signs of corticostriatal neuropathology; functional changes appear first. In fact, the efficacy of corticostriatal transmission begins to decline even before behavioral signs appear (99), and dysfunctional corticostriatal communication is apparent long before neuronal loss (32). Because these functional changes are likely to hold the key for developing an effective therapeutic strategy, they deserve special attention.
The focus here is on how dysregulation of ascorbic acid (AA), an antioxidant vitamin found in high concentrations in the mammalian forebrain, may play a key role in the corticostriatal dysfunction underlying the HD behavioral phenotype. Unlike scurvy, which plagued British sailors well into the 18th century (16), the AA problem in HD is not a simple case of deficiency, but a deficit of release into striatal extracellular fluid that either directly or indirectly involves glutamate, the excitatory amino acid transmitter (EAAT) released by cortical afferents. Together, AA and glutamate exert a critical influence on corticostriatal function (42, 105, 109), and thus an improved understanding of their involvement in HD could lead to new therapeutic approaches.
This forum review begins with an overview of AA—its chemical properties and effects on striatal physiology—and the neurobiology of HD, including the transgenic mouse models that have revealed so much about HD pathogenesis. Subsequent sections highlight the neurobehavioral link between striatal AA release and glutamate uptake and their disruption in HD. Numerous sources provide detailed information on AA biochemistry (13, 121), the molecular genetics of HD (29, 75, 111, 117), and corticostriatal involvement in behavioral control (68, 86).
Biochemistry and Physiology of AA
AA is a hexuronic sugar acid that exists as a deprotonated anion (AA) at physiological pH; for the remainder of this review, therefore, AA is the proper designation for AA. Most animals synthesize AA from glucose through a series of enzymatic steps in the liver (mammals) or kidneys (reptiles). The structural similarity of glucose and AA is shown in Figure 1, although it should be noted that under normal conditions glucose exists as a six-membered glucopyranose. A limited number of animals, including humans, are incapable of synthesizing AA because they lack the final step in this process. For them, AA is obtained by dietary intake, and plants, which produce large amounts of AA to facilitate resistance to oxidative stress (see also below), are the primary source. Animals require AA because of its involvement in key bodily functions, including the synthesis of collagen, the main component of connective tissue, and a contributor to the composition of bone and blood vessels (30). In humans with an AA deficiency, for example, collagen is still produced but in weakened form, resulting in skin lesions, bleeding from mucous membranes, poor wound healing, edema, and other signs of scurvy. AA also plays a role in the synthesis of norepinephrine and neuropeptide hormones (105). In addition, AA facilitates iron absorption from the gut and regulates the ligand-binding properties of some receptor proteins in the plasma membrane (13).
FIG. 1.
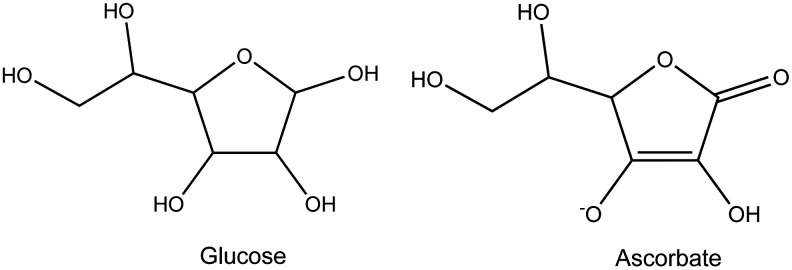
Similar molecular structure of glucose and ascorbate (AA). A series of enzymatic steps in the liver converts glucose to AA in most mammals. Humans, other primates, and a limited number of birds and rodents are unable to make this conversion and obtain AA mainly from plants.
All these diverse functions arise, in large part, from the ability of AA to donate electrons, an ability that also makes AA a powerful antioxidant or reducing agent. As an electron donor, AA can neutralize potentially damaging oxygen radicals (OH•) and nonradical, but highly reactive derivatives of oxygen (hydrogen peroxide [H2O2]), both of which are produced in the course of normal cellular metabolism. In the brain, which has a high rate of metabolic activity, the antioxidant property of AA is one of many mechanisms that can protect against cellular damage caused by highly reactive forms of oxygen. By donating an electron, however, AA itself is oxidized to semi-dehydroascorbate, an ascorbyl radical. But unlike the OH• radical and H2O2, semi-dehydroascorbate is fairly stable and unreactive. By replacing a highly reactive species with a more stable one, AA functions as an effective free radical scavenger (94).
Because AA is water soluble, it scavenges free radicals in the cytoplasm and extracellular fluid. Moreover, although AA is not found in lipids, it can protect against lipid peroxidation by regenerating lipophilic antioxidants such as the tocopherols (vitamin E) to ensure antioxidant protection in cellular membranes (12). If semi-dehydroascorbate loses a second electron and a proton, dehydroascorbate is formed, and both can be reduced back to the reduced form of AA by one of several enzymatic pathways (46). The structures of semi-dehydroascorbate and dehydroascorbate are shown in Figure 2. Alternatively, dehydroascorbate can be hydrolyzed and then metabolized to oxalate. In vulnerable individuals, oxalate accumulation can result in kidney stones (94).
FIG. 2.
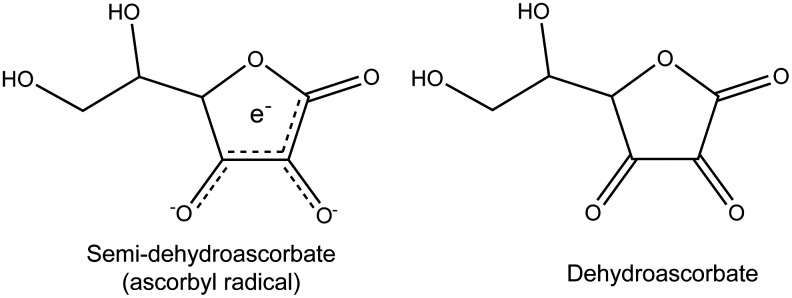
Molecular structure of semi-dehydroascorbate and dehydroascorbate. By acting as an antioxidant, AA loses an electron to become semi-dehydroascorbate, the ascorbyl radical. Unlike oxygen free radicals, semi-dehydroascorbate is relatively stable. When it loses a second electron, semi-dehydroascorbate becomes dehydroascorbate, which can then be metabolized to oxalate for excretion.
The ability to donate electrons also comes in handy for studying AA. In fact, the most widely used technology for detecting, measuring, and quantifying AA in the brain—electrochemistry—relies on oxidation to identify how dynamic fluctuations in the extracellular level of AA can modulate both neuronal function and behavior (101).
Electrochemical assessment
Classical electrochemical methodology uses carbon-based electrodes to measure the current that results when molecules oxidize in response to an applied voltage. One application of this methodology is liquid chromatography with electrochemical detection, which is typically used in conjunction with microdialysis. Extracellular fluid is sampled and collected through a probe equipped with a dialysis membrane, passed through a chromatography column to separate various molecular species, and as they pass over a carbon electrode, a voltage is applied to induce oxidation. The magnitude of the resulting current is directly proportional to the concentration of the species being oxidized. Another application of this methodology, pioneered by Adams (1) and often called voltammetry, dispenses with the dialysis probe and chromatography column and puts the carbon electrode directly in the brain (61, 80). Without the chromatography column to separate various molecular species, voltammetry relies on the ability of different molecules to oxidize at different voltages, thus achieving separation by oxidation. A potential is applied over a voltage range large enough to ensure oxidation of molecules of interest and then back again for their reduction.
Because of growing interest in the catecholamine transmitters, Gonon et al. (41) modified the carbon-fiber electrodes to ensure a clear separation between the oxidation peak for AA and that generated by the catecholamines. Several variations of the modification procedure can be used, but the basic requirement is to modify the carbon surface by electrochemical pretreatment, which shifts the oxidation potential for AA to a value distinct from that for other easily oxidized compounds (101). Figure 3 depicts a sample voltammogram obtained from the striatum of a behaving mouse. Note the distinct peaks for AA, catechols, and a third peak that includes serotonin and uric acid. Although the original goal of electrochemical modification was detection of catechols uncontaminated by AA, the procedure generated a distinct signal for AA that could be used for studying its role in brain function.
FIG. 3.

Representative voltammogram obtained from the striatum of a freely behaving mouse. Note the distinct oxidation peaks for AA, catechols (dopamine, but mainly 3,4-dihydroxyphenyl acetic acid, a dopamine metabolite), and serotonin (5-HT)/uric acid (UA). The 5-HT/UA peak also includes 5-hydroxyindole acetic acid, a 5-HT metabolite.
Accumulation and distribution in brain
In mammals, the brain is among several organs (e.g., adrenal cortex and pituitary gland) with the highest concentration of AA in the body. This is the case whether AA is synthesized endogenously or obtained through the diet. When AA enters the bloodstream, one of two sodium-dependent vitamin C transporters (SVCT1 and SVCT2) in the plasma membrane allows cells to accumulate AA. SVCT1 is found exclusively in peripheral organs, including intestine, lung, liver, and kidney, while SVCT2 is found not only peripherally in some of the same organs, but also in brain (49, 116). Interestingly, both SVCT1 and SVCT2 can regulate the bioavailability of AA by adjusting their rate of transport and even their expression depending on bodily or cellular need. AA supplementation or deprivation, for example, either decreases or increases the intra-cellular accumulation of AA, respectively, making it difficult to adjust brain levels of AA under most conditions. In fact, guinea pigs, which, like humans, cannot synthesize AA, retain a high brain level even when deprived of dietary AA (67) underscoring the importance of AA for brain function. It also is interesting that these transporters play different roles in controlling the internalization of AA. SVCT1 appears to regulate intra- and extracellular homeostasis, whereas SVCT2 targets tissues with the most metabolic activity. In the brain, SVCT2 is located on neuroepithelial cells of the choroid plexus to move AA from plasma to cerebrospinal fluid. SVCT2 also is located on neurons, which can accumulate AA to a concentration up to 10 times higher than extracellular levels. SVCT2 may play a similar role for astrocytes and other glial cells, but the intracellular level of AA in glia is not as high as in neurons (110). Dehydroascorbate, the oxidized form of AA, is removed from blood by one of several hexose transporters and then converted back to AA by intracellular enzymes. Functionally, SVCT1 and SVCT2 have been studied in several experimental models, but kinetic properties have been difficult to characterize because they vary depending on the cell, tissue, or species studied (116).
The distribution of AA in the brain is not uniform (48, 79, 81). Highest levels are found in the forebrain including the entire cortical mantle; the striatum has the highest level in the basal ganglia. Within a given region, posterior areas have less AA than anterior areas. Brainstem structures have the lowest amounts of AA. The level of AA in brain extracellular fluid, however, can change dramatically depending on behavioral state. This is certainly the case in striatum where the level can change by >300 μM in a few minutes.
The Dynamics of AA Release into Striatal Extracellular Fluid
Behavioral activation elevates striatal extracellular AA. In anesthetized animals, the extracellular level in striatum is roughly between 250 and 350 μM depending on the region sampled (6), but in the awake state, the level can double (93). Stimuli that provoke behavioral activation, such as a tail pinch, or systemic injection of amphetamine, a psychomotor stimulant, can increase extracellular AA to even higher levels (44, 89, 97, 133). The increase is not due to release from peripheral stores that are subsequently transported to the brain since adrenalectomy does not alter amphetamine-induced AA release in striatum, whereas administration of para-hydroxy-amphetamine, which has the same sympathomimetic action as amphetamine but does not cross the blood-brain barrier, fails to elicit striatal AA release (130). Moreover, the increase in striatal AA release that occurs when animals are spontaneously active and the decrease that occurs when they are quiet are several hours out of phase with the fluctuations in plasma AA (44). In contrast, cortical lesions lower striatal extracellular AA levels by >70% and also blunt the increase caused by amphetamine (5). In addition, electrical stimulation of cerebral cortex evokes a large increase in striatal AA release (27). Interestingly, these manipulations cause similar changes in striatal glutamate transmission, suggesting a link between glutamate release from cortical afferents and the release of AA into striatal extracellular fluid.
Role of glutamate uptake
Ample evidence not only supports the idea of an AA-glutamate link, but implicates glutamate uptake as the critical event in AA release. The earliest evidence came from studies showing that the addition of L-, but not D-glutamate provoked AA release in striatal tissue (40, 43). Glutamate transport is stereo-selective for the naturally occurring L-isomer. The release of AA, moreover, is not calcium dependent, arguing against a vesicular source (43). Additional research showed that pharmacological blockade of glutamate transport but not glutamate receptors blocked cortically evoked striatal AA release. Collectively, these data suggest that the uptake of glutamate released from cortical afferents triggers a corresponding release of AA that involves a form of heteroexchange: extracellular glutamate is replaced by AA (35, 105). Although the precise relationship between glutamate uptake and AA release remains to be established, the underlying process is controlled, in part, by a neural network that includes dopamine neurons. Dopamine is not required for AA release in striatum and dopamine neurons are not the source of this release, but dopamine can promote striatal AA release by activating the corticostriatal pathway either directly or indirectly via a basal ganglia-thalamic loop (see 100). In either case, the essential requirement is corticostriatal activation to ensure increased glutamate release, the clearance of which triggers the release of AA.
Although glutamate uptake may be the trigger, the source of the released AA is not clear. The corticostriatal projection itself is a likely possibility. SVCT2 is highly expressed in cortical pyramidal neurons, for example, but not in striatum (8, 90), suggesting that cortical afferents are the main source of striatal AA release. In fact, kainic acid lesions of striatum, which destroy cell bodies but not axon terminals, fail to alter the basal level of AA in striatal extracellular fluid (96). Neurons, moreover, accumulate a much higher concentration of AA than glia (110), making neurons a more likely source of AA release. Astrocytes, however, can store AA and release it in a glutamate-dependent manner (129). Moreover, glutamate uptake primarily occurs on astrocytes, and astrocytes have the highest expression of glutamate transporters. Interestingly, although cortical lesions can block AA release in striatum, they also lower the level of striatal extracellular glutamate and thus make less glutamate available for uptake by astrocytes. At this point, it is difficult to rule out either the cortical projection or astrocytes as the source of AA in striatal extracellular fluid.
A more intriguing question is the function of AA after it is released. Both electrophysiological and behavioral evidence suggest that AA modulates striatal neuronal processing.
Modulation of striatal function
The striatum plays a critical role in action selection and the flexibility of behavioral choice (11, 59, 69). Pyramidal neurons in deep layers of cerebral cortex send massive glutamate projections to the striatum where they mainly innervate medium spiny neurons (MSNs), which account for >90% of the striatal neuronal population. Cortical afferents also project to striatal interneurons, which in turn target MSNs. A glutamate projection to MSNs also arises from midline thalamus. Glutamate is the primary driver of MSN activity because without cortical input MSNs are silent (128). In contrast, dopamine, which innervates MSNs from the substantia nigra, plays a modulatory role, enhancing the strength of the glutamate signal relative to background activity (62, 64). As shown in Figure 4, MSNs, which release gamma-amino-butyric acid, are strategically positioned to process behaviorally relevant cortical information and route it to downstream structures for integration into ongoing behavioral activity.
FIG. 4.

Schematic representation of the major neuronal systems that innervate the striatum, including the medium spiny neurons (MSNs), which inhibit (−) downstream basal ganglia nuclei. Cortical pyramidal neurons provide the bulk of excitatory (+) glutamate input along with afferents from midline thalamus. Dopamine, which arises from brainstem neurons, and striatal interneurons exert a modulatory or a net excitatory or inhibitory influence (+/−). In striatum, interneurons are represented by the large circle, while the two smaller flanking circles represent MSNs, which are thought to play a critical integrative role.
Acute systemic injection of a large dose of AA (500–1000 mg/kg) to rats leads to a rapid and prolonged increase in the activity of presumed MSNs (33) and alters their responsiveness to somatosensory stimuli (21). Simultaneous monitoring of striatal extracellular fluid by voltammetry indicates that the neuronal change closely parallels a rise in AA. The increase in AA peaks at a level similar to that caused by amphetamine or other behaviorally activating stimuli. At such high doses, AA is likely to enter the brain by diffusion, although other mechanisms cannot be ruled out. When AA is directly applied to striatal neurons by iontophoresis, dose-dependent increases in striatal activity are commonly observed in awake, unrestrained rats (98). Further assessment of this effect indicates that AA mainly potentiates the excitatory action of glutamate, possibly by interfering with glutamate uptake (63). The role of uptake, however, is difficult to interpret from these data because although AA increases the magnitude of the glutamate response, the duration of the effect is not prolonged, arguing against a decline in uptake. But elevated extracellular glutamate could desensitize some receptors and thus mask an increase in the level of synaptic glutamate. In fact, measurements of striatal extracellular glutamate in response to cortical stimulation indicate that glutamate uptake decreases after intrastriatal infusions of AA (107). Thus, corticostriatal glutamate transmission is sensitive to the level of AA in striatal extracellular fluid. The effect of AA on glutamate transport is consistent with an AA-glutamate heteroexchange mechanism in which the uptake of glutamate released from striatal afferents occurs in conjunction with release of AA (see above). According to this model, manipulations that elevate extracellular AA prolong glutamate transmission by interfering with the corresponding release of AA from cellular stores. Interestingly, this effect may depend on behavioral state since an increase in striatal extracellular AA has a greater effect on glutamate transmission when animals are less behaviorally active (114). It also is noteworthy that the neuronal response to AA cannot be explained by an antioxidant effect since iontophoresis of the D-isomer, which has the same redox potential as the naturally occurring L-form, does not potentiate glutamate signaling (63).
When AA is removed from striatal extracellular fluid by local infusion of AA oxidase in rats, behavioral activity declines dramatically (106). Spontaneous movement in the open field, exploration of novel objects, and interaction with other rats are significantly decreased relative to vehicle infusions. Behavioral responding returns when striatal AA is elevated by a subsequent injection of amphetamine.
In sum, multiple lines of evidence indicate that dynamic changes in extracellular AA play a critical role in both striatal function and behavior by modulating glutamate transmission. An AA interaction with glutamate uptake is especially interesting because the removal of extracellular glutamate is arguably the most important mechanism available for controlling the flow of cortical information to striatal neurons.
Mechanisms of Glutamate Transport
Neurons are highly sensitive to the excitatory effects of glutamate. In fact, prolonged activation of N-methyl-D-aspartate (NMDA) and other ionotropic glutamate receptors, including alpha-amino-3-hydroxy-5-methylisoxazol-4-propionic acid (AMPA) and kainate receptors, can lead to epileptic seizures, increased susceptibility to neuronal injury, and excitotoxicity. These receptors, moreover, are found both inside and outside the synaptic cleft, suggesting that glutamate may have a different action depending on where the receptors are located. Extrasynaptic NMDA receptors, for example, have been implicated in the excitotoxic effects of glutamate and may play a critical role in the neurodegeneration that occurs in HD and possibly other neurodegenerative diseases (88). Thus, maintaining a proper level of synaptic and extrasynaptic glutamate is crucial not only for the transfer of information across the synapse but also for neuronal survival. To ensure the clearance of glutamate after its release, astrocytes are equipped with proteins that transport glutamate from an extra- to an intracellular location (24). These EAATs (in humans but different designations in rodents) comprise a five-member family: EAAT1 (glutamate aspartate transporter [GLAST]), EAAT2 (glutamate transporter 1 [GLT1]), EAAT3 (EAAC1), EAAT4, and EAAT5. All appear to operate by coupling the inward transport of glutamate with that of Na+ and H+ ions, while K+ ions are transported outward (25). The stoichiometry of this transport is shown schematically in Figure 5. Despite the stoichiometry, glutamate transport is not necessarily electrogenic since anion movement through the transporter itself or neighboring channels may balance the charge (24). Surprisingly, glutamate also can be transported out of the cell. This process involves cystine, an amino acid formed from the oxidation of cysteine. In this transport system, known as Xc−, the anionic form of cystine is taken up for use in the synthesis of glutathione, a powerful intracellular antioxidant, while glutamate is transported in an outward direction (115). Most of the glutamate detected in extracellular fluid comes from glutamate-cystine exchange rather than neuronal release (3).
FIG. 5.

Stoichiometry of glutamate transport across the plasma membrane. Uptake of one molecule of glutamate and three Na+ ions and one H+ ion is accompanied by the efflux of one K+ ion. Neighboring channels (not shown) may balance the change in charge by allowing movement of additional anions but the actual mechanism is not clear (24).
In the case of GLT1, the rodent form of EAAT2, glutamate transport is redox sensitive, suggesting that glutamate uptake can be regulated by AA and possibly other antioxidants (122). GLT1 is the most abundant of the glutamate transporters. Accordingly, it is responsible for the removal of ∼90% of extracellular glutamate in most brain regions. GLAST, the rodent equivalent of EAAT1, is the next most abundant and actually predominates in cerebellum and retina where EAAT4 (cerebellum) and EAAT5 (retina) are also expressed (78). Because all glutamate transporters, including the Xc− system, are found on astrocytes, these cells are critical participants in the effectiveness of glutamate transmission. Some inter-related mechanisms of glutamate transmission, glutamate transport, and AA release are shown schematically in Figure 6. A GLT1 isoform is present on presynaptic axon terminals, but its importance for controlling synaptic glutamate is unclear (24).
FIG. 6.

Schematic representation of key events related to AA cycling at a tripartite synapse involving the presynaptic terminal, the postsynaptic spine, and an astrocyte. Release of glutamate (Glu) activates receptors (GluR) both inside and outside the synaptic cleft. The released Glu is cleared by glutamate transporter 1 (GLT1) on the adjacent astrocyte. This promotes the release of AA by heteroexchange involving GLT1 directly or activation of an intervening mechanism on the astrocyte. Events within the dashed lines indicate the presumed relationship between Glu uptake by GLT1 and AA release. It is possible that another source of AA release is a GLT1 isoform on the presynaptic terminal, but the significance of presynaptic GLT1 requires further investigation. Extracellular AA is taken up by SVCT2 on the presynaptic terminal or binds to GluR to modulate neuronal excitability. Fully oxidized AA in the form of dehydroascorbate (DHAA) is recycled back to AA in the astrocyte by glutathione (GSH), an intracellular antioxidant, which itself is oxidized to oxidized glutathione (GSSH). Glu in the astrocyte is either converted to glutamine (Gln) for conversion back to Glu in the presynaptic terminal or released by Xc− in exchange for cystine, which is used to synthesize GSH. The protein(s) responsible for AA efflux is (are) unknown and could include GLT1 itself, reverse operation of SVCT2, or another protein.
GLT1 is the primary mechanism for keeping the extracellular concentration of glutamate in the low micromolar range. When this mechanism fails, the flow of information across the synapse is distorted by overactivation of glutamate receptors, and as the extracellular concentration approaches the millimolar level, activation of extrasynaptic receptors can trigger neuropathology. In HD, dysregulation of AA release appears to coincide with a failure of GLT1, which may explain the deficits in striatal function that underlie the HD behavioral phenotype.
Genetics and Molecular Biology of HD
HD is an autosomal dominant condition caused by an expanded CAG repeat in exon 1 of the huntingtin gene located on chromosome 4 (57). Individuals with a repeat length above 35 CAGs are at risk for HD and full penetrance occurs above 39. Although multiple factors, including environmental influences (123, 127, 131), determine age of onset, HD typically emerges in middle age (40–50 years), but as the length of the repeat increases, the age of onset tends to decrease (47). In juvenile HD, for example, CAG repeat length can easily exceed 50. Moreover, among HD-affected individuals, CAG repeat length tends to increase across generations if transmission is paternal, suggesting an effect of HD on spermatogenesis (134).
The huntingtin protein (HTT) contains over 3000 amino acids with the CAG expansion appearing in the N-terminal region. Because mutant HTT is widely expressed, it triggers cellular pathologies throughout the body, but the most notable of these, insoluble intranuclear and cytoplasmic inclusions, is common in brain tissue (60). The role of these inclusions in HD pathogenesis is far from settled since they could be directly pathogenic or simply represent a protective response as cells try to sequester the abnormal protein from disrupting vital functions. In either case, the inclusions are not present in unaffected brains, indicating that mutant HTT is processed differently from its normal counterpart. Ample research indicates that HTT participates in multiple cellular functions, ranging from regulation of transcription to intracellular trafficking, but the mechanism by which mutant HTT leads to HD is unknown. What is clear is that mutant HTT promotes aberrant protein–protein interactions. This and other valuable insights into the pathogenesis of HD have come from the development of transgenic animal models, which allow for assessments of cellular function that are impossible to perform in human patients (22). Mice have become the most widely used animals for this line of research.
Transgenic mouse models
Identification of the HD gene paved the way for the development of animal models. The first was developed by introducing exon 1 of the huntingtin gene from a patient with juvenile HD into the mouse genome (77). The result was rapid onset of robust neurological signs in what have become known as the R6/1 and R6/2 strains of mice. Although many additional models are now available, they can be grouped into three broad categories based on the genetic manipulation, as shown in Figure 7. Truncated models, represented by the R6 mice, carry only a portion of the mutant gene, typically exon 1 or the N-terminal portion that includes the expanded CAG repeat. These animals, therefore, express only a truncated version of mutant HTT. In an alternative approach, researchers randomly inserted the full-length human mutant huntingtin gene in the form of either bacterial artificial chromosome (BAC) or yeast artificial chromosome (YAC) DNA into the mouse genome (28). These are the so-called BAC and YAC mice, respectively, and are categorized as full-length models. They mimic adult-onset HD, but lack the robust behavioral phenotype of the R6 mice. A third category includes mouse strains in which the expanded CAG repeat is inserted directly or knocked in to the mouse huntingtin gene. These knock-in mice model late adult onset HD, but never completely replicate the severe behavioral impairments and early mortality of HD.
FIG. 7.

Three categories of mouse models based on the human mutant huntingtin gene for studying Huntington's disease (HD). Truncated models include exon 1 (R6) or the relevant portion of exon 1 that contains the expanded CAG repeat. Full-length models (bacterial artificial chromosome [BAC] and yeast artificial chromosome [YAC]) include the entire gene. In both truncated and full-length models, the genetic manipulation is added to the mouse genome. In contrast, the CAG expansion is inserted or “knocked in” to the mouse genome in knock-in models.
Researchers use mice in particular categories to address a specific question or to build on earlier work. The R6/2 line, for example, is widely used because the entire disease course plays out between 6 and 14 weeks of age and the behavioral phenotype is strongly expressed by 9 or 10 weeks of age. These features make R6/2 mice popular as an initial screen for new therapeutics or for identifying the most prominent underlying cellular abnormalities. In contrast, neurological signs in BAC and YAC mice may take several months to develop, but there also is ample opportunity for assessing cellular pathogenesis when animals are still healthy. Knock-in mice provide a model in which the HD mutation occurs in the correct genetic location. Each model, therefore, has a unique set of features that can be exploited for studying a particular aspect of HD. Findings that generalize across models are especially valuable for gaining insight into the underlying neurobiology.
Altered corticostriatal processing
Abnormal patterns of electroencephalogram (EEG) activity have been reported for patients in various, even early, stages of HD (15, 95, 119). A prominent feature of the EEG record is a loss of power in the alpha and theta bands along with deteriorating motor control and mental performance. Abnormal cognitive evoked potentials also have been reported for patients showing early signs of HD (112). Collectively, these recordings provided early confirmation of neuronal processing deficits in HD, and paved the way for more detailed investigations of electrophysiological changes in transgenic HD models. In R6/2, YAC, and knock-in mice, the balance of excitatory and inhibitory inputs that normally controls the membrane properties of cortical pyramidal neurons recorded in vitro shifts toward a hyper-excitable state (23). Similarly, pyramidal neurons recorded from BAC mice show decreased inhibition, consistent with an elevation of cortical excitability (118). In cultured cortical neurons, which express the first 171 amino acids of mutant HTT and are grown on microelectrode arrays, there is evidence of network-wide dysfunction in that the number of spikes, the overall population of spike bursts, and interburst intervals are all decreased (39). The pattern of neuronal spiking, including burst activity, plays a critical role in information transmission and synaptic plasticity (58, 74), making the decrease in bursting especially relevant for HD. In fact, decreases in bursting and in the number of spikes that participate in a burst have been reported for pyramidal neurons recorded from both prelimbic and primary motor areas of cortex in freely behaving R6/2 mice as early as 7 weeks of age (84, 125). Although the burst properties of individual cortical neurons in mildly symptomatic knock-in mice did not change relative to healthy strain controls, neuron pairs recorded simultaneously from knock-in mice showed significantly less correlated firing, including fewer coincident bursts. A decline in correlated firing also has been reported for R6/2 mice (125). The loss of correlated or synchronous firing among groups of cortical neurons across different models is a key point for HD because cooperative interactions among functionally related neurons, which are often manifest as synchronous oscillatory activity, shape behavioral output (9, 14, 84). In support of this view, dysregulation of prelimbic activity occurs in R6/2 mice during abnormalities in the extinction of fear conditioning (126).
Like cortical neurons, striatal MSN electrophysiology is altered in HD models, and again in vitro data point to an increase in neuronal excitability (19, 65, 70, 87). Intracellular recordings from R6/2 and knock-in mice, for example, indicate that MSNs have a depolarized resting membrane potential and enhanced sensitivity of the NMDA glutamate receptor relative to wild-type (18, 71). Interestingly, recordings from behaving, symptomatic HD models indicate increased MSN activity in both R6 lines, but not in knock-ins, which express a relatively mild behavioral phenotype relative to the R6s (84, 85, 104). Presumably, the elevated R6 firing rate reflects the robust HD behavioral phenotype. In contrast, MSN burst activity is decreased in all three models (84, 85). In fact, decrease in correlated firing and coincident bursting between pairs of simultaneously recorded MSNs is a consistent finding across multiple mouse models and the behaving transgenic HD rat (84). Thus, as in cortex, dysregulated MSN activity patterns occur across multiple HD models.
Altered corticostriatal processing is most evident when the activity of large populations of neurons, recorded as local field potentials (LFPs), are recorded simultaneously in both regions (53). Assessments of LFP activity in R6/2 mice behaving in an open-field arena showed high frequency oscillations in both motor cortex and striatum that did not appear in wild-type control mice during quiet rest and grooming. Interestingly, R6/2s most closely resembled wild-type when both groups were engaged in active exploration. In fact, corticostriatal synchrony at high frequencies declined in R6/2s as behavior moved from quiet rest to active exploration, completely opposite to the control pattern. Abnormal cortical and striatal high frequency oscillations also occur in R6/1 mice relative to control during performance of a procedural learning task (17). It appears, therefore, that HD mice fail to modulate corticostriatal communication appropriately to fit behavioral demand. In an intriguing parallel, the decrease in AA release occurs when HD mice are behaviorally active.
Dysfunctional AA release
When symptomatic R6/2 mice and age-matched wild-type controls are anesthetized and assessed for extracellular AA in striatum, the AA level is comparable in both groups (102). As the animals recover from anesthesia and begin spontaneous movement, however, only wild-type mice show the expected increase in AA. By 60 min after recovery from anesthesia, their extracellular AA increases by >50%. In contrast, R6/2 mice show the opposite response, a 25%–50% decline in AA that trends slowly back to the anesthetized level after 100 min but still well below the AA level in the behaving wild-types. Similar results have been reported for striatal AA release evoked by cortical stimulation: a much larger increase in control than R6/2 mice (27). The postanesthesia decrease in R6/2 extracellular AA, moreover, cannot be explained by a loss of intracellular AA since R6/2 and control intracellular levels are comparable (120). In line with the decrease in AA release, R6/2 behavior also is impaired. These mice are less active overall and engage in a more limited repertoire of movements than wild-type, consistent with evidence that a loss of extracellular AA in striatum hampers a wide range of behavioral responses (see above).
Knock-in mice tested in early to mid-adulthood show a similar decrease in striatal AA release upon recovery from anesthesia, but with an interesting sex difference (26). Males, which show greater behavioral deficits than females, also show a greater behavior-related decrease in striatal AA during behavioral episodes. Thus, the problem in male HD mice appears to be a failure of AA release. In females, estrogen may play a neuroprotective role that delays onset of HD neurological signs, including onset of the failure to release AA.
Further assessment of R6/2 behavior indicates that the pattern of movement is also altered (37). This can be assessed in an enclosed plus-shaped maze; the maze is mounted on a force plate and is used to measure turning probability and should not be confused with the elevated, open-arm plus maze used to measure anxiety. When allowed to move freely in the enclosed plus maze, R6/2 mice spend as much time moving as healthy strain controls but are less likely than controls to turn into a perpendicular arm upon reaching the center or choice point of the maze (52, 103). Turning probability is significantly reduced. Four intraperitoneal injections of 300 mg/kg AA spread over 7 days not only reverses the decline in turning but also increases striatal AA release relative to vehicle treatment (103). AA, but not vehicle, injections also increase overall R6/2 locomotion in the open field. Thus, a loss of extracellular AA in striatum may play an important role in the HD behavioral phenotype.
The loss of extracellular AA also impacts striatal neuronal activity (104). MSN firing rate is significantly elevated in R6/2 mice relative to control in both anesthetized and behaving conditions, but with firing rates in the behaving condition that often exceed 15 spikes/s, a rate that rarely appears in wild-type mice. If, as some evidence suggests (108), the striatopallidal projection degenerates early in the course of HD, then it is tempting to speculate that an elevated rate of striatal activity is confined to the striatonigral pathway. Excessive activity in this pathway could lead to the release of motor programs that disrupt ongoing behavior and trigger the HD behavioral phenotype (2). Interestingly, increased bursting has been reported in the substantia nigra reticulata, the main target of the striatonigral projection (91). Although more research is required to identify the downstream pathways that promote the HD behavioral phenotype, administration of AA according to a protocol that elevates extracellular AA in striatum (300 mg/kg/injection/day for several days) significantly lowers striatal activity in R6/2 mice (104). Representative spike rasters, shown in Figure 8, illustrate the ability of AA to slow striatal firing to a wild-type pattern, while also increasing burst firing. This same protocol also improves R6/2 behavior (103).
FIG. 8.

Representative spike rasters recorded from the striatum of freely behaving wild-type (WT) or R6/2 mice. Each row represents an individual neuron. Burst firing, calculated by the burst surprise method (see 85), is designated by a horizontal line through a cluster of spikes. Note the spike bursts present in both the WT mouse treated with saline and the R6/2 mouse treated with AA. The R6/2 mouse treated with saline shows the characteristic increase in firing but lack of burst activity (see 104).
Although the mechanism by which AA injections improve behavioral responding and striatal electrophysiology in HD models remains to be established, a change in glutamate transmission is a likely possibility. One interpretation is that an increase in extracellular AA interferes with the operation of the NMDA glutamate receptor. AA has been shown to occupy a redox site on this receptor to decrease NMDA function (76), but whether systemic injections of AA can elevate the extracellular concentration to the high level (>500 μM) required for this effect is unknown. Interestingly, however, NMDA receptor sensitivity changes in HD mice (20), which may allow for even a small increase in AA to antagonize NMDA activation. An alternative interpretation is that glutamate uptake is impaired in HD mice, and in contrast to what happens in healthy animals, AA injections may actually promote glutamate uptake.
The glutamate uptake problem
In HD, ample evidence points to a problem with EAAT2 in patients and its homologue in HD mice, GLT1. Evaluation of tissue from HD patients indicates a decrease in EAAT2 mRNA and a decrease in EAAT2 expression in both striatum and cortex (34). The loss of EAAT2, moreover, parallels the severity of symptoms and cannot be explained by a loss of astrocytes since these cells may actually increase in HD. In addition, when HD postmortem tissue is evaluated for glutamate uptake, a significant decrease is evident even at relatively early stages (50). Similar results have been obtained from HD mice. Decreases in both mRNA and protein have been reported for GLT1 in R6/2 and other HD models (7, 73), and even before the loss of GLT1 protein, there is a significant decrease in striatal glutamate uptake (83). In fact, the decrease develops just prior to the onset of the behavioral phenotype (92). Collectively, these results suggest that a loss of GLT1 function is a critical step in HD progression and can occur independent of a change in the level of GLT1 protein.
GLT1 function depends on palmitoylation, a process that regulates protein insertion in cellular membranes (38). Palmitoylation is accomplished by a family of palmitoyl-acyl transferases (PATs), and the connection to HD involves HIP14, a HTT-interacting protein that was the first PAT to be characterized (55). Mutant HTT reduces palmitoylation by altering HIP14 activity (132). In fact, YAC model mice show a decrease in both GLT1 palmitoylation and glutamate uptake (54). HIP14 knock-out mice, moreover, show an HD-like phenotype, underscoring the importance of palmitoylation and its role in glutamate transmission. GLT1 dysfunction also points to the direct involvement of astrocytes in HD neuropathology. Moreover, the molecular mechanisms altered by mutant HTT in astrocytes are closely associated with neuronal dysfunction and the development of phenotypic alterations in HD. Consistent with this view, Bradford et al. (10) restricted the expression of mutant HTT to astrocytes and found decreased levels of GLT1, reduced glutamate uptake, and most importantly, development of the HD behavioral phenotype. Over the course of HD, the loss of GLT1 protein leads to increased susceptibility to glutamate-induced neurotoxicity (31). Drug treatments are now available to increase GLT1 expression, and the results of these treatments in HD mice not only confirm a link between glutamate uptake and AA release but suggest a possible therapeutic strategy.
Increasing GLT1 function and striatal AA release
The beta-lactam antibiotic, ceftriaxone, increases GLT1 expression. As a third generation cephalosporin, ceftriaxone crosses the blood-brain barrier, and after several days of treatment, GLT1 levels increase in mouse striatum and other brain regions (113). Ceftriaxone treatment of symptomatic R6/2 mice not only elevates GLT1 expression in striatum but also increases glutamate uptake, indicating that the increased expression is functional (83). More importantly, HD neurological signs in these mice also improve. Daily ceftriaxone for five consecutive days, for example, decreases clasping, a unique response of the fore- and hind limbs of HD mice to tail suspension, and it also increases turning in the enclosed plus maze and climbing behavior in the open field, both of which are suppressed in HD mice. In fact, the R6/2 turning and climbing responses no longer differ from the level of expression of these behaviors in ceftriaxone- or vehicle-treated strain controls. In light of the chronic nature of HD, it would be useful to know whether ceftriaxone treatment remains effective after long-term exposure, but the results to date implicate upregulation of GLT1 as a potential therapeutic target.
If GLT1 upregulation improves striatal glutamate uptake, then it should be accompanied by a corresponding increase in AA release. New evidence supports this view. In R6/2 mice treated with ceftriaxone, electrical stimulation of cortex elicits a rapid increase in striatal AA release that matches the wild-type response, whereas vehicle-treated R6/2s release less AA than similarly treated wild-type mice (82). In addition, local infusion of a GLT1 inhibitor into striatum blocks cortically evoked AA release. Thus, release of AA into striatal extracellular fluid is not only linked to GLT1 function, but dysregulation of this release may play an important role in HD pathophysiology.
Although ceftriaxone also elevates GLT1 expression in wild-type mice (83), the increase in striatal AA is not significant (82). This may be due to compensatory mechanisms that regulate synaptic levels of AA, a process that appears necessary for normal function of corticostriatal activity (114). Alternatively, cortical stimulation may evoke a near-maximal AA release in these mice, which could limit the effectiveness of ceftriaxone, whereas R6/2 mice are well below a maximal release response. It also is interesting that apart from upregulation of GLT1, ceftriaxone may promote glutamate release by increasing expression of the cystine-glutamate exchanger (66, 72). This would make more glutamate available for uptake and thus promote a further increase in AA release. Although this mechanism cannot be ruled out, it seems unlikely because glutamate released by activation of Xc− also acts on presynaptic metabotropic glutamate receptors to inhibit further release (4). Collectively, the data suggest that GLT1 plays a key role in driving AA into striatal extracellular fluid. Recent evidence for differential expression of three GLT1 splice variants (51), however, suggests a need for further investigation of GLT1 involvement in AA release.
Innovation
Dysfunctional communication between cortical and striatal neurons is an early sign of neuropathology in Huntington's disease (HD). Under normal conditions, striatal neurons are driven by glutamate released from cortical afferents and modulated by the release of ascorbic acid (AA). Both glutamate clearance and AA release are impaired in HD owing to a failure of GLT1, a protein primarily found on astrocytes. Growing evidence suggests that by rebalancing striatal extracellular glutamate and AA upregulation of GLT1 could be a potential therapeutic target for HD neuropathology.
Conclusions
Ample evidence indicates that activation of GLT1, which promotes the clearance of extracellular glutamate, results in the release of AA, which in turn modulates the action of glutamate on striatal neurons. The interaction between AA and glutamate, which is tightly regulated by astrocytes, has important implications for understanding both striatal function and behavior. In fact, dysregulation of glutamate uptake and AA release appears to be a critical feature of the corticostriatal neuropathology that underlies HD. The time is right to focus on astrocytes, including their role in striatal neuronal processing and the control of AA release. The results may have important implications for a disease that has so far resisted numerous attempts to devise an effective therapeutic strategy.
Abbreviations Used
- 5-HT
serotonin
- AA
ascorbic acid or ascorbate
- AMPA
alpha-amino-3-hydroxy-5-methylisoxazol-4-propionic acid
- BAC
bacterial artificial chromosome
- DHAA
dehydroascorbate
- EAAT
excitatory amino acid transporter
- GLAST
glutamate aspartate transporter
- Gln
glutamine
- GLT1
glutamate transporter 1
- Glu
glutamate
- GluR
glutamate receptor
- GSH
glutathione
- GSSH
oxidized glutathione
- H2O2
hydrogen peroxide
- HD
Huntington's disease
- HTT
huntingtin protein
- LFP
local field potential
- MSN
medium spiny neuron
- NMDA
N-methyl-D-aspartate
- OH•
hydroxyl radical
- PAT
palmitoyl-acyl transferases
- SVCT
sodium-dependent vitamin C transporter
- UA
uric acid
- WT
wild-type
- Xc−
glutamate/cystine antiporter system
- YAC
yeast artificial chromosome
Acknowledgments
This Forum review was prepared with support from NIH (NS 35663, AG 39818) and CHDI. Scott Barton and Faye Caylor assisted with preparation of figures and article formatting, respectively.
References
- 1.Adams RN. Probing brain chemistry with electroanalytical techniques. Anal Chem 48: 1128A–1138A, 1976 [DOI] [PubMed] [Google Scholar]
- 2.Albin RL, Young AB, and Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366–375, 1989 [DOI] [PubMed] [Google Scholar]
- 3.Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, and Methner A. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol Disord Drug Targets 9: 373–382, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, and Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743–749, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Basse-Tomusk A. and Rebec GV. Corticostriatal and thalamic regulation of amphetamine induced ascorbate release in the neostriatum. Pharmacol Biochem Behav 35: 55–60, 1990 [DOI] [PubMed] [Google Scholar]
- 6.Basse-Tomusk A. and Rebec GV. Regional distribution of ascorbate and 3,4-dihydroxyphenylacetic acid (DOPAC) in rat neostriatum. Brain Res 538: 29–35, 1991 [DOI] [PubMed] [Google Scholar]
- 7.Behrens PF, Franz P, Woodman B, Lindenberg KS, and Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125: 1908–1922, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Berger UV. and Hediger MA. The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. Neuroreport 11: 1395–1399, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Berke JD, Okatan M, Skurski J, and Eichenbaum HB. Oscillatory entrainment of striatal neurons in freely moving rats. Neuron 43: 883–896, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, and Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A 106: 22480–22485, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braun S. and Hauber W. The dorsomedial striatum mediates flexible choice behavior in spatial tasks. Behav Brain Res 220: 288–293, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Brigelius-Flohé R. and Traber MG. Vitamin E: function and metabolism. FASEB J 13: 1145–1155, 1999 [PubMed] [Google Scholar]
- 13.Burns JJ, Rivers JM, and Machlin LJ. (Editors). Third Conference on Vitamin C, Vol 498 New York: Annals of the New York Academy of Sciences, 1987, pp. 1–538 [PubMed] [Google Scholar]
- 14.Buzsáki G. and Chrobak JJ. Synaptic plasticity and self-organization in the hippocampus. Nat Neurosci 8: 1418–1420, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Bylsma FW, Peyser CE, Folstein SE, Folstein MF, Ross C, and Brandt J. EEG power spectra in Huntington's disease: clinical and neuropsychological correlates. Neuropsychologia 32: 137–150, 1994 [DOI] [PubMed] [Google Scholar]
- 16.Carpenter KJ. The History of Scurvy and Vitamin C. New York: Cambridge University Press, 1986, pp. 1–288 [Google Scholar]
- 17.Cayzac S, Delcasso S, Paz V, Jeantet Y, and Cho YH. Changes in striatal procedural memory coding correlate with learning deficits in a mouse model of Huntington disease. Proc Natl Acad Sci U S A 108: 9280–9285, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cepeda C, Ariano MA, Calvert CR, Flores-Hernández J, Chandler SH, Leavitt BR, Hayden MR, and Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res 66: 525–539, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Cepeda C, Cummings DM, André VM, Holley SM, and Levine MS. Genetic mouse models of Huntington's disease: focus on electrophysiological mechanisms. ANS Neuro 2: 103–111, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cepeda C, Wu N, André VM, Cummings DM, and Levine MS. The corticostriatal pathway in Huntington's disease. Prog Neurobiol 81: 253–271, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortright JJ. and Rebec GV. Ascorbate modulation of sensorimotor processing in striatum of freely moving rats. Brain Res 1092: 108–116, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Crook ZR. and Housman D. Huntington's disease: can mice lead the way to treatment? Neuron 69: 423–435, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummings DM, André VM, Uzgil BO, Gee SM, Fisher YE, Cepeda C, and Levine MS. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci 29: 10371–10386, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danbolt NC, Lehre KP, Dehnes Y, and Ullensvang K. Transporters for synaptic transmitter on the glial cell plasma membrane. In: The Tripartite Synapse: Glia in Synaptic Transmission, edited by Volterra A, Magistretti PJ, and Haydon PG. New York: Oxford University Press, 2002, pp. 47–61 [Google Scholar]
- 25.Danbolt NC. Glutamate uptake. Prog Neurobiol 65: 1–105, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Dorner JL, Miller BR, Barton SJ, Brock TJ, and Rebec GV. Sex differences in behavior and striatal ascorbate release in the 140 CAG knock-in mouse model of Huntington's disease. Behav Brain Res 178: 90–97, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorner JL, Miller BR, Klein EL, Murphy-Nakhnikian A, Andrews RL, Barton SJ, and Rebec GV. Corticostriatal dysfunction underlies diminished striatal ascorbate release in the R6/2 mouse model of Huntington's disease. Brain Res 1290: 111–120, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehrnhoefer DE, Butland SL, Pouladi MA, and Hayden MR. Mouse models of Huntington disease: variations on a theme. Dis Model Mech 2: 123–129, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehrnhoefer DE, Sutton L, and Hayden MR. Small changes, big impact: posttranslational modifications and function of Huntingtin in Huntington's disease. Neuroscientist 17: 475–492, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Englard S. and Seifter S. The biochemical functions of ascorbic acid. Ann Rev Nutr 6: 365–406, 1986 [DOI] [PubMed] [Google Scholar]
- 31.Estrada-Sánchez AM, Montiel T, Segovia J, and Massieu L. Glutamate toxicity in the striatum of the R6/2 Huntington's disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiol Dis 34: 78–86, 2009 [DOI] [PubMed] [Google Scholar]
- 32.Estrada-Sánchez AM. and Rebec GV. Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington's disease: interactions between neurons and astrocytes. Basal Ganglia 2: 57–66, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ewing AG, Alloway KD, Curtis SD, Dayton MA, Wightman RM, and Rebec GV. Simultaneous electrochemical and unit recording measurements: characterization of the effects of D-amphetamine and ascorbic acid on neostriatal neurons. Brain Res 261: 101–108, 1983 [DOI] [PubMed] [Google Scholar]
- 34.Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Déglon N, Ferrante RJ, and Bonvento G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet 19: 3053–3067, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fillenz M, O'Neill RD, and Grunewald RA. Changes in extracellular brain ascorbate concentration as an index of excitatory aminoacid release. In: Monitoring Neurotransmitter Release During Behaviour, edited by Joseph MH, Fillenz M, MacDonald IA, and Marsden CA. Chichester: Ellis Norwood, 1986, pp. 144–163 [Google Scholar]
- 36.Forno L. and Norville R. Ultrastructure of the neostriatum in Huntington's and Parkinson's disease. Adv Neurol 23: 123–134, 1979 [Google Scholar]
- 37.Fowler SC, Miller BR, Gaither TW, Johnson MA, and Rebec GV. Force-plate quantification of progressive behavioral deficits in the R6/2 mouse model of Huntington's disease. Behav Brain Res 202: 130–137, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukata Y. and Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci 11: 161–175, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Gambazzi L, Gokce O, Seredenina T, Katsyuba E, Runne H, Markram H, Giugliano M, and Luthi-Carter R. Diminished activity-dependent brain-derived neurotrophic factor expression underlies cortical neuron microcircuit hypoconnectivity resulting from exposure to mutant huntingtin fragments. J Pharmacol Exp Ther 335: 13–22, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Ghasemzadeh B, Cammack J, and Adams RN. Dynamic changes in extracellular fluid ascorbic acid monitored by in vivo electrochemistry. Brain Res 547: 162–166, 1991 [PubMed] [Google Scholar]
- 41.Gonon F, Buda M, Cespuglio R, Jouvet M, and Pujol JF. Voltammetry in the striatum of chronic freely moving rats: detection of catechols and ascorbic acid. Brain Res 223: 69–80, 1981 [DOI] [PubMed] [Google Scholar]
- 42.Grunewald RA. Ascorbic acid in the brain. Brain Res Rev 18: 123–133, 1993 [DOI] [PubMed] [Google Scholar]
- 43.Grunwald RA. and Fillenz M. Release of ascorbate from synaptosomal fraction of rat brain. Neurochem Int 5: 491–500, 1984 [DOI] [PubMed] [Google Scholar]
- 44.Grunewald RA, Fillenz M, and Albery WJ. The origin of circadian and amphetamine-induced changes in the extracellular concentration of brain ascorbate. Neurochem Int 5: 773–778, 1983 [DOI] [PubMed] [Google Scholar]
- 45.Gutekunst CA, Norflus F, and Hersch SM. The neuropathology of Huntington's disease. In: Huntington's Disease, 3rd ed., edited by Bates G, Harper P, and Jones L. New York: Oxford University Press, 2002, pp. 251–275 [Google Scholar]
- 46.Halliwell B. and Gutteridge JMC. Free Radicals in Biology and Medicine, 3rd ed. New York: Oxford University Press, 1999, p. 851 [Google Scholar]
- 47.Harper PS. Huntington's disease: a historical background. In: Huntington's Disease, 3rd ed., edited by Bates G, Harper P, and Jones L. New York: Oxford University Press, 2002, pp. 3–27 [Google Scholar]
- 48.Harrison FE, Green RJ, Dawes SM, and May JM. Vitamin C distribution and retention in the mouse brain. Brain Res 1348: 181–186, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrison FE. and May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol Med 46: 719–730, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hassel B, Tessler S, Faull RL, and Emson PC. Glutamate uptake is reduced in prefrontal cortex in Huntington's disease. Neurochem Res 33: 232–237, 2008 [DOI] [PubMed] [Google Scholar]
- 51.Holmseth S, Scott HA, Real K, Leher KP, Leergaard TB, Bjaalie JG, and Danbolt NC. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience 162: 1055–1071, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Hong SL, Barton SJ, and Rebec GV. Altered neural and behavioral dynamics in Huntington's Disease: An entropy conservation approach. PLoS One 7: e30879, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong SL, Cossyleon D, Hussain WA, Walker LJ, Barton SJ, and Rebec GV. Dysfunctional behavioral modulation of corticostriatal communication in the R6/2 mouse model of Huntington's disease. PLoS One 7: e47026, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang K, Kang MH, Askew C, Kang R, Sanders SS, Wan J, Davis NG, and Hayden MR. Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol Dis 40: 207–215, 2010 [DOI] [PubMed] [Google Scholar]
- 55.Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst CA, Hayden MR, and El-Husseini A. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron 44: 977–986, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Huntington G. On Chorea. Med Surg Reporter Philadelphia 26: 317–321, 1872 [Google Scholar]
- 57.Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983, 1993 [DOI] [PubMed] [Google Scholar]
- 58.Izhikevich EM, Desai NS, Walcott EC, and Hoppensteadt FC. Bursts as a unit of neural information: selective communication via resonance. Trends Neurosci 26: 161–167, 2003 [DOI] [PubMed] [Google Scholar]
- 59.Jin X. and Costa RM. Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature 466: 457–462, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones L. The cell biology of Huntington's disease. In: Huntington's Disease, 3rd Ed., edited by Bates G, Harper P, and Jones L. New York: Oxford University Press, 2002, pp. 348–386 [Google Scholar]
- 61.Justice JB, Jr., Introduction to in vivo voltammetry. In: Voltammetry in the Neurosciences, Principles, Methods, and Applications, edited by Justice JB., Jr.Clifton, NJ: Humana Press, 1987, pp. 3–101 [Google Scholar]
- 62.Kiyatkin EA. and Rebec GV. Dopaminergic modulation of glutamate-induced excitations of neurons in the neostriatum and nucleus accumbens of awake unrestrained rats. J Neurophysiol 75: 142–153, 1996 [DOI] [PubMed] [Google Scholar]
- 63.Kiyatkin EA. and Rebec GV. Ascorbate modulates glutamate-induced excitations of striatal neurons. Brain Res 812: 14–22, 1998 [DOI] [PubMed] [Google Scholar]
- 64.Kiyatkin EA. and Rebec GV. Striatal neuronal activity and responsiveness to dopamine and glutamate after selective blockade of D1 and D2 dopamine receptors in freely moving rats. J Neurosci 19: 3594–3609, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, and Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol 86: 2667–2677, 2001 [DOI] [PubMed] [Google Scholar]
- 66.Knackstedt LA, Melendez RI, and Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry 67: 81–84, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kratzing CC, Kelly JD, and Oelrichs BA. Ascorbic acid in neural tissues. J Neurochem 39: 625–627, 1982 [DOI] [PubMed] [Google Scholar]
- 68.Kreitzer AC. and Berke JD. Investigating striatal function through cell-type-specific manipulations. Neuroscience 198: 19–26, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kreitzer AC. and Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron 60: 543–554, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, and Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci 21: 9112–9123, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, and Chesselet MF. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res 58: 515–532, 1999 [PubMed] [Google Scholar]
- 72.Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, Wiedau-Pazos M, Schubert D, Maher P, and Methner A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem 111: 332–343, 2009 [DOI] [PubMed] [Google Scholar]
- 73.Liévens JC, Woodman B, Mahal A, Spasic-Boscovic , Samuel D, Kerkerian-Le Goff , and Bates GP. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis 8: 807–821, 2001 [DOI] [PubMed] [Google Scholar]
- 74.Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci 20: 38–43, 1997 [DOI] [PubMed] [Google Scholar]
- 75.Lo DC. and Hughes RE. (Eds). Neurobiology of Huntington's Disease: Applications to Drug Discovery. Boca Raton, FL: CRC Press, 2011, p. 312. [PubMed] [Google Scholar]
- 76.Majewska MD, Bell JA, and London ED. Regulation of the NMDA receptor by redox phenomenon: inhibitory role of ascorbate. Brain Res 537: 328–332, 1990 [DOI] [PubMed] [Google Scholar]
- 77.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, and Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493–506, 1996 [DOI] [PubMed] [Google Scholar]
- 78.Maragakis NJ. and Rothstein JD. Glutamate transporters in neurologic disease. Arch Neurol 58: 365–370, 2001 [DOI] [PubMed] [Google Scholar]
- 79.Mefford IN, Oke A, and Adams RN. Regional distribution of ascorbate in human brain. Brain Res 212: 223–226, 1981 [DOI] [PubMed] [Google Scholar]
- 80.Michael AC. and Borland LM. (Eds). Electrochemical Methods for Neuroscience. Boca Ratan, FL: CRC Press, 2007, p. 544. [PubMed] [Google Scholar]
- 81.Milby K, Oke A, and Adams RN. Detailed mapping of ascorbate distribution in rat brain. Neurosci Lett 28: 15–20, 1982 [DOI] [PubMed] [Google Scholar]
- 82.Miller BR, Dorner JL, Bunner KD, Gaither TW, Klein EL, Barton SJ, and Rebec GV. Upregulation of GLT1 reverses the deficit in cortically evoked striatal ascorbate efflux in the R6/2 mouse model of Huntington's disease. J Neurochem 121: 629–638, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, and Rebec GV. Upregulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience 153: 329–337, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miller BR, Walker AG, Barton SJ, and Rebec GV. Dysregulated neuronal activity patterns implicate corticostriatal circuit dysfunction in multiple rodent models of Huntington's disease. Front Syst Neurosci 5: 1–10, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miller BR, Walker AG, Shah AS, Barton SJ, and Rebec GV. Dysregulated information processing by medium-spiny neurons in striatum of freely behaving mouse models of Huntington's disease. J Neurophysiol 100: 2205–2216, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller R. and Wickens JR. Brain Dynamics and the Striatal Complex. Amsterdam, The Netherlands: Taylor and Francis Publishers, 2000, p. 330 [Google Scholar]
- 87.Milnerwood AJ. and Raymond LA. Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load. J Physiol 585: 817–831, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Milnerwood AJ. and Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci 33: 513–523, 2010 [DOI] [PubMed] [Google Scholar]
- 89.Mueller K. and Haskett C. Effects of haloperidol on amphetamine-induced increases in ascorbic acid as determined by voltammetry in vivo. Pharmacol Biochem Behav 27: 231–234, 1987 [DOI] [PubMed] [Google Scholar]
- 90.Mun GH, Kim MJ, Lee JH, Kim HJ, Chung YH, Chung YB, Kang JS, Hwang YI, Oh SH, Kim JG, Hwang DH, Shin DH, and Lee WJ. Immunohistochemical study of the distribution of sodium-dependent vitamin C transporters in adult rat brain. J Neurosci Res 83: 919–928, 2006 [DOI] [PubMed] [Google Scholar]
- 91.Murphy-Nakhnikian JM, Dorner JL, Fischer BI, Bower-Bir ND, and Rebec GV. Abnormal burst patterns of single neurons recorded in the substantia nigra reticulata of behaving 140 CAG Huntington's disease mice. Neurosci Lett 512: 1–5, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nicniocaill B, Haraldsson B, Hansson O, O'Connor WT, and Brundin P. Altered striatal amino acid neurotransmitter release monitored using microdialysis in R6/1 Huntington transgenic mice. Eur J Neurosci 13: 206–210, 2001 [DOI] [PubMed] [Google Scholar]
- 93.O'Neill RD. and Fillenz M. Circadian changes in extracellular ascorbate in rat cortex, accumbens, striatum and hippocampus: correlations with motor activity. Neurosci Lett 60: 331–336, 1985 [DOI] [PubMed] [Google Scholar]
- 94.Padayatty S, Katz A, Wang Y, Eck P, Kwon O, Lee J, Chen S, Corpe C, Dutta A, Dutta S, and Levine M. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr 22: 18–35, 2003 [DOI] [PubMed] [Google Scholar]
- 95.Painold A, Anderer P, Holl AK, Letmaier M, Saletu-Zyhlarz GM, Saletu B, and Bonelli RM. EEG low-resolution brain electromagnetic tomography (LORETA) in Huntington's Disease. J Neurol 258: 840–854, 2011 [DOI] [PubMed] [Google Scholar]
- 96.Pierce RC, Miller DW, Reising DB, and Rebec GV. Unilateral neostriatal kainate, but not 6-OHDA, lesions block dopamine agonist-induced ascorbate release in the neostriatum of freely moving rats. Brain Res 597: 138–143, 1992 [DOI] [PubMed] [Google Scholar]
- 97.Pierce RC. and Rebec GV. Stimulation of both D1 and D2 dopamine receptors increases behavioral activation and ascorbate release in the neostriatum of freely moving rats. Eur J Pharmacol 191: 295–302, 1990 [DOI] [PubMed] [Google Scholar]
- 98.Pierce RC. and Rebec GV. Iontophoresis in the neostriatum of awake unrestrained rats: differential effects of dopamine glutamate and ascorbate on motor- and nonmotor-related neurons. Neuroscience 67: 313–324, 1995 [DOI] [PubMed] [Google Scholar]
- 99.Raymond LA, André VM, Cepeda C, Gladding CM, Milnerwood AJ, and Levine MS. Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function. Neuroscience 198: 252–273, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rebec GV. Ascorbate: an antioxidant neuroprotectant and extracellular neuromodulator. In: Metals and Oxidative Damage in Neurological Disorders, edited by Connor JR. New York: Plenum Press, 1997, pp. 149–173 [Google Scholar]
- 101.Rebec GV. From interferant anion to neuromodulator: ascorbate oxidizes its way to respectability. In: Electrochemical Methods for Neuroscience, edited by Michael AC. and Borland LM. Boca Raton, FL: CRC Press, 2007, pp. 149–165 [PubMed] [Google Scholar]
- 102.Rebec GV, Barton SJ, and Ennis MD. Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington's disease gene. J Neurosci 22: RC202, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rebec GV, Barton SJ, Marseilles AM, and Collins K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. NeuroReport 14: 1263–1265, 2003 [DOI] [PubMed] [Google Scholar]
- 104.Rebec GV, Conroy SK, and Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience 137: 327–336, 2006 [DOI] [PubMed] [Google Scholar]
- 105.Rebec GV. and Pierce RC. A vitamin as neuromodulator: ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog Neurobiol 43: 537–565, 1994 [DOI] [PubMed] [Google Scholar]
- 106.Rebec GV. and Wang Z. Behavioral activation in rats requires endogenous ascorbate release in striatum. J Neurosci 21: 668–675, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rebec GV, Witokski SR, Sandstrom MI, Rostand RD, and Kennedy RT. Extracellular ascorbate modulates cortically evoked glutamate dynamics in rat striatum. Neurosci Lett 378: 166–170, 2005 [DOI] [PubMed] [Google Scholar]
- 108.Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, and Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A 85: 5733–5737, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rice ME. Ascorbate and its neuroprotective role in the brain. Trends Neurosci 23: 209–216, 2000 [DOI] [PubMed] [Google Scholar]
- 110.Rice ME. and Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 82: 1213–1223, 1998 [DOI] [PubMed] [Google Scholar]
- 111.Roos RAC. Huntington's disease: a clinical review. Orphanet J Rare Dis 5: 40 (1–8), 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosenberg C, Nudleman K, and Starr A. Cognitive evoked potentials (P300) in early Huntington's disease. Arch Neurol 42: 984–987, 1985 [DOI] [PubMed] [Google Scholar]
- 113.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, and Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433: 73–77, 2005 [DOI] [PubMed] [Google Scholar]
- 114.Sandstrom MI. and Rebec GV. Extracellular ascorbate modulates glutamate dynamics: Role of behavioral activation. BMC Neurosci 8: 32, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sato H, Tamba M, Ishii T, and Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274: 11455–11458, 1999 [DOI] [PubMed] [Google Scholar]
- 116.Savini I, Rossi A, Pierro C, Avigliano L, and Catani MV. SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids 34: 347–355, 2008 [DOI] [PubMed] [Google Scholar]
- 117.Seredenina T. and Luthi-Carter R. What have we learned from gene expression profiles in Huntington's disease? Neurobiol Dis 45: 83–98, 2012 [DOI] [PubMed] [Google Scholar]
- 118.Spampanato J, Gu X, Yang XW, and Mody I. Progressive synaptic pathology of motor cortical neurons in a BAC transgenic mouse model of Huntington's disease. Neuroscience 157: 606–620, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Streletz LJ, Reyes PF, Zalewska M, Katz L, and Fariello RG. Computer analysis of EEG activity in dementia of the Alzheimer's type and Huntington's disease. Neurobiol Aging 11: 15–20, 1990 [DOI] [PubMed] [Google Scholar]
- 120.Tkac I, Dubinsky JM, Keene CD, Gruetter R, and Low WC. Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. J Neurochem 100: 1397–1406, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tolbert BM. and Ward JB. Dehydroascorbic acid. In: Ascorbic Acid: Chemistry, Metabolism, and Uses, edited by Seib PA. and Tolbert GM. Washington, DC: American Chemical Society, 1982, pp. 101–123 [Google Scholar]
- 122.Trotti D, Rizzini BL, Rossi D, Haugeto O, Racagni G, Danbolt NC, and Volterra A. Neuronal and glial glutamate transporters possess an SH-based redox regulatory mechanism. Eur J Neurosci 9: 1236–1243, 1997 [DOI] [PubMed] [Google Scholar]
- 123.van Dellen A, Blakemore C, Deacon R, York D, and Hannan AJ. Delaying the onset of Huntington's in mice. Nature 404: 721–722, 2000 [DOI] [PubMed] [Google Scholar]
- 124.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, and Richardson EP. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44: 559–577, 1985 [DOI] [PubMed] [Google Scholar]
- 125.Walker AG, Miller BR, Fritsh JN, Barton SJ, and Rebec GV. Altered information processing in the proforntal cortex of Huntington's Disease mouse models. J Neurosci 28: 8973–8982, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Walker AG, Ummel JR, and Rebec GV. Reduced expression of conditioned fear in the R6/2 mouse model of Huntington's disease is related to abnormal activity in prelimbic cortex. Neurobiol Dis 43: 379–387, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wexler NS. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A 101: 3498–3503, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wilson CJ. and Kawaguchi Y. The origins of the two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci 16: 2397–2410, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wilson JX, Peters CE, Sitar SM, Daoust P, and Gelb AW. Glutamate stimulates ascorbate transport by astrocytes. Brain Res 858: 61–66, 2000 [DOI] [PubMed] [Google Scholar]
- 130.Wilson RL. and Wightman RM. Systemic and nigral application of amphetamine both cause an increase in extracellular concentration of ascorbate in the caudate nucleus of the rat, Brain Res 339: 219–226, 1985 [DOI] [PubMed] [Google Scholar]
- 131.Wood NI, Glynn D, and Morton AJ. “Brain training” improves cognitive performance and survival in a transgenic mouse model of Huntington's disease. Neurobiol Dis 42: 427–437, 2011 [DOI] [PubMed] [Google Scholar]
- 132.Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, Drisdel RC, Green WN, Ravikumar B, Rubinsztein DC, El-Husseini A, and Hayden MR. Palmitoylation of Huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci 9: 824–831, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zetterström T, Wheeler DB, Boutelle MG, and Fillenz M. Striatal ascorbate and its relationship to dopamine receptor stimulation and motor activity. Eur J Neurosci 3: 940–946, 1992 [DOI] [PubMed] [Google Scholar]
- 134.Zühlke Ch, Reiss O, Bockel B, Lange H, and Thies U. Mitotic stability and meiotic variability of the (CAG)n repeat in the Huntington disease gene. Hum Mol Genet 2: 2063–2067, 1993 [DOI] [PubMed] [Google Scholar]