

Abstract
Mouse models are useful tools for developing potential therapies for human inherited retinal diseases, such as retinitis pigmentosa (RP), since more strains are being identified with the same mutant genes and phenotypes as humans with corresponding retinal degenerative diseases. Mutations in the beta subunit of the human rod phosphodiesterase (PDE6B) gene are a common cause of autosomal recessive RP (arRP). This article focuses on two well-established naturally occurring mouse models of arRP caused by spontaneous mutations in Pde6b, their discovery, phenotype, mechanism of degeneration, strengths and limitations, and therapeutic approaches to restore vision and delay disease progression. Viral vector, especially adeno-associated viral vector (AAV) -mediated gene replacement therapy, pharmacological treatment, cell-based therapy and other approaches that extend the therapeutic window of treatment, is a potentially promising strategy for improving photoreceptor function and significantly slowing the process of retinal degeneration.
Introduction
Retinitis pigmentosa (RP) is a family of inherited diseases with clinical and genetic heterogeneity causing retinal dysfunction and eventual photoreceptor cell death [1-3]. RP can be either autosomal dominant, autosomal recessive, or X-linked [4-6]. Mutations in the phosphodiesterase 6B, cyclic guanosine monophosphate-specific, rod, beta (PDE6B) gene encoding the beta subunit of phosphodiesterase have been linked to autosomal recessive RP (arRP) in humans [7-9]. Retinitis pigmentosa resulting from mutations in the PDE6B gene is one of the earliest onset and most aggressive forms of this disease, accounting for up to 5% of arRP [7,9]. Rod PDE is a membrane-associated protein composed of two distinct catalytic subunits (PDE6Α, PDE6Β) of approximately 99 kDa, and two identical gamma inhibitory subunits (approximately 10 kDa). Both catalytic subunits contain two high-affinity non-catalytic cyclic guanosine monophosphate (cGMP) binding sites and a C-terminal half representing the catalytic domain [10,11]. PDE is an essential part of the phototransduction cascade, playing a role in hydrolyzing the cGMP second messenger and resulting in channel closure in response to light [12]. Mutations in Pde6b result in a nonfunctional PDE and an accumulation of cGMP [13-15]. In cells with the defective PDE6B enzyme, increased levels of cGMP lead to photoreceptor cell death [3,15-17]. In this review, we describe the role of two well-characterized, naturally occurring mouse lines with defects in Pde6b as ocular models for the human disease [18,19], particularly focusing on various therapeutic studies to compare the potential for treating this form of RP.
Naturally occurring mouse models of Pde6b retinitis pigmentosa
The rd1 (rodless) mouse model of arRP is characterized by severe, early onset, rapid retinal degeneration caused by mutations in Pde6b [13,20]. The mutant Pde6b gene in rd1 mice, mapped on chromosome 5 [21], contains a murine leukemia provirus insertion in intron 1 and a point mutation, which introduces a stop codon in exon 7 (Figure 1) [22,23]. A rodless retina (gene symbol, r, rd, rd1) was discovered in mice by Keeler and was first reported in 1924 as an autosomal recessive mutation leading to the absence of visual cells (rods), including the outer nuclear layer [24,25]. This animal-related work continued in the United States and Europe over the next decade, but Keeler’s stock was lost by the end of World War II [26]. In 1951, Bruckner reported a similar retinal abnormality that he first recognized in wild mice with ophthalmoscopy [27]. Using PCR analysis of DNA from archival retinal sections, Pitler et al. demonstrated that this line of rd mice contained a homozygous nonsense point mutation in exon 7 (codon 347) and intronic polymorphisms in the Pde6b gene identical to those in the rodless strain initially discovered by Keeler [28]. Histological analysis showed that the outer segments (OSs) and inner segments (ISs) of the photoreceptors were never well developed in rd mice [13,29]. At P10, the OS discs showed signs of disruption, the chromatin was fragmented, and TUNEL-positive photoreceptor cells increased with a rapid loss of rods by P14. In all regions of the eye, rapid rod degeneration preceded cone degeneration. Only about 2% of the rods remained in the posterior region at P17, and none by P36. In contrast, at least 75% of the cone nuclei remained at P17 in rd mice. As the retinal degeneration developed, the outer nuclear layer (ONL) became rapidly thinner but left a single row of cone perikarya at 18 months of age [29,30].
Figure 1.
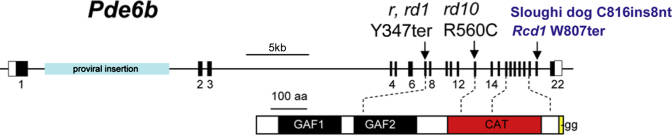
Schematic representation of the mouse PDE6B gene and protein, and the localization of spontaneous mutations in animal models. The rd1 mouse contains a murine leukemia provirus insertion in intron 1 and a point mutation, which introduces a stop codon in exon 7. The rd10 mouse carries a missense mutation (R560C) in exon 13. Two canine models, the rcd1 Irish setter and the Sloughi dog, contain a nonsense amber mutation at codon 807 (W807ter) and an 8 bp insertion after codon 816, respectively. The PDE6B protein contains two high-affinity non-catalytic cGMP binding sites (GAF domains) and a catalytic domain in which the majority of human mutations are located. Reprinted from Vision Research, vol. 49(22), Baehr W. and Frederick J.M., Naturally occurring animal models with outer retina phenotypes, 2636–2652, 2009, with permission from Elsevier.
In addition to the established role as an animal model for recessive RP, the rd1 mouse, as a source of rodless retinas, has been used for cDNA microarray gene expression studies to elucidate the molecular pathways underlying photoreceptor cell death [31], and to determine the endogenous source of mRNA transcripts for proteins whose cellular localization is unknown [32,33]. Comparative studies use real-time quantitative reverse transcription (RT)–PCR using cDNA samples from rd1 retinas devoid of photoreceptor cells and wild-type controls have confirmed either the rod-specific expression of genes or whether a particular transcript originates mainly from the inner retina [32,33]. Rodless mice have also been used to study circadian entrainment, pupillary constriction, and intrinsically sensitive melanopsin-positive ganglion cells [34-37].
The rd10 mouse, first described by Chang et al. in 2002, carries a missense mutation (R560C) in exon 13 of the Pde6b gene, and represents another useful natural model of recessive retinal degeneration [20,38]. In contrast to rd1, in which PDE6B protein expression and activity are undetectable, rd10 mice are characterized by a relatively slower onset of retinal degeneration, with decreased PDE activity. PDE6B protein can be detected early in rd10 mouse retinas (P10) with western blotting and immunostaining, although the level of expression was significantly reduced compared to that of age-matched wild-type controls [38]. In rd1 mice, peak photoreceptor cell death occurs before full development of the retinal structures, whereas most cells in rd10 mouse retinas have terminally matured before degeneration occurs [38,39]. Histological examination reveals progressive ONL degeneration in rd10 mice, starting from the central retina at P16 and spreading to the peripheral retina by P20. Rapid degeneration occurs between P18 and P25, and by P60, most of the photoreceptors disappear, although the thickness of the INL and the ganglion cell layer is not yet affected. Sclerotic retinal vessels appear at 4 weeks of age in rd10 mice, and retinal degeneration can be easily distinguished at 2 months of age with fundoscopy [38,39]. Dark-rearing rd10 mice further slows the retinal degeneration rate by as much as 3 months [38,40].
The maximal electroretinogram (ERG) response in cyclic light-reared rd10 occurs at 3 weeks and is undetectable at 2 months of age [38]. In contrast, rd1 mice do not generate a scotopic ERG response at any age due to a complete lack of functional PDE6B. Furthermore, aggressive rd1 retinal degeneration cannot be delayed by rearing the mice in darkness (Figure 2) [38]. The residual activity of PDE6b in rd10 retinas may prevent a toxic increase in cGMP early in the mouse’s life, thus delaying the onset of photoreceptor cell death, and extending the window for therapeutic interventions. Many missense pathogenic human mutations in PDE6B leading to autosomal recessive retinitis pigmentosa are located within the catalytic domain [9], potentially resulting in partial loss of function and reduced PDE6B enzymatic activity, as seen in rd10. Thus, the rd10 represents a better mouse model than rd1 for developing strategies for treating human patients with recessive RP.
Figure 2.
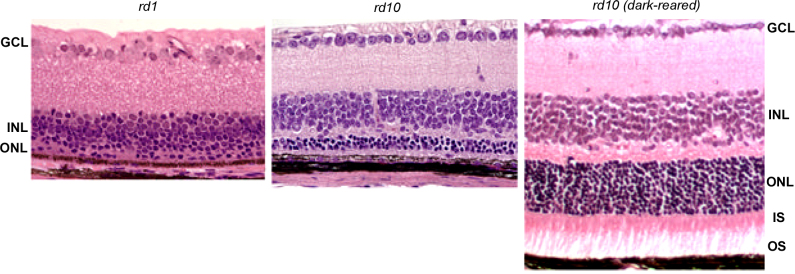
Retinal morphology of rodless rd1 and rd10 mice, at 24 days of age. Both rodless rd1 (left panel) and rd10 (middle panel) retinas exhibit marked thinning of the photoreceptor layer. Note the normal ONL thickness of the dark-reared rd10 mouse retina (right panel), in contrast to that of the cyclic light-reared rd10 (middle panel); ONL, outer nuclear layer; IS, inner segments; INL, inner nuclear layer; GCL, ganglion cell layer; Reprinted from Vision Research, vol. 47(5), Chang B, et al., Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene, 624-33, 2007, with permission from Elsevier.
Therapeutic approaches
Pharmacological treatment
Several pharmacology-based treatments were developed to temporarily delay the photoreceptor degeneration in mouse models caused by PDE6B deficiency [41]. In 1999, one study showed that D-cis-diltiazem, a calcium-channel blocker that also acts on light-sensitive cGMP-gated channels, rescued photoreceptors and preserved visual function in the rd1 mouse [42]. The dihydropyridine, diltiazem, is a competitive antagonist of L-type calcium channels. The D-cis stereoisomer is the active compound in several commonly used prescription drugs. Since rd1 rods begin to degenerate due to apoptosis caused by Ca2+ overload around P10 [43,44], intraperitoneal injections of D-cis-diltiazem was applied at P9. Retinal whole mounts and sections were labeled with an antibody against rhodopsin and showed more positive staining in treated mice, which were supported by a measurable scotopic ERG b-wave (approximately 20 μV) at P36. Additionally, the diltiazem-treated cohort at P36 had approximately 18,600 surviving rods, a two- to threefold increase compared to 7,500 rods in the untreated eyes; a modest protective effect on cone survival was also observed [42]. Other laboratories also concluded that D-cis-diltiazem could partially protect photoreceptor cells for the short term in the rd1 mouse [45]. These studies provided evidence that suitable pharmacological approaches to vision protection could be obtained through managing Ca2+ overload [46,47]. Controversially, Pawlyk et al. concluded that there was no significant difference between treated and untreated rd1 mice following D-cis-diltiazem treatment [48].
LaVail et al. have shown that intravitreal delivery of survival factors, such as brain-derived neurotrophic factor (BDNF), neurotrophin-3, neurotrophin-4, and ciliary neurotrophic factor (CNTF), in several animal models of retinal degeneration, including rd1, can slow the progression of photoreceptor cell death [49]. CNTF, especially, has been shown to increase photoreceptor survival as a neuroprotective factor [50,51]. A study by Cayouette et al. used an adenoviral vector to deliver CNTF intravitreally to rd1 mouse eyes, resulting in increased photoreceptor survival [52]. Treatment with CNTF in combination with BDNF was also shown to rescue photoreceptors in cultured rd1 retinal explants [53]. The therapeutic efficacy of encapsulated CNTF-secreting cell therapy was demonstrated in the rcd1 canine model of RP [51,54].
Frasson et al. attempted to treat rd1 degeneration by delivering glial cell line–derived neurotrophic factor (GDNF) into the rd1 eyes at 13 and 17 days of age via sub-retinal delivery [55]. GDNF provided histological and functional neuroprotective benefits on rod photoreceptors in the rd1 mouse, although the effects were partial and temporary. Ohnaka et al. have also shown that GDNF can temporarily slow photoreceptor cell loss in rd10 mice [56].
Systemic administration of recombinant erythropoietin (Epo), an oxygen-regulated hormone, has also been shown to have protective effects on photoreceptor cells in either light-induced or certain inherited forms of retinal degeneration [57,58]. Rex et al. have shown that Epo protein expression following systemic adeno-associated viral (AAV)–mediated gene delivery resulted in improved retinal morphology in the rds/peripherin (Prph2), but not the rd10 mouse model of retinal degeneration [58]. Another study indicated that activation of bone marrow–derived microglia following systemic injections with Epo and granulocyte colony-stimulating factor slowed retinal degeneration in rd10 mice, as indicated by increased outer nuclear layer thickness at P30 [59].
An indirect way of extending photoreceptor survival without pharmacological treatment is environmental enrichment, which may lead to an increase in the endogenous levels of potentially beneficial trophic factors. Barone et al. have shown that prolonged exposure of rd10 mice to an enriched environment from birth had protective effects on retinal function and morphology, leading to a decrease in their retinal degeneration rate and increased expression of retinal CNTF mRNA [60].
Tauroursodeoxycholic acid (TUDCA), the active antioxidant component in bear bile, has been used in traditional Chinese medicine for thousands of years. TUDCA acts via a phosphatidylinositol 3-kinase (PI3K)-dependent signaling pathway to block neuronal death triggered by amyloid-β peptide [61]. In 2006, Boatright et al. showed that more photoreceptors survived and better photoreceptor morphology was maintained, and the dark-adapted ERG a- and b-wave amplitudes were significantly greater by P18 when TUDCA was injected subcutaneously in the nape of P6 rd10 mice [62]. TUDCA-treated retinas showed almost no TUNEL signal and much less immunoreactivity for activated caspase-3. These findings clearly indicate that TUDCA injections suppress apoptosis in rd10 mice. Similar studies also found that the overall morphology of the photoreceptor cells was better preserved in the TUDCA-treated rd10 eyes at P30 and P38 [63-65].
Komeima et al. have demonstrated the beneficial effects of antioxidants in slowing the photoreceptor degeneration process in rd1 and rd10 mice [66,67]. Systemic delivery of combined antioxidants, including alpha-tocopherol, ascorbic acid, and alpha-lipoic acid, reduced cone cell death in both models. The rd10 mouse displayed substantial therapeutic effects, such as better preservation of cone function and retinal morphology, due to the later onset of photoreceptor cell death in this model compared to rd1. Another study showed that a combination of antioxidants, such as lutein, zeaxanthin, and alpha lipoic acid, slowed rd1 rod photoreceptor degeneration following early administration [68].
Cell-based therapy and optogenetic approaches
Stem cell-based therapy and optogenetic approaches have the potential to restore visual function for late stages of retinal degeneration, by either focusing on replacing the lost photoreceptor cells or by rendering the remaining bipolar and ganglion cells photosensitive [69-72]. Adult bone marrow contains a population of hematopoietic stem cells (HSCs) that can be divided into lineage positive (Lin+) and lineage negative (Lin–) cells. The Lin– subpopulation contains various progenitor cells capable of becoming vascular endothelial cells [73]. These endothelial progenitor cells can mobilize in response to various signaling molecules and target sites of angiogenesis in ischemic peripheral vasculature. Otani et al. reported that intravitreal injection of adult bone marrow–derived Lin– HSCs at P6 could completely prevent retinal vascular degeneration in the rd1 mouse [74]. Furthermore, this vascular rescue correlated with neuronal rescue: The INL remained nearly normal, and the ONL was partially preserved, with the rescued cells predominantly cones [74]. This study suggests that the rescue effect of Lin– HSCs can last for as long as 6 months if the cells are injected just before the onset of degeneration, but the ERG amplitudes were only 8–10 μV at 2 months after treatment. The ERG amplitudes in the rescued eyes were considerably lower than that of the gene therapy-based rescue studies (see below). Sasahara et al. later showed that endogenous bone marrow–derived microglia played a protective role in vascular and neural degeneration in rd1 and rd10 mice [59].
In addition to stem cells, Barber et al. have shown that photoreceptor transplantation is feasible in several mouse models at different stages of retinal degeneration, including rd1, and the recipient microenvironment affects the transplanted receptor morphology [75]. In another recent study, Singh et al. showed that transplanted rod precursors can form an outer nuclear layer in degenerated rd1 mouse retinas [76], and this approach may hold the potential to recreate a new functional photoreceptor layer in advanced retinal degeneration.
The rd1 mouse represents a useful model for testing optogenetic tools for treating late stage RP [77]. Studies have shown that exogenous expression of opsins, such as channelrhodopsin 2 (ChR2) or melanopsin, in surviving retinal neurons can render them photosensitive, and restored some visual function in rd1 mice in which all photoreceptor cells were lost [34,78]. Bi et al. found that following intravitreal delivery of AAV2-ChR2-GFP in the rd1 mouse, expression of ChR2 was achieved in retinal ganglion cells, leading to restoration of visually evoked cortical responses [79]. ChR2 gene delivery in ON bipolar cells was also achieved, leading to restored photosensitivity and behavioral responses in animal models [80,81]. Caporale et al. used an AAV2-mediated intravitreal delivery of an engineered light-gated ionotropic glutamate receptor to restore light responsiveness to the retinal ganglion cells of adult rd1 mice [82].
Gene replacement therapy
Due to the early and rapid rate of photoreceptor cell loss in the rd1 mouse, providing effective gene therapy in a timely manner to rescue visual function in this animal model have proved difficult. In the early 1990s, Bennett et al. found that the photoreceptors of the rd1 mouse could be partially rescued with in vivo gene therapy, thus supporting the feasibility of treating inherited retinal degeneration with somatic gene therapy [83]. Bennett et al. conducted an experiment with sub-retinal injection of a recombinant replication-defective adenovirus that contained murine cDNA for β-PDE, Ad-CMV-βPDE. Sub-retinal injection of Ad-CMV-βPDE resulted in β-PDE transcripts, increased PDE activity, and delayed photoreceptor cell death by 6 weeks. However, this early generation of adenoviral vectors had limitations, including short-term expression of the transduced gene due to immune response against the adenoviral vector. Additionally, the rescue effect was not long lasting. Subsequently, other researchers switched to an encapsidated adenovirus minichromosome (EAM), which showed relatively longer transgene expression [84,85]. EAM-mediated delivery of the β-subunit of cGMP phosphodiesterase cDNA to rd1 mice showed prolonged β-PDE expression and rescue of rod photoreceptor cells [84]. RT–PCR analysis from the injected retina indicated that transgene products were present for up to 18 weeks post-injection. Examination of outer nuclear thickness showed significant differences until 12 weeks post-injection [84].
Jomary et al. also evaluated the efficacy of a recombinant AAV vector for delivering and expressing the β-PDE gene in the retinas of rd1 mice [86]. Following intravitreal injection of AAV2-β-PDE, increased retinal expression of immunoreactive PDE protein was observed, including expression within photoreceptor cell bodies. Compared with the age-matched untreated controls, the treated eyes showed only a modest delay in photoreceptor degeneration. A major limitation of that study was the use of a low-titer traditional AAV2 vector in an animal model of rapid retinal degeneration. Since then, major progress has been made in vector purification methods and capsid engineering, resulting in high-titer AAVs of different serotypes with faster onset of expression and improved transduction efficiency [87-89]. Sub-retinal delivery of these optimized vectors in the rd10 mouse with a slower degeneration rate, as described below, has led to significant recovery of retinal function and morphological preservation by providing therapeutic levels of transgene expression before the photoreceptor cell loss.
Researchers also showed that lentiviral vectors based on human immunodeficiency virus (HIV) type 1 can achieve stable and efficient gene transfer into retinal cells [90,91]. Takahashi et al. tried the HIV1-based lentiviral vector to rescue retinal degeneration in rd1 mice [92]. Lentiviral vector containing Pde6b under the control of the cytomegalovirus promoter or rhodopsin promoter was injected into the sub-retinal space of rd1 eyes between P2 and P5. One to three rows of photoreceptor nuclei were observed in the eyes for at least 24 weeks post-injection. In summary, sub-retinal injection of adenoviral, adeno-associated viral, or lentiviral vectors encoding the Pde6b gene in neonatal rd1 mice resulted in partial preservation of photoreceptor structure although retinal function with ERG was never well restored.
The rd10 mouse is more amenable to successful gene replacement therapy than rd1, since the rd10 mouse displays a later onset and slower retinal degeneration process, thus providing a longer therapeutic window for intervention [38]. In 2008, Pang et al. reported a successful gene replacement therapy using AAV5 to rescue rd10 mice [93]. Since light exposure may speed up retinal degeneration, dark-reared P14 rd10 mice were injected sub-retinally with an AAV-5-smCBA-PDEß vector under dim light, resulting in prolonged photoreceptor survival and improved ERG and vision-guided performance for at least 3 weeks after sub-retinal injection. However, the therapeutic effects faded 6 weeks after treatment. Since photoreceptor degeneration starts around P16 in rd10 mice, and the AAV5-PDE6b vector takes at least 1 week to express enough functional protein following sub-retinal injection, treatment earlier than P9 in the rd10 mouse could lead to even more rescue. In reality, it is difficult to detach a significant fraction of the mouse retina following trans-cornea sub-retinal injection with minimal injection-related damage before the mouse eyes open around P14 [94]. Although trans-sclera sub-retinal injection can be used for neonatal mouse treatment, the injection transduces only a small part of the retina with extensive sub-retinal injection-related complications [95,96]. For example, in rd10 mice, AAV5-mediated rescue following P14 treatment [93] is more robust than that following P2 treatment [97] with a similar AAV5 vector when both cases are evaluated at P35. An optimal stable rescue could be related to the extent of retinal coverage by the vector and might be offset by injection-related damage to the neonatal mouse eye [98]. Yao et al. have shown that an AAV5-mediated delivery of the X-linked inhibitor of apoptosis at P4 can significantly slow photoreceptor degeneration in combination with gene-replacement therapy and may extend the window of treatment [99].
A tyrosine-capsid mutant AAV8 (Y733F) vector capable of transducing most of the photoreceptors within several days following sub-retinal injection represents a useful tool for treatment in animal models with early onset of retinal degeneration [100]. Consequently, a more significant rescue was achieved by using this capsid mutant serotype, compared to an AAV5 vector containing the same Pde6b gene [98]. Sub-retinal injection of the tyrosine-capsid mutant AAV8 (Y733F)-smCBA-PDE6b in the same dark-reared P14 rd10 mice led to restored retinal function and improved visual behavioral performance. Additionally, more than half of the photoreceptors were preserved for at least 6 months, as determined with ERG, optomotor behavioral tests, spectral domain optical coherence tomography (SD-OCT), and histology. Secondary retinal degeneration and remodeling in older rd10 mice were also prevented in rd10 eyes treated at P14, which lasted for at least 6 months [98]. Although the cause of the protective effect of dark-rearing on the progression of retinal degeneration is not completely understood, the effect may have implications for treating human patients with PDE-based mutations [40]. The long-term rescue effect in rd10 mice using the potent capsid mutant AAV8 vector provides a promising approach for gene therapy for patients with RP and paves the way for potential clinical trials in the future.
Mouse models of recessive RP have demonstrated that gene replacement therapy holds great promise for treating monogenic disorders [101-104]. Although both naturally occurring mouse models presented here mimic certain features of the clinical phenotype of recessive RP found in human patients with Pde6b mutations, the therapeutic findings may not fully extend to the human disease [105]. Mouse eyes are not similar to those of humans, due to the small size, low cone to rod ratio, and absence of a macula. The successful restoration of retinal function in mouse models of arRP, such as rd10, can be attributed in part to the ability to transduce the entire retina following a single sub-retinal delivery of a small volume of vector. We noticed the rescue effect fades within several months if the treated retinal area is less than 50% [98]. The widespread distribution of the vector following intraocular delivery in mouse models allows the majority of mutant photoreceptors to receive the missing functional gene. In contrast, transduction in human patients is restricted to a small area (10%–15%) of the retina following a single sub-retinal injection, and the remaining untreated photoreceptors may exert a negative effect on the neighboring cells during the course of degeneration. Consequently, one major goal of current gene therapy studies is to develop highly efficient and penetrating AAV vectors that can transduce photoreceptor cells over the entire retina following intravitreal delivery [100,106], and avoid the damage associated with sub-retinal surgical procedures, especially around the sensitive foveal region [107]. As demonstrated by experiments in animal models of recessive RP, gene therapy is most effective when treatment is initiated early, before the onset of photoreceptor cell death [108,109]. A recent study of human patients with RPE65-associated Leber congenital amaurosis (LCA2) has shown that although gene therapy led to substantial visual improvement, retinal degeneration continued to progress in the treated and untreated regions of the retina [110]. To better evaluate the treatment options for human patients with arRP caused by mutations in PDE6B, more studies are needed to explore the effects of therapeutic interventions in larger animal models at various stages of photoreceptor degeneration. Two naturally occurring canine models with recessive mutations in Pde6b have previously been described, the rcd1 Irish setter, which contains a nonsense amber mutation at codon 807 (W807ter) causing a truncation of the PDE β-subunit, and the Sloughi dog, caused by an 8 bp insertion after codon 816 [111,112]. A recent study identified a three base pair deletion in exon 21 in crd1 dogs, leading to partial loss of PDE6B function and a relatively slower rate in photoreceptor cell loss, similar to human patients [113]. These larger animal models more closely mimic the human eye and can provide more translational knowledge of disease progression and therapeutic interventions for arRP caused by Pde6b mutations.
Conclusions
This review focused on the pathogenesis of natural mouse models of recessive RP caused by Pde6b mutations, and the current available therapeutic options. Although some of the treatments discussed lead to partial protection of the morphology and function of photoreceptors, gene therapy is the only currently available technology that halts apoptosis and maintains long-term functional rescue, when treatment is provided before photoreceptor cell death is initiated. The rd10 mouse model of recessive RP, with later onset of retinal degeneration than rd1, provides an invaluable tool for studying the effects of various therapeutic interventions on slowing or preventing progressive photoreceptor loss caused by a deficiency in functional PDE6B.
A combination treatment would perhaps provide more benefits to human patients with different stages of retinal degeneration, by focusing on replacing the defective gene as well as extending the therapeutic window and the survival of remaining photoreceptor cells. Optogenetic tools may represent a potential strategy for treating patients in advanced stages of RP. Following more preclinical and clinical studies, gene replacement therapy using intravitreal injections of highly efficient and penetrating AAV vectors combined with either antioxidants, growth factors, or other pharmacological reagents, could become an important strategy for treating autosomal recessive RP in the future.
Acknowledgments
We acknowledge retinal gene therapy study grant from the National Natural Science Foundation of China (81371060), Wenzhou Medical University, Wenzhou, China (QTJ11018), a grant from Eye Hospital, School of Ophthalmology & Optometry, Wenzhou Medical University, Wenzhou, China (YNKT0507), a grant from MSTC: 2011ZX09301–001, an NIH grant R21EY023543, and a grant from Research to Prevent Blindness to UF. We also thank Shiyi S. Pang and J. Hugh McDowell (University of Florida) for assistance with English grammar.
References List
- 1.Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84:132–41. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrar GJ, Kenna PF, Humphries P. On the genetics of retinitis pigmentosa and on mutation-independent approaches to therapeutic intervention. EMBO J. 2002;21:857–64. doi: 10.1093/emboj/21.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sancho-Pelluz J, Arango-Gonzalez B, Kustermann S, Romero FJ. van VT, Zrenner E, EkstroP, Paquet-Durand F. Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol Neurobiol. 2008;38:253–69. doi: 10.1007/s12035-008-8045-9. [DOI] [PubMed] [Google Scholar]
- 4.Humphries P, Farrar GJ, Kenna P, McWilliam P. Retinitis pigmentosa: genetic mapping in X-linked and autosomal forms of the disease. Clin Genet. 1990;38:1–13. doi: 10.1111/j.1399-0004.1990.tb03541.x. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan J, Bonneau D, Frezal J, Munnich A, Dufier JL. Clinical and genetic heterogeneity in retinitis pigmentosa. Hum Genet. 1990;85:635–42. doi: 10.1007/BF00193589. [DOI] [PubMed] [Google Scholar]
- 6.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danciger M, Blaney J, Gao YQ, Zhao DY, Heckenlively JR, Jacobson SG, Farber DB. Mutations in the PDE6B gene in autosomal recessive retinitis pigmentosa. Genomics. 1995;30:1–7. doi: 10.1006/geno.1995.0001. [DOI] [PubMed] [Google Scholar]
- 8.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4:130–4. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA. 1995;92:3249–53. doi: 10.1073/pnas.92.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cote RH. Characteristics of photoreceptor PDE (PDE6): similarities and differences to PDE5. Int J Impot Res. 2004;16(Suppl 1):S28–33. doi: 10.1038/sj.ijir.3901212. [DOI] [PubMed] [Google Scholar]
- 11.Mou H, Cote RH. The catalytic and GAF domains of the rod cGMP phosphodiesterase (PDE6) heterodimer are regulated by distinct regions of its inhibitory gamma subunit. J Biol Chem. 2001;276:27527–34. doi: 10.1074/jbc.M103316200. [DOI] [PubMed] [Google Scholar]
- 12.Farber DB. From mice to men: the cyclic GMP phosphodiesterase gene in vision and disease. The Proctor Lecture. Invest Ophthalmol Vis Sci. 1995;36:263–75. [PubMed] [Google Scholar]
- 13.Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–80. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- 14.Farber DB, Lolley RN. Enzymic basis for cyclic GMP accumulation in degenerative photoreceptor cells of mouse retina. J Cyclic Nucleotide Res. 1976;2:139–48. [PubMed] [Google Scholar]
- 15.Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–51. doi: 10.1126/science.186.4162.449. [DOI] [PubMed] [Google Scholar]
- 16.Lolley RN, Farber DB, Rayborn ME, Hollyfield JG. Cyclic GMP accumulation causes degeneration of photoreceptor cells: simulation of an inherited disease. Science. 1977;196:664–6. doi: 10.1126/science.193183. [DOI] [PubMed] [Google Scholar]
- 17.Marigo V. Programmed cell death in retinal degeneration: targeting apoptosis in photoreceptors as potential therapy for retinal degeneration. Cell Cycle. 2007;6:652–5. doi: 10.4161/cc.6.6.4029. [DOI] [PubMed] [Google Scholar]
- 18.Baehr W, Frederick JM. Naturally occurring animal models with outer retina phenotypes. Vision Res. 2009;49:2636–52. doi: 10.1016/j.visres.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pennesi ME, Michaels KV, Magee SS, Maricle A, Davin SP, Garg AK, Gale MJ, Tu DC, Wen Y, Erker LR, Francis PJ. Long-term characterization of retinal degeneration in rd1 and rd10 mice using spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2012;53:4644–56. doi: 10.1167/iovs.12-9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–25. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- 21.Sidman RL, Green MC. Retinal degeneration in the mouse: location of the rd locus in linkage group xvii. J Hered. 1965;56:23–9. doi: 10.1093/oxfordjournals.jhered.a107364. [DOI] [PubMed] [Google Scholar]
- 22.Bowes C, Li T, Frankel WN, Danciger M, Coffin JM, Applebury ML, Farber DB. Localization of a retroviral element within the rd gene coding for the beta subunit of cGMP phosphodiesterase. Proc Natl Acad Sci USA. 1993;90:2955–9. doi: 10.1073/pnas.90.7.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pittler SJ, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci USA. 1991;88:8322–6. doi: 10.1073/pnas.88.19.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keeler CE. The Inheritance of a Retinal Abnormality in White Mice. Proc Natl Acad Sci USA. 1924;10:329–33. doi: 10.1073/pnas.10.7.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keeler CE. On the Occurrence in the House Mouse of Mendelizing Structural Defect of the Retina Producing Blindness. Proc Natl Acad Sci USA. 1926;12:255–8. doi: 10.1073/pnas.12.4.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keeler C. Retinal degeneration in the mouse is rodless retina. J Hered. 1966;57:47–50. doi: 10.1093/oxfordjournals.jhered.a107462. [DOI] [PubMed] [Google Scholar]
- 27.Bruckner R. Slit-lamp microscopy and ophthalmoscopy in rat and mouse. Doc Ophthalmol. 1951;5–6:452–554. doi: 10.1007/BF00143667. [DOI] [PubMed] [Google Scholar]
- 28.Pittler SJ, Keeler CE, Sidman RL, Baehr W. PCR analysis of DNA from 70-year-old sections of rodless retina demonstrates identity with the mouse rd defect. Proc Natl Acad Sci USA. 1993;90:9616–9. doi: 10.1073/pnas.90.20.9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978;17:489–98. [PubMed] [Google Scholar]
- 30.LaVail MM, Matthes MT, Yasumura D, Steinberg RH. Variability in rate of cone degeneration in the retinal degeneration (rd/rd) mouse. Exp Eye Res. 1997;65:45–50. doi: 10.1006/exer.1997.0308. [DOI] [PubMed] [Google Scholar]
- 31.Hackam AS, Strom R, Liu D, Qian J, Wang C, Otteson D, Gunatilaka T, Farkas RH, Chowers I, Kageyama M, Leveillard T, Sahel JA, Campochiaro PA, Parmigiani G, Zack DJ. Identification of gene expression changes associated with the progression of retinal degeneration in the rd1 mouse. Invest Ophthalmol Vis Sci. 2004;45:2929–42. doi: 10.1167/iovs.03-1184. [DOI] [PubMed] [Google Scholar]
- 32.Geller SF, Guerin KI, Visel M, Pham A, Lee ES, Dror AA, Avraham KB, Hayashi T, Ray CA, Reh TA, Bermingham-McDonogh O, Triffo WJ, Bao S, Isosomppi J, Vastinsalo H, Sankila EM, Flannery JG. CLRN1 is nonessential in the mouse retina but is required for cochlear hair cell development. PLoS Genet. 2009;5:e1000607. doi: 10.1371/journal.pgen.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao H, Tummala P, Guzman E, Mali RS, Gregorski J, Swaroop A, Mitton KP. The transcription factor neural retina leucine zipper (NRL) controls photoreceptor-specific expression of myocyte enhancer factor Mef2c from an alternative promoter. J Biol Chem. 2011;286:34893–902. doi: 10.1074/jbc.M111.271072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin B, Koizumi A, Tanaka N, Panda S, Masland RH. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci USA. 2008;105:16009–14. doi: 10.1073/pnas.0806114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucas RJ, Douglas RH, Foster RG. Characterization of an ocular photopigment capable of driving pupillary constriction in mice. Nat Neurosci. 2001;4:621–6. doi: 10.1038/88443. [DOI] [PubMed] [Google Scholar]
- 36.Lucas RJ. Mammalian inner retinal photoreception. Curr Biol. 2013;23:R125–33. doi: 10.1016/j.cub.2012.12.029. [DOI] [PubMed] [Google Scholar]
- 37.Semo M, Peirson S, Lupi D, Lucas RJ, Jeffery G, Foster RG. Melanopsin retinal ganglion cells and the maintenance of circadian and pupillary responses to light in aged rodless/coneless (rd/rd cl) mice. Eur J Neurosci. 2003;17:1793–801. doi: 10.1046/j.1460-9568.2003.02616.x. [DOI] [PubMed] [Google Scholar]
- 38.Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT, Nusinowitz S, Rengarajan K, Boyd AP, Sidney SS, Phillips MJ, Stewart RE, Chaudhury R, Nickerson JM, Heckenlively JR, Boatright JH. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 2007;47:624–33. doi: 10.1016/j.visres.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. J Comp Neurol. 2007;500:222–38. doi: 10.1002/cne.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cronin T, Lyubarsky A, Bennett J. Dark-rearing the rd10 mouse: implications for therapy. Adv Exp Med Biol. 2012;723:129–36. doi: 10.1007/978-1-4614-0631-0_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sahni JN, Angi M, Irigoyen C, Semeraro F, Romano MR, Parmeggiani F. Therapeutic challenges to retinitis pigmentosa: from neuroprotection to gene therapy. Curr Genomics. 2011;12:276–84. doi: 10.2174/138920211795860062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frasson M, Sahel JA, Fabre M, Simonutti M, Dreyfus H, Picaud S. Retinitis pigmentosa: rod photoreceptor rescue by a calcium-channel blocker in the rd mouse. Nat Med. 1999;5:1183–7. doi: 10.1038/13508. [DOI] [PubMed] [Google Scholar]
- 43.Doonan F, Donovan M, Cotter TG. Activation of multiple pathways during photoreceptor apoptosis in the rd mouse. Invest Ophthalmol Vis Sci. 2005;46:3530–8. doi: 10.1167/iovs.05-0248. [DOI] [PubMed] [Google Scholar]
- 44.Sanges D, Comitato A, Tammaro R, Marigo V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci USA. 2006;103:17366–71. doi: 10.1073/pnas.0606276103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takano Y, Ohguro H, Dezawa M, Ishikawa H, Yamazaki H, Ohguro I, Mamiya K, Metoki T, Ishikawa F, Nakazawa M. Study of drug effects of calcium channel blockers on retinal degeneration of rd mouse. Biochem Biophys Res Commun. 2004;313:1015–22. doi: 10.1016/j.bbrc.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 46.Barabas P, Cutler PC, Krizaj D. Do calcium channel blockers rescue dying photoreceptors in the Pde6b (rd1) mouse? Adv Exp Med Biol. 2010;664:491–9. doi: 10.1007/978-1-4419-1399-9_56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakazawa M. Effects of calcium ion, calpains, and calcium channel blockers on retinitis pigmentosa. J Ophthalmol. 2011;xx:292040. doi: 10.1155/2011/292040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pawlyk BS, Li T, Scimeca MS, Sandberg MA, Berson EL. Absence of photoreceptor rescue with D-cis-diltiazem in the rd mouse. Invest Ophthalmol Vis Sci. 2002;43:1912–5. [PubMed] [Google Scholar]
- 49.LaVail MM, Yasumura D, Matthes MT, Lau-Villacorta C, Unoki K, Sung CH, Steinberg RH. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602. [PubMed] [Google Scholar]
- 50.Wen R, Tao W, Li Y, Sieving PA. CNTF and retina. Prog Retin Eye Res. 2012;31:136–51. doi: 10.1016/j.preteyeres.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tao W, Wen R, Goddard MB, Sherman SD, O'Rourke PJ, Stabila PF, Bell WJ, Dean BJ, Kauper KA, Budz VA, Tsiaras WG, Acland GM, Pearce-Kelling S, Laties AM, Aguirre GD. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43:3292–8. [PubMed] [Google Scholar]
- 52.Cayouette M, Gravel C. Adenovirus-mediated gene transfer of ciliary neurotrophic factor can prevent photoreceptor degeneration in the retinal degeneration (rd) mouse. Hum Gene Ther. 1997;8:423–30. doi: 10.1089/hum.1997.8.4-423. [DOI] [PubMed] [Google Scholar]
- 53.Azadi S, Johnson LE, Paquet-Durand F, Perez MT, Zhang Y, Ekstrom PA, van Veen T. CNTF+BDNF treatment and neuroprotective pathways in the rd1 mouse retina. Brain Res. 2007;1129:116–29. doi: 10.1016/j.brainres.2006.10.031. [DOI] [PubMed] [Google Scholar]
- 54.Zeiss CJ, Allore HG, Towle V, Tao W. CNTF induces dose-dependent alterations in retinal morphology in normal and rcd-1 canine retina. Exp Eye Res. 2006;82:395–404. doi: 10.1016/j.exer.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 55.Frasson M, Picaud S, Leveillard T, Simonutti M, Mohand-Said S, Dreyfus H, Hicks D, Sabel J. Glial cell line-derived neurotrophic factor induces histologic and functional protection of rod photoreceptors in the rd/rd mouse. Invest Ophthalmol Vis Sci. 1999;40:2724–34. [PubMed] [Google Scholar]
- 56.Ohnaka M, Miki K, Gong YY, Stevens R, Iwase T, Hackett SF, Campochiaro PA. Long-term expression of glial cell line-derived neurotrophic factor slows, but does not stop retinal degeneration in a model of retinitis pigmentosa. J Neurochem. 2012;122:1047–53. doi: 10.1111/j.1471-4159.2012.07842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grimm C, Wenzel A, Groszer M, Mayser H, Seeliger M, Samardzija M, Bauer C, Gassmann M, Reme CE. HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nat Med. 2002;8:718–24. doi: 10.1038/nm723. [DOI] [PubMed] [Google Scholar]
- 58.Rex TS, Allocca M, Domenici L, Surace EM, Maguire AM, Lyubarsky A, Cellerino A, Bennett J, Auricchio A. Systemic but not intraocular Epo gene transfer protects the retina from light-and genetic-induced degeneration. Mol Ther. 2004;10:855–61. doi: 10.1016/j.ymthe.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 59.Sasahara M, Otani A, Oishi A, Kojima H, Yodoi Y, Kameda T, Nakamura H, Yoshimura N. Activation of bone marrow-derived microglia promotes photoreceptor survival in inherited retinal degeneration. Am J Pathol. 2008;172:1693–703. doi: 10.2353/ajpath.2008.080024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barone I, Novelli E, Piano I, Gargini C, Strettoi E. Environmental enrichment extends photoreceptor survival and visual function in a mouse model of retinitis pigmentosa. PLoS ONE. 2012;7:e50726. doi: 10.1371/journal.pone.0050726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Solá S, Castro RE, Laires PA, Steer CJ, Rodrigues CM. Tauroursodeoxycholic acid prevents amyloid-beta peptide-induced neuronal death via a phosphatidylinositol 3-kinase-dependent signaling pathway. Mol Med. 2003;9:226–34. doi: 10.2119/2003-00042.rodrigues. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boatright JH, Moring AG, McElroy C, Phillips MJ, Do VT, Chang B, Hawes NL, Boyd AP, Sidney SS, Stewart RE, Minear SC, Chaudhury R, Ciavatta VT, Rodrigues CM, Steer CJ, Nickerson JM, Pardue MT. Tool from ancient pharmacopoeia prevents vision loss. Mol Vis. 2006;12:1706–14. [PubMed] [Google Scholar]
- 63.Drack AV, Dumitrescu AV, Bhattarai S, Gratie D, Stone EM, Mullins R, Sheffield VC. TUDCA slows retinal degeneration in two different mouse models of retinitis pigmentosa and prevents obesity in Bardet-Biedl syndrome type 1 mice. Invest Ophthalmol Vis Sci. 2012;53:100–6. doi: 10.1167/iovs.11-8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oveson BC, Iwase T, Hackett SF, Lee SY, Usui S, Sedlak TW, Snyder SH, Campochiaro PA, Sung JU. Constituents of bile, bilirubin and TUDCA, protect against oxidative stress-induced retinal degeneration. J Neurochem. 2011;116:144–53. doi: 10.1111/j.1471-4159.2010.07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Phillips MJ, Walker TA, Choi HY, Faulkner AE, Kim MK, Sidney SS, Boyd AP, Nickerson JM, Boatright JH, Pardue MT. Tauroursodeoxycholic acid preservation of photoreceptor structure and function in the rd10 mouse through postnatal day 30. Invest Ophthalmol Vis Sci. 2008;49:2148–55. doi: 10.1167/iovs.07-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Komeima K, Rogers BS, Lu L, Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2006;103:11300–5. doi: 10.1073/pnas.0604056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Komeima K, Rogers BS, Campochiaro PA. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J Cell Physiol. 2007;213:809–15. doi: 10.1002/jcp.21152. [DOI] [PubMed] [Google Scholar]
- 68.Sanz MM, Johnson LE, Ahuja S, Ekstrom PA, Romero J, van Veen T. Significant photoreceptor rescue by treatment with a combination of antioxidants in an animal model for retinal degeneration. Neuroscience. 2007;145:1120–9. doi: 10.1016/j.neuroscience.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 69.Busskamp V, Picaud S, Sahel JA, Roska B. Optogenetic therapy for retinitis pigmentosa. Gene Ther. 2012;19:169–75. doi: 10.1038/gt.2011.155. [DOI] [PubMed] [Google Scholar]
- 70.Rowland TJ, Buchholz DE, Clegg DO. Pluripotent human stem cells for the treatment of retinal disease. J Cell Physiol. 2012;227:457–66. doi: 10.1002/jcp.22814. [DOI] [PubMed] [Google Scholar]
- 71.Huang Y, Enzmann V, Ildstad ST. Stem cell-based therapeutic applications in retinal degenerative diseases. Stem Cell Rev. 2011;7:434–45. doi: 10.1007/s12015-010-9192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ramsden CM, Powner MB, Carr AJ, Smart MJ. da CL, Coffey PJ. Stem cells in retinal regeneration: past, present and future. Development. 2013;140:2576–85. doi: 10.1242/dev.092270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Asahara T. Cell therapy and gene therapy using endothelial progenitor cells for vascular regeneration. Handb Exp Pharmacol 2007:181–194. [DOI] [PubMed] [Google Scholar]
- 74.Otani A, Dorrell MI, Kinder K, Moreno SK, Nusinowitz S, Banin E, Heckenlively J, Friedlander M. Rescue of retinal degeneration by intravitreally injected adult bone marrow-derived lineage-negative hematopoietic stem cells. J Clin Invest. 2004;114:765–74. doi: 10.1172/JCI21686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barber AC, Hippert C, Duran Y, West EL, Bainbridge JW, Warre-Cornish K, Luhmann UF, Lakowski J, Sowden JC, Ali RR, Pearson RA. Repair of the degenerate retina by photoreceptor transplantation. Proc Natl Acad Sci USA. 2013;110:354–9. doi: 10.1073/pnas.1212677110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh MS, Charbel IP, Butler R, Martin C, Lipinski DM, Sekaran S, Barnard AR, Maclaren RE. Reversal of end-stage retinal degeneration and restoration of visual function by photoreceptor transplantation. Proc Natl Acad Sci USA. 2013;110:1101–6. doi: 10.1073/pnas.1119416110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cronin T, Bennett J. Switching on the lights: the use of optogenetics to advance retinal gene therapy. Mol Ther. 2011;19:1190–2. doi: 10.1038/mt.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y, Ivanova E, Bi A, Pan ZH. Ectopic expression of multiple microbial rhodopsins restores ON and OFF light responses in retinas with photoreceptor degeneration. J Neurosci. 2009;29:9186–96. doi: 10.1523/JNEUROSCI.0184-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bi A, Cui J, Ma YP, Olshevskaya E, Pu M, Dizhoor AM, Pan ZH. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron. 2006;50:23–33. doi: 10.1016/j.neuron.2006.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doroudchi MM, Greenberg KP, Liu J, Silka KA, Boyden ES, Lockridge JA, Arman AC, Janani R, Boye SE, Boye SL, Gordon GM, Matteo BC, Sampath AP, Hauswirth WW, Horsager A. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther. 2011;19:1220–9. doi: 10.1038/mt.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lagali PS, Balya D, Awatramani GB, Munch TA, Kim DS, Busskamp V, Cepko CL, Roska B. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci. 2008;11:667–75. doi: 10.1038/nn.2117. [DOI] [PubMed] [Google Scholar]
- 82.Caporale N, Kolstad KD, Lee T, Tochitsky I, Dalkara D, Trauner D, Kramer R, Dan Y, Isacoff EY, Flannery JG. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011;19:1212–9. doi: 10.1038/mt.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bennett J, Tanabe T, Sun D, Zeng Y, Kjeldbye H, Gouras P, Maguire AM. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat Med. 1996;2:649–54. doi: 10.1038/nm0696-649. [DOI] [PubMed] [Google Scholar]
- 84.Kumar-Singh R, Farber DB. Encapsidated adenovirus mini-chromosome-mediated delivery of genes to the retina: application to the rescue of photoreceptor degeneration. Hum Mol Genet. 1998;7:1893–900. doi: 10.1093/hmg/7.12.1893. [DOI] [PubMed] [Google Scholar]
- 85.Kumar-Singh R, Yamashita CK, Tran K, Farber DB. Construction of encapsidated (gutted) adenovirus minichromosomes and their application to rescue of photoreceptor degeneration. Methods Enzymol. 2000;316:724–43. doi: 10.1016/s0076-6879(00)16759-x. [DOI] [PubMed] [Google Scholar]
- 86.Jomary C, Vincent KA, Grist J, Neal MJ, Jones SE. Rescue of photoreceptor function by AAV-mediated gene transfer in a mouse model of inherited retinal degeneration. Gene Ther. 1997;4:683–90. doi: 10.1038/sj.gt.3300440. [DOI] [PubMed] [Google Scholar]
- 87.Binny CJ, Nathwani AC. Vector systems for prenatal gene therapy: principles of adeno-associated virus vector design and production. Methods Mol Biol. 2012;891:109–31. doi: 10.1007/978-1-61779-873-3_6. [DOI] [PubMed] [Google Scholar]
- 88.Davidoff AM, Ng CY, Sleep S, Gray J, Azam S, Zhao Y, McIntosh JH, Karimipoor M, Nathwani AC. Purification of recombinant adeno-associated virus type 8 vectors by ion exchange chromatography generates clinical grade vector stock. J Virol Methods. 2004;121:209–15. doi: 10.1016/j.jviromet.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 89.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–28. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 90.Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci USA. 1997;94:10319–23. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takahashi M. Delivery of genes to the eye using lentiviral vectors. Methods Mol Biol. 2004;246:439–49. doi: 10.1385/1-59259-650-9:439. [DOI] [PubMed] [Google Scholar]
- 92.Takahashi M, Miyoshi H, Verma IM, Gage FH. Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J Virol. 1999;73:7812–6. doi: 10.1128/jvi.73.9.7812-7816.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pang JJ, Boye SL, Kumar A, Dinculescu A, Deng W, Li J, Li Q, Rani A, Foster TC, Chang B, Hawes NL, Boatright JH, Hauswirth WW. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation. Invest Ophthalmol Vis Sci. 2008;49:4278–83. doi: 10.1167/iovs.07-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pang JJ, Lauramore A, Deng WT, Li Q, Doyle TJ, Chiodo V, Li J, Hauswirth WW. Comparative analysis of in vivo and in vitro AAV vector transduction in the neonatal mouse retina: effects of serotype and site of administration. Vision Res. 2008;48:377–85. doi: 10.1016/j.visres.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 95.Kong F, Li W, Li X, Zheng Q, Dai X, Zhou X, Boye SL, Hauswirth WW, Qu J, Pang JJ. Self-complementary AAV5 vector facilitates quicker transgene expression in photoreceptor and retinal pigment epithelial cells of normal mouse. Exp Eye Res. 2010;90:546–54. doi: 10.1016/j.exer.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pang J, Cheng M, Haire SE, Barker E, Planelles V, Blanks JC. Efficiency of lentiviral transduction during development in normal and rd mice. Mol Vis. 2006;12:756–67. [PubMed] [Google Scholar]
- 97.Allocca M, Manfredi A, Iodice C, Di VU, Auricchio A. AAV-mediated gene replacement, either alone or in combination with physical and pharmacological agents, results in partial and transient protection from photoreceptor degeneration associated with betaPDE deficiency. Invest Ophthalmol Vis Sci. 2011;52:5713–9. doi: 10.1167/iovs.10-6269. [DOI] [PubMed] [Google Scholar]
- 98.Pang JJ, Dai X, Boye SE, Barone I, Boye SL, Mao S, Everhart D, Dinculescu A, Liu L, Umino Y, Lei B, Chang B, Barlow R, Strettoi E, Hauswirth WW. Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol Ther. 2011;19:234–42. doi: 10.1038/mt.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yao J, Jia L, Khan N, Zheng QD, Moncrief A, Hauswirth WW, Thompson DA, Zacks DN. Caspase inhibition with XIAP as an adjunct to AAV vector gene-replacement therapy: improving efficacy and prolonging the treatment window. PLoS ONE. 2012;7:e37197. doi: 10.1371/journal.pone.0037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Petrs-Silva H, Dinculescu A, Li Q, Min SH, Chiodo V, Pang JJ, Zhong L, Zolotukhin S, Srivastava A, Lewin AS, Hauswirth WW. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther. 2009;17:463–71. doi: 10.1038/mt.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chang B, Hurd R, Wang J, Nishina P. Survey of common eye diseases in laboratory mouse strains. Invest Ophthalmol Vis Sci. 2013;54:4974–81. doi: 10.1167/iovs.13-12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fletcher EL, Jobling AI, Vessey KA, Luu C, Guymer RH, Baird PN. Animal models of retinal disease. Prog Mol Biol Transl Sci. 2011;100:211–86. doi: 10.1016/B978-0-12-384878-9.00006-6. [DOI] [PubMed] [Google Scholar]
- 103.Sahel JA, Roska B. Gene therapy for blindness. Annu Rev Neurosci. 2013;36:467–88. doi: 10.1146/annurev-neuro-062012-170304. [DOI] [PubMed] [Google Scholar]
- 104.Smith AJ, Bainbridge JW, Ali RR. Gene supplementation therapy for recessive forms of inherited retinal dystrophies. Gene Ther. 2012;19:154–61. doi: 10.1038/gt.2011.161. [DOI] [PubMed] [Google Scholar]
- 105.Buch PK, Bainbridge JW, Ali RR. AAV-mediated gene therapy for retinal disorders: from mouse to man. Gene Ther. 2008;15:849–57. doi: 10.1038/gt.2008.66. [DOI] [PubMed] [Google Scholar]
- 106.Petrs-Silva H, Dinculescu A, Li Q, Deng WT, Pang JJ, Min SH, Chiodo V, Neeley AW, Govindasamy L, Bennett A, Agbandje-McKenna M, Zhong L, Li B, Jayandharan GR, Srivastava A, Lewin AS, Hauswirth WW. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther. 2011;19:293–301. doi: 10.1038/mt.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ, Calcedo R, Pang JJ, Erger KE, Olivares MB, Mullins CL, Swider M, Kaushal S, Feuer WJ, Iannaccone A, Fishman GA, Stone EM, Byrne BJ, Hauswirth WW. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Colella P, Auricchio A. Gene therapy of inherited retinopathies: a long and successful road from viral vectors to patients. Hum Gene Ther. 2012;23:796–807. doi: 10.1089/hum.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rolling F. Recombinant AAV-mediated gene transfer to the retina: gene therapy perspectives. Gene Ther. 2004;11(Suppl 1):S26–32. doi: 10.1038/sj.gt.3302366. [DOI] [PubMed] [Google Scholar]
- 110.Cideciyan AV, Jacobson SG, Beltran WA, Sumaroka A, Swider M, Iwabe S, Roman AJ, Olivares MB, Schwartz SB, Komaromy AM, Hauswirth WW, Aguirre GD. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA. 2013;110:E517–25. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dekomien G, Runte M, Godde R, Epplen JT. Generalized progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene. Cytogenet Cell Genet. 2000;90:261–7. doi: 10.1159/000056785. [DOI] [PubMed] [Google Scholar]
- 112.Suber ML, Pittler SJ, Qin N, Wright GC, Holcombe V, Lee RH, Craft CM, Lolley RN, Baehr W, Hurwitz RL. Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase beta-subunit gene. Proc Natl Acad Sci USA. 1993;90:3968–72. doi: 10.1073/pnas.90.9.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goldstein O, Mezey JG, Schweitzer PA, Boyko AR, Gao C, Bustamante CD, Jordan JA, Aguirre GD, Acland GM. IQCB1 and PDE6B mutations cause similar early onset retinal degenerations in two closely related terrier dog breeds. Invest Ophthalmol Vis Sci. 2013;54:7005–19. doi: 10.1167/iovs.13-12915. [DOI] [PMC free article] [PubMed] [Google Scholar]