

Abstract
High-throughput transcriptomic experiments have made it possible to classify genes that are ubiquitously expressed as housekeeping (HK) genes and those expressed only in selective tissues as tissue-specific (TS) genes. Although partitioning a transcriptome into HK and TS genes is conceptually problematic owing to the lack of precise definitions and gene expression profile criteria for the two, information whether a gene is an HK or a TS gene can provide an initial clue to its cellular and/or functional role. Consequently, the development of new and novel HK (TS) classification methods has been a topic of considerable interest in post-genomics research. Here, we report such a development. Our method, called HKera, differs from the others by utilizing a novel property of HK genes that we have previously uncovered, namely that the ranking order of their expression levels, as opposed to the expression levels themselves, tends to be preserved from one tissue to another. Evaluated against multiple benchmark sets of human HK genes, including one recently derived from second generation sequencing data, HKera was shown to perform significantly better than five other classifiers that use different methodologies. An enrichment analysis of pathway and gene ontology annotations showed that HKera-predicted HK and TS genes have distinct functional roles and, together, cover most of the ontology categories. These results show that HKera is a good transcriptome partitioner that can be used to search for, and obtain useful expression and functional information for, novel HK (TS) genes.
Introduction
Transcriptomics, which investigates patterns of gene expression across different tissues and different experimental conditions on a genome-wide scale, is a key component of post-genomics research. Genes that are ubiquitously expressed over a wide range of tissues and experimental conditions are usually called housekeeping (HK) genes, while those that are not are called tissue-specific (TS) or tissue-selective genes [1], [2]. To study a complex transcriptome, such as that of the human genome, it is often useful to determine which genes of the genome are HK genes and which are TS genes in order to understand their roles in cellular functions or disease processes [3]–[5]. Many bioinformatics tools have been developed for this purpose (e.g., [6]–[9]), although classification as HK and TS genes is not unambiguous, as it depends on the classification criteria and methodologies used [10].
At least six different methodologies have been used to partition a transcriptome into HK and TS genes, namely those that classify the genes based on the 1) magnitude of expression (Exp), 2) number of present calls of expression (PCall), 3) fraction present weighted expression intensity (FPEI), 4) tissue specificity index (TSI), 5) biophysical properties (Phy), or 6) Fourier analysis of expression data obtained at different time points in the cell cycle (Fourier analysis). Exp identifies genes as HK genes based on the criterion of high [2] or fairly constant [11], [12] expression, whereas PCall does not focus on the magnitude of expression, but, instead, uses a certain number of “present calls” as a threshold [13], [14], and FPEI [15] is a combination of the two. In contrast, TSI [16] uses a quantitative measure of variation in expression profiles in different tissues to evaluate the tendency of a gene to be HK (little tissue-wide variation) or TS (high variation). Different from all of the above, Phy [17], [18] ignores the expression data completely and uses the observations that, compared to TS genes, HK genes tend to be shorter [2], be flanked with more short repeats [17], have fewer protein domains [19], show lower promoter sequence conservation [20], and have simpler transcriptional regulation and slower rates of evolution [21], [22] to distinguish between HK and TS genes. Finally, Fourier analysis [23] transforms time-series gene expression data into Fourier spectra for a support vector machine (SVM, a machine learning method) to classify genes as HK or TS.
Despite their proven usefulness in numerous studies, all of these methods have shortcomings. For instance, Exp, PCall, FPEI, and TSI all tend to identify HK genes that are expressed at a high and/or fairly constant level and therefore may miss those expressed at a low level or at significantly different levels in different tissues [14], [24], [25]. As for Phy, despite the appeal of not using expression data, there have been conflicting results for the properties of HK genes (e.g. whether the gene structures of HK genes are compact [2], [26] or not [27]), which is not surprising, since there is a significant overlap in static gene properties between HK and non-HK genes [28]. Finally, the main limitation of the more sophisticated Fourier analysis is its use of time-series expression data and the resultant higher cost.
We have previously shown that the ranking order of expression levels for HK genes tends to be preserved from one tissue to another and that dispersion, stableness, and co-expression are the three factors making the greatest contribution to this novel property of HK genes that can be decomposed into a composite of 16 tensor components [28]. Here, we describe the development of an SVM classifier for designating a given human gene as an HK or TS gene based on the tensor structure of tissue-wide gene expression profiles. We have named this classifier HKera, ‘era’ being an abbreviation for ‘expression ranking assessment’. HKera is similar to Fourier analysis in that they both utilize a mathematical transformation of an underlying structure of gene expression data, but HKera does not require time-series data.
To evaluate the performance of an HK gene classifier, a so-called ‘gold-standard’ set of HK genes is required, and several such sets have been derived and used as the benchmark to evaluate HK (TS) gene classifiers [2], [6], [10], [14]. In this work, HKera and five other HT (TS) prediction methods were evaluated using three widely-used HK gene sets and one very large HK set derived recently from RANseq experiments as benchmark. The results showed that HKera performed significantly better than the five other methods evaluated (a comparison with Fourier analysis was not made because we did not use time-series data). Furthermore, an analysis using the functional annotations of the Kyoto Encyclopedia of Genes and Genomics (KEGG) [29], Protein Information Resource (PIR) [30], and Gene Ontology (GO) [31] revealed that functional categories enriched in HK genes are distinct from those enriched in TS genes, supporting the notion that, by and large, the two have distinct functional roles in the cell.
Materials and Methods
Datasets
The GSE2361 Affymetrix microarray data for human genes compiled by Ge et al. [32] was downloaded from GEO depositories [33] and processed using previously described procedures [28]. This GSE2361 dataset contains gene expression profiles for 13,075 genes in 36 normal human tissues. As previously [28], this dataset was divided into three gene sets, namely HK, TS, and MR (“middle-ranged”), the HK set consisting of a set of 388 genes present in the 408 HK gene set manually curated by Zhu et al. [10], the TS set consisting of 734 genes that satisfy four stringent criteria for being TS genes [28], [32], and the MR set consisting of the remaining 11,953 genes. Three hundred HK genes and 300 TS genes were then randomly selected from the HK or TS set to train and test the SVM models of HKera described below.
We treated the set of 388 HK genes curated by Zhu et al. [10] and two other HK sets derived from microarray data [2], [14] as ‘gold-standard’ HK genes, since they have been used as such in the past (e.g. [15], [18], [28]). The HK set identified by Ramskold et al. [6] was also treated as gold-standard, since it was derived from RNAseq experiments, which can detect expression signals more comprehensively and at a higher resolution than conventional microarray experiments [34]. These four HK gene sets were denoted, respectively, as HK388, HK383, HK557, and HK6121, the subscript indicating the size (number of genes with expression data in GSE2361) of the set, while the set of 734 TS genes used to train/test HKera was denoted as TS734.
Development of HKera
HKera is an SVM model from a five-fold cross validation, in which the 300 HK (and 300 TS) genes randomly selected from the HK388 and TS734 sets were arbitrarily partitioned into five subgroups of equal size, each of which was alternately used for testing, while the remaining four subgroups were used for training (Figure 1A). This resulted in five models and, although fairly similar results were obtained for all five (see Results), for simplicity, the one with the best numerical performance was chosen as the HKera classifier.
Figure 1. Developing the HKera classifier.

(A) The HKera classifier is one of five SVM models resulting from 5-fold cross validation of SVM learning on 300 ‘gold-standard’ HK genes and 300 ‘gold-standard’ TS genes (see Methods). (B) To create the attributes for the SVM learning, each query gene used for training and test (Qi) was paired with each of 50 reference genes (Rj) (see Methods) to compute the Kendall’s  of their tissue-wide gene expression profile, which was then decomposed into 16 tensor components (A1, A2, …, A16) following our previously described procedure [28]. The mean of each tensor component averaged over a set of query genes (e.g. HK1, TS1, etc. in (A)) provided one of the16 attributes used to train/test the SVM models.
of their tissue-wide gene expression profile, which was then decomposed into 16 tensor components (A1, A2, …, A16) following our previously described procedure [28]. The mean of each tensor component averaged over a set of query genes (e.g. HK1, TS1, etc. in (A)) provided one of the16 attributes used to train/test the SVM models.
The attributes used to train the SVM models were the 16 components (Figure 1B) derived by tensor decomposition of Kendall’s τ, a measure of tissue-wide concordance in the ranking order of expression levels between any two genes [28]. The meanings of these 16 attributes are schematically illustrated and explained in supplemental Figure S1 in File S1, along with an example of actual data for a specific gene pair in Figure S2 in File S1. Since this method requires data for gene pairs, it was necessary to have a set of reference genes with which to pair any query gene (training or testing). In principle, any gene can serve as a reference gene. Indeed, similar performances were obtained when three very different reference gene sets were used (see supplemental Table S1 in File S1). These three reference gene sets contained, respectively, 50 HK genes, 50 TS genes, or 25 HK and 25 TS genes randomly selected from the HK388 and TS734 sets described above after excluding those already selected to be included in the training and test sets. To demonstrate it was not necessary to use only HK genes as reference genes to derive HKera, in this study, we report only the results of the HKera built using the reference set of 50 TS genes. We employed the bioinformatics toolbox of the Matlab software (version 7.6.0.324, release R2008a) [35] available at http://www.mathworks.com, particularly its “svmdecision” command, to build the HKera classifier. Using HKera, every gene in the training set received a score from −1 to +1, indicating the extent of its tendency to be an HK gene (a more positive score) or a TS gene (a more negative score), but some test genes may receive a score slightly beyond the −1 or the +1 limit. In this work, those with a positive score were regarded as HK genes and those with a negative score as TS genes. Using a threshold of zero, the GSE2361 data set (13,075 genes) was partitioned into 8,072 HK genes (61.7%) and 5,003 TS genes (38.3%).
Performance Evaluation
To evaluate HKera and other HK prediction methods, we calculated the following performance measures:
| (1) |
| (2) |
| (3) |
| (4) |
where TP denotes true positive, FP false positive, TN true negative, and FN false negative. When evaluating the five-fold cross validation, TP was the number of correctly predicted HK genes for the 300 genes chosen from the HK388 set to be included in the training and testing sets and TN was the number of correctly predicted TS genes for the 300 genes chosen from the TS734 set; when computing the receiver operating characteristic (ROC) curves [36] for HKera and the other methods to compare their performance, TP was the number of correctly predicted HK genes in each of the four benchmark HK sets, i.e. HK388 [10], HK383 [14], HK557 [10], and HK6121 [6], and TN was the number of correctly predicted TS genes in the TS734 set. For both computations, FP was the number of TS734 genes predicted to be HK and FN the number of benchmark HK genes predicted to be TS. A few genes present in both the TS734 set and the benchmark HK set (3 using HK383, 5 using HK557, and 103 using HK6121) were excluded from the computation of the performance measures. The criteria used to define HK genes in the GSE2361 dataset using the various methods compared in this study are listed in Table 1.
Table 1. HK criterion and the resulting number of HK genes in the GSE2361 set using different methods.
| Method | HK criterion | Number ofHK genes | Reference |
| Exp |
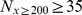 a
a
|
1,114 | [2] |
| PCall |
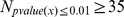 b
b
|
1,685 | [13], [14] |
| FPEI |
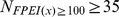 c
c
|
2,064 | [15] |
| TSI |
TSI 0.1d 0.1d
|
1,039 | [16] |
| Phy |
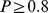 e
e
|
1,219 | [18] |
| RNAseq |
 f
f
|
6,121 | [6] |
| HKera |
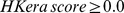
|
7,761 | This work |
Any gene is an HK gene if it has an expression intensity (x) > = 200, as recommended by [2], in at least 35 tissues. N is the number of tissues.
The p value for gene expression intensity needs to be less than 0.01 to make a detection call of ‘Present’ [13], and a gene needs to have a ‘Present’ call in at least 35 tissues to be considered an HK gene. N is the number of tissues.
Following [15], genes with an FPEI score above 100 in at least 35 tissues were defined as HK genes. N is the number of tissues.
TSI [16] is bounded between 0 and 1. A lower TSI indicates a lower tendency for the gene to be TS (or a higher tendency for it to be HK). The 0.1 threshold was chosen following [16].
For each gene, the Näive Bayes classifier [18] calculates a probability (P) of it being an HK gene; in this study, we choose those with a P value greater than 0.8 to be classified as HK genes.
According to [6], those genes with an RPKM (reads per kilobase of exon model per million mapped reads) score greater than 0.3 were classified as HK genes.
Functional Annotation and Enrichment Analysis
About two-thirds of the MR genes were scored as positive by HKera, yielding thousands of predicted HK genes. To investigate what functional roles of these genes might differ from those of the genes scored negatively and thus classified as TS genes, genes of the MR set were sorted by their HKera score and divided into four sets of putative HK genes and two sets of putative TS genes, each containing about 2,000 genes. The choice of 2,000 as a cut-off was arbitrary, but was based on the consideration that a choice of 1,000 would result in each group having too few members for enrichment analysis (data not shown). We employed the DAVID Bioinformatics Resource [37], [38] to compute the p value for genes in each set and for genes in the HK388 set and the TS734 set to be associated with a specific pathway category of KEGG [29] and the p value for the likelihood of their being ubiquitous according to PIR [30]. We then extended the enrichment analysis to GO categories [31], in which comparisons were made with gene sets derived from FPEI [15] and RNAseq experiments [6].
Results
Performance of SVM Models
Table 2 summarizes the performance for each of the five SVM models resulting from five-fold cross validation on training/test data of randomly selected HK and TS genes (see Methods). The three measures (accuracy, recall, and precision) were nearly all >99% perfect for training and remained very good (the worst being 87% recall for one model) for the test. We chose model 3 to be HKera because it gave the best test result for all three performance measures. Note that although the 16 attributes used by HKera were derived from ranking order data [28], they themselves are not ordinal data and therefore machine learning methods for ordinal classifications [39]–[41] may not be easily applied, not to mention that, to our knowledge, these methods tend to classify data into ordinal classes (i.e. the output end of the learning) but do not classify data with ordinal features (i.e. the input end). When five other machine leaning methods (decision tree, neural network, rule learner, naïve Bayes, and instance-based learning algorithm) representing different categories of classification methodologies [42] were used instead of SVM, the results showed that HKera (i.e. SVM) was among the best performers (Figure S3 in File S1).
Table 2. Performance of HKera’s SVM models derived from 5-fold cross validation on training/test data.
| Training (%) | Test (%) | |||||
| Model | Accuracy | Recall | Precision | Accuracy | Recall | Precision |
| 1 | 99.2 | 98.8 | 99.6 | 92.0 | 87.7 | 96.1 |
| 2 | 99.2 | 99.0 | 99.4 | 92.3 | 89.7 | 94.9 |
| 3 * | 99.3 | 98.7 | 99.9 | 95.2 | 93.3 | 96.9 |
| 4 | 99.5 | 99.3 | 99.8 | 91.7 | 87.3 | 95.8 |
| 5 | 99.0 | 98.2 | 99.7 | 93.5 | 90.0 | 96.9 |
Model 3 was chosen to represent HKera in this study.
Comparisons with Other Methods using Different Benchmark HK Sets
The abilities of HKera and five other methods (PCall, Exp, FPEI, Phy, and TSI) to identify HK genes from the GSE2361 dataset were compared using the ROC evaluation of four benchmark HK sets (HK388 [10], HK383 [14], HK557 [10], and HK6121 [6]). ROC is a measure of sensitivity (i.e. recall ability) as a function of specificity, in which a larger area under the ROC curve (conventionally known as the AUC) indicates a better performance. As shown in Figure 2, when the sensitivity, or the percentage of recall, was not required to be high, most of the HK genes recalled were correct (i.e. specificity high, or 1-specificity close to zero) for all methods, but when greater sensitivity was required, differences in performance between the methods became apparent. With all sets, HKera exhibited the best performance, producing not only an almost perfect ROC curve for HK388 on which the classifier was trained (see Methods), but also an excellent ROC curve for the other three benchmark sets. Given its simplicity, TSI, which, like HKera, employs a mathematical transformation, albeit a much simpler one [16], performed surprisingly well. In contrast, PCall, Exp, and FPEI all exhibited an unbalanced performance, being accurate at low sensitivity, but bad at high sensitivity, except when using the HK383 set. Compared to PCall, Exp, and FPEI, Phy had a prediction accuracy that was relatively insensitive to the HK set used for evaluation, but a much smaller AUC, presumably owing to its use of static gene properties and not expression data, as mentioned above.
Figure 2. Performance of six HK prediction methods on four benchmark HK sets.

The plots are ROC curves of sensitivity vs. 1-specificity, where sensitivity (i.e. recall) and specificity are defined, respectively, by Equation (1) and Equation (3) in the Methods. The six HK prediction methods compared are PCall [13], [14], Exp [2], TSI [16], Phy [18], FPEI [15], and HKera (this work), and the four benchmark HK sets are (A) HK388 [10], (B) HK383 [14], (C) HK557 [2], and (D) HK6121 (RANseq) [6].
HKera Scores and Coverage of Benchmark HK Sets
Figure 3 shows the distribution of HKera scores using various benchmark gene sets and the MR set, which, together with HK388 and TS734, contain the entire 13,075 genes of the GSE2361 set. Overlaps of the HKera scores were seen among the various gene sets, including between the HK388 set, the TS734 set, and the MR set (11,953 genes), reinforcing the notion that HK and TS genes are largely distinguished based on qualitative descriptions and different quantitative measures will yield different HK/TS genes. Nevertheless, numerous MR genes appeared to have expression characteristics similar to those of expert-curated HK genes, as suggested by their similar high HKera scores, and can therefore be considered as HK genes in the subsequent functional analysis. Although a more positive HKera score indicates a higher tendency of having canonical HK expression characteristics, 0.0 was chosen as the threshold to partition the human transcriptome (Figure 3), because it produced a balanced cross-coverage between HKera-predicted HK genes and those determined from RNAseq data (Figure 4). An Excel file containing a complete listing of the 13,075 GSE2361 human genes ordered by HKera score is provided as a supplement (Table S2 in File S2).
Figure 3. Distribution of HKera scores for various gene sets.

Using the HKera classifier, every gene in each gene set was scored by a numerical value that indicated its tendency to be HK (more positive) or TS (more negative). Note that the entire GSE2361 set was scored, as it was divided into the three sets HK388, TS734, and MR (see Methods). In this work, 0.0 was the threshold used to divide the MR set into HK genes and TS genes.
Figure 4. Cross-coverage between HKera and benchmark HK sets at various thresholds of the HKera score.

At each score threshold, genes with a score greater than the threshold were classified as HK genes, while the same number of genes was randomly selected from the whole pool (GSE2361) to form a random set. The percentages of HKera-predicted HK genes (solid symbols) and randomly selected genes (empty symbols) that were present in the benchmark set considered were computed at various thresholds of the HKera score (top panel); the percentage of the reverse coverage was also computed (bottom panel).
Enriched KEGG Pathways and GO Categories
Of the 13,075 genes for which expression data is given in GSE2361 that were analyzed in this work, only 3,729 (28.5%) had pathway information in KEGG and only 4,190 (32.0%) had tissue specificity data in PIR. For those genes with available pathway or tissue specificity data, those annotated with different KEGG pathways and PIR tissue specificity categories that were enriched (p<0.05) in the HK (TS) genes grouped according to HKera scores (see Methods) are presented in Table 3. Consistent with the notion that HK genes are expressed in a wide range of tissues, while TS genes are not, genes annotated with the PIR categories of “ubiquitous” and related terms were enriched only in the HK groups, while those annotated with tissue-specific expression (e.g. for liver and testis) were enriched only in the TS groups. In addition, apart from the seven pathways of molecular biology’s central functions that we have previously shown to be enriched in the HK388 set [28], several others indispensable to cells, such as DNA repair, energy production (oxidative phosphorylation), RNA degradation, and cell waste management (lysosome), were also enriched in the HK groups. Many disease- and infection-involved pathways were enriched in the HK groups, suggesting that many of the predicted HK genes are important for cell viability and that defects in these genes often lead to disease. In contrast, pathways involving biosynthesis, metabolism of sex and reproduction hormones, and metabolism of retinol and drugs (a reaction that takes place in liver [43]), were enriched in the TS groups.
Table 3. Number and percentage of genes annotated with the indicated KEGG pathway or PIR tissue specificity term enriched in different HK and TS gene sets.
| Gene seta | enriched/annotatedgenes in this set | KEGG pathway or PIRtissue specificity term | Number of KEGG(PIR) genes | % of KEGG (PIR)genes in this set | p valueb | |
| HK388 | KEGG: 270/276 | hsa03010 Ribosome | 84 | 94.1 | 2.0E-91 | |
| hsa03050 Proteasome | 42 | 92.9 | 1.8E-39 | |||
| hsa03040 Spliceosome | 113 | 46.9 | 1.1E-32 | |||
| hsa03022 Basal transcription factors | 33 | 69.7 | 1.7E-18 | |||
| hsa04120 Ubiquitin-mediated proteolysis | 117 | 34.2 | 2.2E-17 | |||
| hsa00970 Aminoacyl-tRNA biosynthesis | 30 | 63.3 | 2.2E-11 | |||
| hsa03020 RNA polymerase | 11 | 100.0 | 3.7E-05 | |||
| hsa03420 Nucleotide excision repair | 44 | 22.7 | 1.7E-02 | |||
| PIR: 26/61 | (PIR) Ubiquitous | 247 | 4.5 | 1.3E-04 | ||
| (PIR) Expressed ubiquitously | 13 | 23.1 | 7.0E-03 | |||
| (PIR) Widely expressed | 165 | 3.6 | 2.3E-02 | |||
| (PIR) Ubiquitously expressed | 118 | 4.2 | 2.9E-02 | |||
| HKI (1∼2,000) | KEGG: 202/686 | hsa00190 Oxidative phosphorylation | 130 | 56.9 | 3.3E-29 | |
| hsa05012 Parkinson's disease | 128 | 57.0 | 7.6E-29 | |||
| hsa05010 Alzheimer's disease | 163 | 46.0 | 6.2E-22 | |||
| hsa05016 Huntington's disease | 180 | 43.9 | 1.3E-21 | |||
| hsa04722 Neurotrophin signaling pathway | 124 | 33.1 | 7.5E-06 | |||
| hsa05110 Vibrio cholerae infection | 56 | 42.9 | 4.9E-05 | |||
| hsa04142 Lysosome | 117 | 29.9 | 1.1E-03 | |||
| hsa05120 Epithelial cell signaling inHelicobacter pylori infection | 68 | 33.8 | 8.4E-03 | |||
| hsa05220 Chronic myeloid leukemia | 75 | 32.0 | 1.5E-02 | |||
| PIR: 134/621 | (PIR) Ubiquitous | 247 | 28.3 | 9.7E-16 | ||
| (PIR) Ubiquitously expressed | 118 | 23.7 | 4.1E-05 | |||
| (PIR) Widely expressed | 165 | 21.2 | 4.5E-05 | |||
| HKII (2,001∼4,000) | KEGG: 51/595 | hsa03018 RNA degradation | 57 | 36.8 | 5.6E-04 | |
| hsa04142 Lysosome | 117 | 24.8 | 2.4E-02 | |||
| PIR: 49/685 | (PIR) Ubiquitous | 247 | 19.8 | 9.2E-05 | ||
| (PIR) Widely expressed | 165 | 20.0 | 1.4E-03 | |||
| (PIR) Ubiquitously expressed | 118 | 21.2 | 2.8E-03 | |||
| HKIII (4,001∼6,000) | KEGG: 0/571 | – | – | – | – | |
| PIR: 40/685 | (PIR) Ubiquitous | 247 | 16.2 | 1.8E-02 | ||
| HKIV (6,001∼7,761) | KEGG: 51/511 | hsa04060 Cytokine-cytokine receptorinteraction interaction | 262 | 19.5 | 6.5E-04 | |
| PIR: 0/578 | – | – | – | – | ||
| TSI (7,762∼9,953) | KEGG: 110/638 | hsa04080 Neuroactive ligand-receptorinteraction | 256 | 21.1 | 9.7E-04 | |
| hsa04060 Cytokine-cytokine receptorinteraction | 262 | 20.6 | 3.5E-02 | |||
| PIR: 0/761 | – | – | – | – | ||
| TSII (9,954∼11,953) | KEGG: 77/728 | hsa04610 Complement and coagulationcascades | 69 | 49.3 | 3.9E-08 | |
| hsa00830 Retinol metabolism | 54 | 38.9 | 1.6E-02 | |||
| hsa00982 Drug metabolism | 62 | 35.5 | 4.6E-02 | |||
| hsa00140 Steroid hormone biosynthesis | 46 | 43.5 | 4.1E-03 | |||
| hsa00591 Linoleic acid metabolism | 28 | 46.4 | 4.0E-02 | |||
| PIR_TS: 13/860 | (PIR) Expressed by the liver and secretedinto the plasma | 23 | 56.5 | 1.8E-05 | ||
| TS734 | KEGG: 12/222 | hsa00140 Steroid hormone biosynthesis | 46 | 26.1 | 4.4E-04 | |
| hsa00150 Androgen and estrogenmetabolism | 37 | 24.3 | 2.2E-02 | |||
| PIR_TS: 14/357 | (PIR) Testis-specific | 39 | 35.9 | 1.6E-07 | ||
HKera score-sorted MR genes were divided into 4 HK sets (HKI-HKIV) and 2 TS sets (TSI and TSII), each containing ∼2,000 genes (see Methods).
The p values for the KEGG pathway were estimated using the Boferroni correction method by controlling the family-wide false discovery rate (FDR) under 5%. An additional criterion, gene number >10, was used to screen for genes enriched in the gene set with PIR tissue specificity annotations [37].
Similarly, the GO terms showing up in the enrichment analysis were markedly different for HK and TS genes: this was especially evident for biological process (BP) (Figure 5) and cellular component (CC) (Figure S4 in File S1), but was also seen for molecular function (MF) (Figure S5 in File S1). Furthermore, with the exception of “binding” in MF (Figure S5 in File S1), those GO terms enriched in both HS and TS genes, i.e. “localization” in BP (Figure 5, left panel) and “structural molecule activity” in MF (Figure S5 in File S1), were separable at the next level of GO annotation (Figure 5, right panel and Figure S6 in File S1, respectively). This marked difference is generally in accordance with HK genes being involving in fundamental cellular processes and functional activities executed by various components of the cell and in different locations in the cell, and with TS genes being involving in regulation, immune, and other cellular responses, such as cell mobility.
Figure 5. Percentage of genes annotated with the indicated GO term in different HK and TS sets.

The figure shows the percentages of genes annotated with the indicated enriched GO category (determined using FDR-corrected p<0.05) in the indicated HK or TS gene set. Genes not associated with any enriched GO term were not used to compute the percentage. At GO’s level 2 (left graph), ‘Localization’ (GO: 0051179) was the only enriched BP category common to both the HK and TS sets; however, at the next level (level 3) of this category (right graph), the enriched GO terms for the two gene sets were different. Note that, “cellular localization (GO: 0051641)” and “macromolecule localization (GO: 0033036)” denote that a protein or macromolecule is transported to a specific location in a cell, while “localization of cell (GO: 0051674)” denotes that a cell is transported to a specific location [40]. Similar results were obtained for the analysis using GO Cellular Component (CC) (Figure S4 in File S1) and Molecular Function (MF) categories (Figure S5 in File S1).
Discussion
By definition, HK genes are expressed for functions that are common to all cells and TS genes are expressed for functions specific to certain types of cells. Consequently, the criterion of “ubiquitous expression” has commonly been employed to identify HK genes. Using microarray expression data, the number of HK genes identified has ranged from scores to a few hundreds [2], [10], [14], while, using FPEI predictions [15], it increases to ∼2,000. However, even this seemly large number of 2,000 is a gross underestimate compared to that of >6,000 obtained in experiments using RNAseq [6], so-called next generation sequencing capable of producing transcriptomes of a finer resolution than microarray technology [34]. In the present study, using the tensor structure of gene expression profiles, rather than expression levels or number of present calls, we showed that HKera was capable of identifying thousands of HK genes from microarray data with a good coverage of RNAseq-derived HK genes (Figure 4). Furthermore, compared to several other HK classifiers, HKera gave a significantly better performance against a number of benchmark HK sets derived from both microarray and RNAseq studies (Figure 2). It is noteworthy that the 16 ranking order-derived tensor components of gene expression profile were fairly orthogonal between the TP (HK388) and TN (TS734) data used to derive HKera (Figure 6), explaining its success. Indeed, HKera performed significantly better than SVM models trained on features used by the other HK classification methods compared, and including those features altogether achieved little, if any, improvement on HKera’s performance (Figure S7 in File S1). This is because the 16 attributes of HKera are much more significant features than those used by Exp, TSI, FPEI, PCall, and Phy, and had in fact captured almost all the information needed to classify the HK/TS genes in the benchmark training set, as indicated by the results of the information gain [44] analysis (Figure S8 in File S1). Leave-one(feature)-out analysis also showed that, of the 16 attributes (A1–A16), those with ranking presence (A1–A8) were slightly more important than those with ranking absence (A9–A16) in their impact on HKera performance, with A8 being the most significant feature (see Figures S1 and S2 in File S1 for explanations for the meaning of each of the 16 features). However, the differences were small, and leaving any feature out would all decrease, albeit not significantly, the accuracy of HKera predictions (Figure S9 in File S1). Since preservation of expression ranking order of HK genes has been previously observed using several different expression datasets and in data from different expression platforms [28], we can expect the HKera approach to be applicable to other large-scale gene expression data.
Figure 6. Contributions of the 16 tensor components to Kendall’s  (TS734 vs. HK388).
(TS734 vs. HK388).

The equations for computing the 16 tensor components have been reported previously [28].
It has been noted that the consensus between different HK gene sets identified by different methods, including those often used as benchmark, is not very good (10%–80%) [10], [15]. In comparison, the agreement between HKera and RNAseq was better: the percentage of genes designated as HK genes by RNAseq and predicted as such by HKera (using the threshold of a 0.0 HKera score) was 83.1%, while the converse coverage of HKera-predicted HK genes by RNAseq was 60.7% (Figure 7). The HKera scores (Figure 7, right panel) also showed that method-consensus genes (e.g. those common to the HKera, RNAseq, and FPEI sets or those only common to the HKera and RNAseq sets) generally had a better HK/TS-distinguishing HKera score than either HKera-unique or RNAseq-unique genes. Using a consensus from multiple prediction methods or a high HKera threshold would therefore be advisable practice for finding HK genes with high confidence. Nevertheless, many method-unique genes did have a good HKera score, some even as good as those of the benchmark genes (Figure 3), suggesting that different transcriptome-partitioning methods examine, to some extent, different features of the transcripome. These high HKera-score, method-unique genes are good candidates for novel HK genes.
Figure 7. Common and unique HK/TS genes predicted by HKera, RNAseq, or FPEI, and their HKera-score distributions.

Here, the canonical HK (HK388) and TS (TS734) genes were excluded, leaving only the MR genes for the analysis. Note that, whereas FPEI assigned a small subset of the transcriptome as HK or TS, HKera and RNAseq divided the transcriptome into two sets, i.e. those that were not predicted as HK genes were placed in the TS set.
Interestingly, although the sets of HK and TS genes classified by HKera, RNAseq, or FPEI were not highly concordant (Figure 7), they all contained essentially the same enriched GO terms, most of which would, in fact, have been captured by two much smaller gold-standard sets (HK388 and TS734, see Figure 5); moreover, the GO terms enriched in either HK or TS genes were highly complementary, such that, together, they covered most of the GO landscape (Figures 5, S4 (in File S1), and S5 (in File S1)). Perhaps the most telling observation for a distinct role of HK and TS genes is that, for BP, genes annotated as ‘cellular localization’ (“a localization process that takes place at the cellular level” [45]) were enriched in the HK genes, while those annotated as ‘localization of cell’ (“any process in which a cell is transported to, and/or maintained in, a specific location” [45]) were enriched in the TS genes (Figure 5, right panel). This was further demonstrated in a complete listing of HK- and TS-enriched CC terms (Table S3 and S4 in File S1, respectively), in which all GO levels were considered: namely, for example, genes annotated as ‘intracellular’ or ‘extracellular’ were enriched, respectively, in the HK genes or TS genes and, while genes annotated as ‘cytoplasm’ and ‘membrane’ were enriched in both the HK and TS genes, a more specific cell type (muscle for ‘cytoplasm’) or cell component (plasma membrane for ‘membrane’) was enriched in the TS genes.
In conclusion, we have developed a novel transcriptome partitioner and shown that it is superior to several other methods in reproducing ‘gold-standard’ HK gene sets. The large number (>7,000) of predicted HK genes is similar to that derived from RNAseq experiments and, as indicated by the enrichment analysis results, the human transcriptome can be partitioned into HK and TS gene sets that occupy distinct parts of the GO spectrum, reinforcing the notion that they have distinct cellular and functional roles.
Supporting Information
Figure S1. Schematic illustrations of the 16 attributes used to derive HKera. (A) A schematic illustration of tissue-wide gene expression ranking (R) for a pair of genes and the three main contributing factors (Stableness (S), Co-expression (C) and Dispersion (D)) of its ranking order concordance and discordance [28]. The up and down arrows respectively point to increasing presence (+) and increasing absence (−) of the indicated variable. (B) Illustrations for each of the 16 attributes (A1–A16), which are products of a tensor operation on an equation that relates the expression ranking order (Kendall’s τ) and the three factors [28]. Figure S2. The distribution of attribute value for a gene pair and its tissue-wide expression and expression ranking profiles. (A) (Left) The fractional values for the presence (+) and absence (−) contribution of Stableness, Co-expression, Dispersion and Ranking as computed by tensor decomposition of gene expression ranking data [28], for a specific pair of genes (NCBI Entrez gene id 5143 and 55142). (Right) A wheel presentation of the composite values of the 16 attributes (A1–A16) used to derive HKera, showing that component A13 dominates for this gene pair. (B) Tissue-wide profile of gene expression levels (left) and rankings (right) for the two genes, showing that they have a high value on Stableness and Co-expression, but a low value on concordant rankings and Dispersion. NI>0 is the number of tissue pairs in which the rank of gene 5143 is higher than that of gene 55142, and NI<0 is the number of tissue pairs in which the rank of gene 5143 is lower than that of gene 55142. Note that there are a total 630 tissue pairs for 36 tissues, and ranking presence (R+) is low because NI>0 and NI<0 are about equal. Figure S3. Mean performance of HKera (SVM) and five other machine learning methods. Like HKera, models of the other machine learning methods were derived using the 16 tensor components of expression rankings as attributes. The performance was evaluated on the same training (A) and test (B) set data described in Methods. The performance represents the average of the results (for accuracy, recall, and precision; see Methods) obtained from five-fold cross validations. Error bars are standard deviations of the five-fold cross validation models. The six machine learning methods compared are: HKera (SVM), J48 (J48 decision tree, a logic-based algorithm), MLP (multilayer perception, a perceptron-based technique), OneR (one rule, a rule learning algorithm), NaïveBayes (a statistical learning algorithm), and KNN (k nearest neighbor, an instance-based learning algorithm). We used the Weka software (http://www.cs.waikato.ac.nz/ml/weka/) to derive these models. Figure S4. Percentage of genes annotated with the indicated CC term in different HK and TS sets. The figure shows the percentages of genes annotated with the indicated enriched GO category (determined using FDR-corrected p<0.05) in the indicated HK or TS gene set. Genes not associated with any enriched GO term were not used to compute the percentage. At GO’s level 2, the enriched GO terms for the two gene sets were different. Figure S5. Percentage of genes annotated with the indicated MF term in different HK and TS sets. The figure shows the percentages of genes annotated with the indicated enriched GO category (determined using FDR-corrected p<0.05) in the indicated HK or TS gene set. Genes not associated with any enriched GO term were not used to compute the percentage. At GO’s level 2, ‘Structural molecule activity’ (GO: 0005198) and ‘binding’ (GO: 0005488) were the two enriched MF categories common to both the HK and TS sets; however, at the next level (level 3) of these categories, the enriched GO terms for the two gene sets were mostly different (see Figure S6 in File S1 for the result of ‘Structural molecule activity’ at level 3; data not shown for ‘binding’). Figure S6. Percentage of genes annotated with ‘Structural molecule activity’ (GO:0005198) in different HK and TS sets. The figure shows the percentages of genes annotated with the indicated enriched GO category (determined using FDR-corrected p<0.05) in the indicated HK or TS gene set. Genes not associated with any enriched GO term were not used to compute the percentage. At GO’s level 3, the enriched GO terms of ‘structural molecule activity’ for the two gene sets were different. Figure S7. Mean performance of SVMHKera, SVMConv and SVMAll. The mean performance (average of accuracy, recall and precision rates from five-fold cross validation) of SVM models derived using different features: SVMHKera used HKera scores, SVMConv used scores computed from the HK criteria (Table 1 in the main text) of the five conventional HK classification methods (Exp, PCall, FPEI, TSI, and Phy) compared in this study, and SVMAll used all these scores. For TSI and Phy, the score was assigned to be the TSI index value and the Phy probability value, respectively; for Exp, PCall and FPEI, the score was the fraction of 36 tissues in which the gene in question was regarded as expressed by the method (e.g. expression intensity > = 200 for Exp, see Table 1). RNAseq was excluded because microarray gene expression data were used in this comparison. The performance was evaluated on the same training (A) and test (B) set data described in Methods. Error bars are standard deviations of the five-fold cross validation models. Figure S8. The information gain of the six HK classification features used to derive SVMAll. The information gain, which ranges between 0 and 1 and can be computed based on theory of information entropy [44], is a measure of the capability of a feature to distinguish between HK class and TS class, based on the feature’s presence or absence in the HK388 and TS734 benchmark set (see Methods for the benchmark dataset and Figure S7 in File S1 for the derivation of SVMAll). Error bars are standard deviations of the five-fold cross validation models. Figure S9. Leave-one(feature)-out accuracies of HKera. These accuracies were computed by leaving the indicated feature (one of the 16 tensor component attributes) out in HKera predictions of the training (A) and test (B) set data described in Methods. Arrow points to the accuracy of HKera using all of the 16 attributes (see Table 2). Error bars are standard deviations of the five-fold cross validation models. Table S1. Performance of HKera derived using different sets of reference genes. Table S3. Number of genes annotated with the indicated enriched cellular component GO terms in all levels in the HK genes predicted by HKera. Table S4. Number of genes annotated with the indicated enriched cellular component GO terms in all GO levels in the TS genes predicted by HKera.
(DOC)
Table S2. List of all 13,075 genes (in GSE2361) with their HKera score and GO, KEGG and PIR annotations. (This table is provided in a separate, Excel file).
(XLS)
Acknowledgments
We thank Dr. Hsuan-Tien Lin of the Department of Computer Science and Information Engineering, National Taiwan University, for helpful discussion on ordinal classification methods. We thank Dr. T. Barkas for English editing.
Funding Statement
This work was supported in part by a grant from the National Science Council of Taiwan (NSC grant no. 101-2311-B-001-026-MY3). No additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Watson JD (1987) The functioning of higher eukaryotic genes. Molecular biology of the gene. Menlo Park, Calif.: Benjamin/Cummings. 704 p. [Google Scholar]
- 2. Eisenberg E, Levanon EY (2003) Human housekeeping genes are compact. Trends Genet 19: 362–365. [DOI] [PubMed] [Google Scholar]
- 3. Butte AJ, Dzau VJ, Glueck SB (2001) Further defining housekeeping, or “maintenance,” genes Focus on “A compendium of gene expression in normal human tissues”. Physiol Genomics 7: 95–96. [DOI] [PubMed] [Google Scholar]
- 4. Tu Z, Wang L, Xu M, Zhou X, Chen T, et al. (2006) Further understanding human disease genes by comparing with housekeeping genes and other genes. BMC Genomics 7: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hwang PI, Wu HB, Wang CD, Lin BL, Chen CT, et al. (2011) Tissue-specific gene expression templates for accurate molecular characterization of the normal physiological states of multiple human tissues with implication in development and cancer studies. BMC Genomics 12: 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramskold D, Wang ET, Burge CB, Sandberg R (2009) An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol 5: e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Szabo A, Perou CM, Karaca M, Perreard L, Palais R, et al. (2004) Statistical modeling for selecting housekeeper genes. Genome Biol 5: R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chikina MD, Huttenhower C, Murphy CT, Troyanskaya OG (2009) Global prediction of tissue-specific gene expression and context-dependent gene networks in Caenorhabditis elegans. PLoS Comput Biol 5: e1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kadota K, Ye J, Nakai Y, Terada T, Shimizu K (2006) ROKU: a novel method for identification of tissue-specific genes. BMC Bioinformatics 7: 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu J, He F, Song S, Wang J, Yu J (2008) How many human genes can be defined as housekeeping with current expression data? BMC Genomics 9: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szabo A, Perou CM, Karaca M, Perreard L, Quackenbush JF, et al. (2004) Statistical modeling for selecting housekeeper genes. Genome Biol 5: R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee S, Jo M, Lee J, Koh SS, Kim S (2007) Identification of novel universal housekeeping genes by statistical analysis of microarray data. J Biochem Mol Biol 40: 226–231. [DOI] [PubMed] [Google Scholar]
- 13. Warrington JA, Nair A, Mahadevappa M, Tsyganskaya M (2000) Comparison of human adult and fetal expression and identification of 535 housekeeping/maintenance genes. Physiol Genomics 2: 143–147. [DOI] [PubMed] [Google Scholar]
- 14. Hsiao LL, Dangond F, Yoshida T, Hong R, Jensen RV, et al. (2001) A compendium of gene expression in normal human tissues. Physiol Genomics 7: 97–104. [DOI] [PubMed] [Google Scholar]
- 15. Chang CW, Cheng WC, Chen CR, Shu WY, Tsai ML, et al. (2011) Identification of human housekeeping genes and tissue-selective genes by microarray meta-analysis. PLoS One 6: e22859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, et al. (2005) Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21: 650–659. [DOI] [PubMed] [Google Scholar]
- 17. Eller CD, Regelson M, Merriman B, Nelson S, Horvath S, et al. (2007) Repetitive sequence environment distinguishes housekeeping genes. Gene 390: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Ferrari L, Aitken S (2006) Mining housekeeping genes with a Naive Bayes classifier. Bmc Genomics 7: 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lehner B, Fraser AG (2004) Protein domains enriched in mammalian tissue-specific or widely expressed genes. Trends Genet 20: 468–472. [DOI] [PubMed] [Google Scholar]
- 20. Farre D, Bellora N, Mularoni L, Messeguer X, Alba MM (2007) Housekeeping genes tend to show reduced upstream sequence conservation. Genome Biol 8: R140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Williams T, Yon J, Huxley C, Fried M (1988) The mouse surfeit locus contains a very tight cluster of four “housekeeping” genes that is conserved through evolution. Proc Natl Acad Sci U S A 85: 3527–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang L, Li WH (2004) Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol Biol Evol 21: 236–239. [DOI] [PubMed] [Google Scholar]
- 23. Dong B, Zhang P, Chen X, Liu L, Wang Y, et al. (2011) Predicting housekeeping genes based on Fourier analysis. PLoS One 6: e21012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. She X, Rohl CA, Castle JC, et al. (2009) Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genomics 10: 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greer S, Honeywell R, Geletu M, Arulanandam R, Raptis L (2010) Housekeeping genes; expression levels may change with density of cultured cells. J Immunol Methods 355: 76–79. [DOI] [PubMed] [Google Scholar]
- 26. Vinogradov AE (2004) Compactness of human housekeeping genes: selection for economy or genomic design? Trends Genet 20: 248–253. [DOI] [PubMed] [Google Scholar]
- 27. Zhu J, He F, Hu S, Yu J (2008) On the nature of human housekeeping genes. Trends Genet 24: 481–484. [DOI] [PubMed] [Google Scholar]
- 28. Shaw GT, Shih ES, Chen CH, Hwang MJ (2011) Preservation of ranking order in the expression of human Housekeeping genes. PLoS One 6: e29314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu CH, Yeh LS, Huang H, Arminski L, Castro-Alvear J, et al. (2003) The Protein Information Resource. Nucleic Acids Research 31: 345–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ge X, Yamamoto S, Tsutsumi S, Midorikawa Y, Ihara S, et al. (2005) Interpreting expression profiles of cancers by genome-wide survey of breadth of expression in normal tissues. Genomics 86: 127–141. [DOI] [PubMed] [Google Scholar]
- 33. Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henson R, Cetto L (2005) The MATLAB bioinformatics toolbox. Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics.: The MathWorks, Inc., Natick, MA, USA.
- 36.Green DM, Swets JA (1966) Signal detection theory and psychophysics: Wiley New York.
- 37. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 38. Huang da W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cardoso JS, Da Costa JFP (2007) Learning to Classify Ordinal Data: The Data Replication Method. Journal of Machine Learning Research 8 (1393–1429) 6. [Google Scholar]
- 40. Chu W, Zoubin G (2005) Gaussian processes for ordinal regression. Journal of Machine Learning Research 6: 1019–1041. [Google Scholar]
- 41.Cardoso JS, Ricardo S, Inês D (2012) Ordinal Data Classification Using Kernel Discriminant Analysis: A Comparison of Three Approaches. Machine Learning and Applications (ICMLA), Vol. 1. IEEE, 2012.
- 42. Kotsiantis SB (2007) Supervised Machine Learning: A Review of Classification Techniques. Informatica 31: 249–268. [Google Scholar]
- 43. Rinn JL, Rozowsky JS, Laurenzi IJ, Petersen PH, Zou KY, et al. (2004) Major molecular differences between mammalian sexes are involved in drug metabolism and renal function. Developmental Cell 6: 791–800. [DOI] [PubMed] [Google Scholar]
- 44. Hild KEII, Erdogmus D, Torkkola K, Principe JC (2006) Feature extraction using information-theoretic learning, Trans on Pattern Analysis and Machine Intelligence. 28: 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, et al. (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25: 288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic illustrations of the 16 attributes used to derive HKera. (A) A schematic illustration of tissue-wide gene expression ranking (R) for a pair of genes and the three main contributing factors (Stableness (S), Co-expression (C) and Dispersion (D)) of its ranking order concordance and discordance [28]. The up and down arrows respectively point to increasing presence (+) and increasing absence (−) of the indicated variable. (B) Illustrations for each of the 16 attributes (A1–A16), which are products of a tensor operation on an equation that relates the expression ranking order (Kendall’s τ) and the three factors [28]. Figure S2. The distribution of attribute value for a gene pair and its tissue-wide expression and expression ranking profiles. (A) (Left) The fractional values for the presence (+) and absence (−) contribution of Stableness, Co-expression, Dispersion and Ranking as computed by tensor decomposition of gene expression ranking data [28], for a specific pair of genes (NCBI Entrez gene id 5143 and 55142). (Right) A wheel presentation of the composite values of the 16 attributes (A1–A16) used to derive HKera, showing that component A13 dominates for this gene pair. (B) Tissue-wide profile of gene expression levels (left) and rankings (right) for the two genes, showing that they have a high value on Stableness and Co-expression, but a low value on concordant rankings and Dispersion. NI>0 is the number of tissue pairs in which the rank of gene 5143 is higher than that of gene 55142, and NI<0 is the number of tissue pairs in which the rank of gene 5143 is lower than that of gene 55142. Note that there are a total 630 tissue pairs for 36 tissues, and ranking presence (R+) is low because NI>0 and NI<0 are about equal. Figure S3. Mean performance of HKera (SVM) and five other machine learning methods. Like HKera, models of the other machine learning methods were derived using the 16 tensor components of expression rankings as attributes. The performance was evaluated on the same training (A) and test (B) set data described in Methods. The performance represents the average of the results (for accuracy, recall, and precision; see Methods) obtained from five-fold cross validations. Error bars are standard deviations of the five-fold cross validation models. The six machine learning methods compared are: HKera (SVM), J48 (J48 decision tree, a logic-based algorithm), MLP (multilayer perception, a perceptron-based technique), OneR (one rule, a rule learning algorithm), NaïveBayes (a statistical learning algorithm), and KNN (k nearest neighbor, an instance-based learning algorithm). We used the Weka software (http://www.cs.waikato.ac.nz/ml/weka/) to derive these models. Figure S4. Percentage of genes annotated with the indicated CC term in different HK and TS sets. The figure shows the percentages of genes annotated with the indicated enriched GO category (determined using FDR-corrected p<0.05) in the indicated HK or TS gene set. Genes not associated with any enriched GO term were not used to compute the percentage. At GO’s level 2, the enriched GO terms for the two gene sets were different. Figure S5. Percentage of genes annotated with the indicated MF term in different HK and TS sets. The figure shows the percentages of genes annotated with the indicated enriched GO category (determined using FDR-corrected p<0.05) in the indicated HK or TS gene set. Genes not associated with any enriched GO term were not used to compute the percentage. At GO’s level 2, ‘Structural molecule activity’ (GO: 0005198) and ‘binding’ (GO: 0005488) were the two enriched MF categories common to both the HK and TS sets; however, at the next level (level 3) of these categories, the enriched GO terms for the two gene sets were mostly different (see Figure S6 in File S1 for the result of ‘Structural molecule activity’ at level 3; data not shown for ‘binding’). Figure S6. Percentage of genes annotated with ‘Structural molecule activity’ (GO:0005198) in different HK and TS sets. The figure shows the percentages of genes annotated with the indicated enriched GO category (determined using FDR-corrected p<0.05) in the indicated HK or TS gene set. Genes not associated with any enriched GO term were not used to compute the percentage. At GO’s level 3, the enriched GO terms of ‘structural molecule activity’ for the two gene sets were different. Figure S7. Mean performance of SVMHKera, SVMConv and SVMAll. The mean performance (average of accuracy, recall and precision rates from five-fold cross validation) of SVM models derived using different features: SVMHKera used HKera scores, SVMConv used scores computed from the HK criteria (Table 1 in the main text) of the five conventional HK classification methods (Exp, PCall, FPEI, TSI, and Phy) compared in this study, and SVMAll used all these scores. For TSI and Phy, the score was assigned to be the TSI index value and the Phy probability value, respectively; for Exp, PCall and FPEI, the score was the fraction of 36 tissues in which the gene in question was regarded as expressed by the method (e.g. expression intensity > = 200 for Exp, see Table 1). RNAseq was excluded because microarray gene expression data were used in this comparison. The performance was evaluated on the same training (A) and test (B) set data described in Methods. Error bars are standard deviations of the five-fold cross validation models. Figure S8. The information gain of the six HK classification features used to derive SVMAll. The information gain, which ranges between 0 and 1 and can be computed based on theory of information entropy [44], is a measure of the capability of a feature to distinguish between HK class and TS class, based on the feature’s presence or absence in the HK388 and TS734 benchmark set (see Methods for the benchmark dataset and Figure S7 in File S1 for the derivation of SVMAll). Error bars are standard deviations of the five-fold cross validation models. Figure S9. Leave-one(feature)-out accuracies of HKera. These accuracies were computed by leaving the indicated feature (one of the 16 tensor component attributes) out in HKera predictions of the training (A) and test (B) set data described in Methods. Arrow points to the accuracy of HKera using all of the 16 attributes (see Table 2). Error bars are standard deviations of the five-fold cross validation models. Table S1. Performance of HKera derived using different sets of reference genes. Table S3. Number of genes annotated with the indicated enriched cellular component GO terms in all levels in the HK genes predicted by HKera. Table S4. Number of genes annotated with the indicated enriched cellular component GO terms in all GO levels in the TS genes predicted by HKera.
(DOC)
Table S2. List of all 13,075 genes (in GSE2361) with their HKera score and GO, KEGG and PIR annotations. (This table is provided in a separate, Excel file).
(XLS)