

Abstract
Due to its bone anabolic activity, methods to increase Wnt activity, such as inhibitors of dickkopf-1 and sclerostin, are being clinically explored. Glycogen synthase kinase (GSK3β) inhibits Wnt signaling through inducing β-catenin degradation. Therefore, AR79, an inhibitor of GSK3β, is being evaluated as a bone anabolic agent. However, Wnt activation has potential to promote tumor growth. The goal of this study was to determine if AR79 impacted progression of prostate cancer (PCa). PCa tumors were established in subcutaneous and bone sites of mice followed by AR79 administration. Tumor growth, β-catenin activation, proliferation (Ki67 expression) and apoptosis (caspase 3 activity) were measured. Additionally, PCa and osteoblast cell lines were treated with AR79 and β-catenin status, proliferation (with β-catenin knocked down in some cases) and proportion of the ALDH+CD133+ stem-like cells was determined. AR79 promoted PCa growth, decreased phospho-β-catenin expression and increased total and nuclear β-catenin expression in tumors and increased tumor-induced bone remodeling. Additionally, it decreased caspase 3 and increased Ki67 expression. In addition, AR79 increased bone formation in normal mouse tibiae. AR79 inhibited β-catenin phosphorylation, increased nuclear β-catenin accumulation in PCa and osteoblast cell lines and increased proliferation of PCa cells in vitro through β-catenin. Furthermore, AR79 increased the ALDH+CD133+ cancer stem cell-like proportion of the PCa cell lines. We conclude that AR79, while being bone anabolic, promotes PCa cell growth through Wnt pathway activation.
Keywords: Wnt, glycogen synthase kinase 3β, prostate cancer, bone metastases, cancer stem cell
Introduction
Bone loss is a common pathology that accompanies multiple diseases. Therefore, extensive research efforts have been aimed towards minimizing bone loss and has resulted in therapies that effectively decrease bone loss through inhibition of bone resorption. In addition to minimizing bone resorption, methods to promote bone formation (i.e., bone anabolic activity) have received great attention but clinical therapeutics in this area lag behind anti-resorptives (1). For example, in the United States, teriparatide (parathyroid hormone, PTH 1–34) is the only United States Food and Drug Administration approved bone anabolic agent. However, its use is limited to two years due to pre-clinical findings of osteosarcoma in the rat model. Thus, continued effort to identify bone anabolic agents is necessary to enhance efficacy and minimize their potential.
The Wnt pathway mediates bone development (2) and thus its manipulation serves as a potential bone anabolic therapy. Wnts mediate signaling through binding cell surface transmembrane low-density lipoprotein receptor-related (LRP5/6) and frizzled proteins, which results in β-catenin nuclear translocation and target gene activation (3). In the absence of Wnts, β-catenin is degraded in the cytoplasm by interacting with a protein complex consisting of axin, adenomatous polyposis coli (APC) and glycogen synthase kinase 3β (GSK3β). Axin and APC are scaffold proteins that facilitate binding of β-catenin to GSK3β. GSK3β then phosphorylates β-catenin which targets it for ubiquitination and degradation. Due to the key role of GSK3β in degrading β-catenin, inhibiting its activity should result in increased β-catenin activity. Thus, GSK3β has been considered as a therapeutic target for induction of β-catenin activity which could ultimately result in bone anabolic activity. Along these lines, other compounds that promote Wnt activity through blocking inhibitors of Wnt signaling, such as sclerostin and dickkopf-1 (4), are being evaluated for their bone anabolic effects including the use of an anti-sclerostin antibody in clinical trials (5).
While induction of bone anabolic activity through increased Wnt pathway signaling has potential therapeutic benefit, a major concern that should be considered is the impact of chronic Wnt activation on cancer progression. Increased β-catenin expression has been associated with multiple cancers (6, 7). Salmon calcitonin, which mediates degradation of GSK-3β (8) and has been used therapeutically for bone anabolic activity, has been associated with increased cancer incidence leading advisory panels of both the United States Food and Drug Administrations to European regulatory committees to recommend discontinuing its use for osteoporosis. The majority of Wnt cancer-related action has been associated with early tumorigenesis. However, Wnt signaling may also play a role in established tumors, including maintenance of cancer stem cells (9). We have previously shown that modulation of the Wnt pathway with Wnt inhibitors, such as dickkopf-1 (DKK1) promotes prostate cancer (PCa) growth in soft tissue and bone (10). Due to the potential for modulation of the Wnt pathway to promote cancer, it is critical to evaluate any Wnt-pathway therapies for impact on cancer biology.
AR79 (AstraZeneca U.K.) is a potent inhibitor of GSK3β that was developed as a potential bone anabolic agent. AR79 upregulates β-catenin and bone mineralization in cell assays and produces an increase in bone mass in vivo (11). As AR79 modulates the Wnt pathway, we sought to determine if it could impact the progression of PCa in soft tissue and bone.
Materials and Methods
Cell Culture
Human prostate cancer cell lines DU145 and PC3 were obtained from the American Type Culture Collection (ATCC; Rockville, MD) and cultured in RPMI 1640 (Invitrogen Co., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Life Technologies, Inc.). The C4-2B cell line, which is an LNCaP subline, (kindly provided by Dr. Leland Chung, Cedars Sinai, Hollywood, CA) were maintained in T medium [80% DMEM (Life Technologies, Inc.), 20% F12 (Invitrogen), 100 units/liter penicillin G, 100 Ag/mL streptomycin, 5μg/mL insulin, 13.6 pg/mL triiodothyronine, 5μg/mL transferrin, 0.25μg/mL biotin, and 25 μg/mL adenine] supplemented with 10% FBS. The human colorectal adenocarcinoma cell line HCT116 was purchased from ATCC and maintained in McCoy's 5a Medium (Gibco Technology, USA) supplemented with 10% heat-inactivated FBS (HyClone, USA), 100U/mL penicillin, 100μg/mL streptomycin (Invitrogen Life Technologies, USA), 2 mmol/L L-glutamine (Invitrogen). The MC3T3-E1 (clone MC-4) cell line (kindly provided by Dr. Renny Franceschi, University of Michigan, Ann Arbor, MI), a pre-osteoblast cell line derived from murine calvariae that, when treated with ascorbate, expresses osteoblast-specific markers and produces a mineralized matrix was routinely maintained in α-MEM containing 10% FBS and 1% penicillin-streptomycin (Life Technologies, Inc.). The ST-2 cell line, a mouse bone marrow stromal cell line, was obtained from RIKEN Cell Bank (Ibaraki, Japan) and maintained in Minimal Essential Medium Alpha (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (Life Technologies, Inc.) and 2mM L-glutamine (Invitrogen). All cultures were maintained at 37°C, 5% CO2, and 100% humidity. Luiferase containing variants of the prostate cancer cell lines were made as previously described (12). Briefly, C4-2B and DU145 were transduced with retrovirus encoding the luciferase gene and selected using G418. Stable expression of luciferase was confirmed using bioluminescent imaging (BLI). Cell identities were confirmed using short tandem repeat (STR) mapping (Supplement Table 1).
siRNA Transfection
C4-2B and DU145 were plated at a density of 5×105 on 100mm plates and then transfected with 100nM two different sequences of β-catenin siRNAs (Cell Signal, signalSilence® β-Catenin siRNAI&II,6225,6238) or scrambled control siRNA (Cell Signal, signalSilence® Control siRNA,6568) using Lipofectamine® RNAiMAX Reagent (Invitrogen,13778). Transfection conditions were adjusted according to the manufacturer's guide. After transfection for 72h, the cells were treated with AR79 (3μg/ml) and rhWnt3a (60ng/ml) (R&D Systems, Minneapolis, MN) for 4 hours. Nuclear and cytoplasmic protein was extracted using NE-PER® Nuclear and Cytoplasmic Extraction Reagents (Thermo scientific, 78835) following the manufacturer's instructions.
Cell Viability assay
DU145 and C4-2B cells and cells transfected with β-catenin siRNA were cultured in 96-well plates for overnight and then cells were treated with AR79 (3μg/ml) and rhWnt3a (60ng/ml) (R&D Systems, Minneapolis, MN) or vehicle (DMSO or PBS) for 24hr, 48hr and 72hr. Cell proliferation reagent WST-1 (Roche, 1644807) was added and incubated at 37°C and 5% CO2 for 4hr. Absorbance was then measured at 440 nm with a plate reader (Multi-Mode Microplate Reader, SpectraMax M5, Molecular Devices MDS Analytical Technologies).
TRACP 5b and Osteocalcin Assays
Whole blood was obtained and centrifuged to obtain serum which was frozen at −80°C until assayed. Mouse TRACP 5b and osteocalcin in mouse serum were measured using the MouseTRAP™ Assay (Immunodiagnostics Systems Inc., Scottsdale, AZ) and Mouse Osteocalcin Assay (Biomedical Technologies Inc., Stoughton, MA), respectively, as recommended by the manufacturer.
Caspase-3/7 Assay
C4-2B and DU145 were cultured into black 96-well plates at a concentration of 1×104 (2×105 cell/ml, 50ul) per well. Cells were treated with 50ul AR79 (0.03μg/ml, 0.3μg/ml, 3μg/ml) or vehicle (DMSO) or etoposide (100nM) (Sigma) as a positive control after 12 hours and were allowed to grow for 24 hours at which point 100ul of Apo-ONE Caspase-3/7 Reagent (Promega, Madison, WI) was added to each well and the cells were incubated for extended periods (>4 hours). Absorbance was determined at 499 and 521 nm using a plate reader (Multi-Mode Microplate Reader, SpectraMax M5, Molecular Devices MDS Analytical Technologies).
Bioluminescence imaging (BLI) in vitro
C4-2Bluc and DU145luc cells were plated into black, clear bottom 96-well plates (Costar, Corning, New York) at a concentration of 3 × 103 (for 24 hours and 48 hours test) or 2 × 103 (for 72 hours test) per well in RPMI medium with 3% FBS. In some instances, after 12 hours, ST2 cells were added and plated with C4-2Bluc or DU145luc at a 1:6 or 1:3 ratio and co-cultured for 12 hours. C4-2Bluc or DU145luc without ST2 were utilized as vehicle control group. The cells were treated with 0.03μg/ml, 0.3μg/ml and 3μg/ml AR79 and vehicle (DMSO), and were allowed to grow for 24 hours, 48 hours and 72 hours at which time 5μl luciferin (40 mg/ml, Regis Technologies,Inc.) was added to each well 5–10 min prior to imaging. BLI signal was quantified (IVIS System, Caliper Life Sciences). To determine the correlation between cell number and BLI signal, C4-2Bluc and DU145luc were seeded at the concentration from 5×103 to 2×104 into black, clear bottom 96-well plates in RPMI with 3% FBS for 24 hours, and BLI signal was measured.
Protein Extraction and Western Blot Analyses
HT116, C4-2B, ST2 and MC3T3-E1 cell lines (1×106 or 5×106 cells/dish) were cultured in 100 mm dishes (1×106 cells/dish for whole cell protein, or 5×106 for nuclear protein extraction). After 12 hours, the culture medium was changed to FBS-free medium for 12 hours and then the cells were treated with AR79 (0.03, 0.3 and 3μg/ml) for 0, 15, 30, 60 and 120min. Also, rhWnt3a (60ng/ml) was used as positive control (R&D Systems, Minneapolis, MN). Whole cell protein was isolated as described previously (13). Nuclear protein was extracted using NEPER® Nuclear and Cytoplasmic Extraction Reagents (Thermo scientific, 78835) following the manufacturer's instructions. For tissue protein extraction, the tumor tissues were homogenized in ice-cold RIPA lysis buffer (Millipore, Billerica, MA). The tissue lysates were incubated at 4°C for 1 hour with rotation, followed by clarification of tissue debris by centrifugation at 12,000 rpm for 10 minutes. The protein concentration of tumor extracts was determined using BCA Protein Assay Kit (Pierce). The level of phosphorylated β-catenin and β-catenin were analyzed using Western blot analysis, which was done as previously described (13), with rabbit anti-human p-β-catenin monoclonal Ab (1:1000, Invitrogen), mouse anti-human β-catenin monoclonal Ab (1:1000, Santa Cruz Biotechnology), mouse anti-human α-Tubulin monoclonal Ab (1:2500, Sigma-Aldrich), rabbit anti-hnRNP polyclonal Ab (1:1000, Santa Cruz Biotechnology). The Ab binding was revealed using an HRP-conjugated anti rabbit IgG (1:3000, Cell Signaling), or anti-mouse IgG (1:3000, Amersham Pharmacia Biotech, Piscataway, New Jersey, USA). Antibody complexes were detected using SuperSignal West Pico Chemiluminescent Substrate or SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposure to X-Omat film (Kodak, Rochester, NY). The densities of the Western blot bands were quantified using Image J software (version 1.40; National Institutes of Health, Bethesda, MD).
HIPK2 analysis
For immunoprecipitation of HIPK2, total protein (500μg) was incubated with a polyclonal rabbit anti-human HIPK2 antibody (1μl) (Abcam, Cambridge, MA) for a minimum of 1 hr at 4°C. Antibody-bound protein complexes were recovered via binding to mixed 50 μl protein A and protein G-Sepharose beads ((Millipore, Billerica, MA) and subsequently incubated overnight at 4°C with gentle agitation. Afterwards, the sepharose-bound immunoprecipitates were centrifuged at 8000 rpm for 2 min followed by several washing steps with immunoprecipitation buffer. Immune complexes were eluted from beads into 30 μl of PBS and subsequent heating to 95°C for 10 min. After centrifugation, the supernatant containing the immunoprecipitated proteins was separated by SDS-PAGE (8%) and blotted onto PVDF membrane (Bio-Rad, Hercules, CA). After blocking for 1 h in 5%BSA in TBS-T (TBS containing 0.05% Tween 20), blots were incubated with monoclonal mouse anti-human phosphor-Tyrosine antibody (1:1000, Cell Signaling) overnight at 4°C followed by detection of binding by anti-mouse IgG (1:3000, Amersham Pharmacia Biotech, Piscataway, New Jersey, USA). Antibody complexes were detected using SuperSignal West Pico Chemiluminescent Substrate or SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposure to X-Omat film (Kodak, Rochester, NY).
In vivo prostate cancer models
Seven-week-old male NOD.CB17-Prkdcscid/NCrCrl (NOD/SCID) mice (Charles River, Wilmington, MA) were used and housed under pathogen-free conditions. All experimental protocols were approved by the University of Michigan Animal Care and Use Committee. For subcutaneous injection, single cell suspensions (1×106cells) of C4-2Bluc cells in T medium were injected in the flank at 100μl/site using a 27-G3/8-inch needle under anesthesia with 2.5% isofluorane/air. Single-cell suspensions (3×105cells) of DU145luc cells with Matrigel (BD Biosciences, San Jose, CA) were injected in the flank at 100μl/site using a 27-G3/8-inch needle. Subcutaneous tumor growth was monitored by palpation. Subcutaneous tumors were measured using BLI weekly. The tumor weights of the DU145luc injection group were measured at the end of the study. Animals in C4-2Bluc injection group were sacrificed at the end of 6 weeks and animals in DU145luc injection group were sacrificed at the end of 9 weeks at which point tumors were harvested. Half of each tumor was kept for histological analysis and the other half flash frozen for molecular analysis. For intratibial injection, mice were anesthetized with 2.5% isofluorane/air, and both legs were cleaned with betadine and 70% ethanol. The knee was flexed, and a 27-G3/8-inch needle was inserted into the proximal end of right tibia followed by injection of 20μl single-cell suspensions of C4-2Bluc cells (5×105 cells). Tumor development in bone was evaluated weekly using BLI (methodology described in BLI section below) and radiography. All animals were sacrificed at the end of 9 weeks. At the time of euthanasia, serum was collected and necropsy was performed to evaluate for gross tumor lesions. Both tumor-injected and contralateral tibiae were collected, placed in formalin, radiographed and subject to dual-energy X-ray absorptiometry, followed by processing for histological analysis.
Immunohistochemistry
Both tibiae and subcutaneous tumors were fixed in 10% neutral buffed formalin for 24 hours. The tibiae were then decalcified for 48 hours in 10% EDTA and then both tibiae and subcutaneous tumors were processed for paraffin embedding. Five-micron (5μM) sections were used for H&E and IHC. Nonstained sections were deparaffinized and rehydrated then stained for Ki67 (1:500, Santa Cruz Biotechnology), Caspase 3 (1:200, Cell Signaling Biotechnology) and β-catenin (1:500, BD Biosciences).
Bioluminescence imaging (BLI) in vivo
Mice were injected intraperitoneally with 100μl luciferin (40 mg/ml in PBS), anesthetized with 1.5% isoflurane and imaged 15 minutes post-luciferin injection on the IVIS BLI system (Caliper Life Sciences) as previously described (14). Mice were imaged weekly after tumor injection to monitor tumor development. Bioluminescence generated by the luciferin/ luciferase reaction served as a locator for cancer growth and was used for quantification using the Living Image software on a red (high intensity/cell number) to blue (low intensity/cell number) visual scale. A digital grayscale animal image was acquired followed by acquisition and overlay of a pseudocolor image representing the spatial distribution of detected photon counts emerging from active luciferase within the animal. Signal intensity was quantified as the sum of all detected photons within the region of interest during a 1-minute luminescent integration time.
Dual-energy X-ray absorptiometry (DEXA) measurement
Bone mineral content (BMC) and Bone mineral density (BMD) of the excised tibiae were measured using dual-energy X-ray absorptiometry (DEXA) on an Eclipse peripheral DEXA as previously described (13). Briefly, excised bones were subject to DEXA using pDEXA Sabre software version 3.9.4 in research mode (Norland Medical Systems, Fort Atkinson, WI). Bones were scanned at 2 mm/s with a resolution of 0.1×0.1 mm. Three 0.5-cm regions of interest were randomly selected for each fragment to determine BMC and BMD. Short-term precision (% coefficient of variation) was ~3% for this technique.
Radiographic Analysis
Magnified flat radiographs of hind limbs were obtained using a Faxitron machine (Faxitron X-ray Corp., Wheeling, IL).
Flow Cytometry Analysis of ALDH and CD133 Expression
C4-2B and DU145 cells were dissociated with Accutase (Innovative Cell Technologies, Inc.) For ALDH staining, cells were subjected to ALDEFLUOR® assay kit (STEMCELL Technologies, Inc.) according to the manufacturer's protocol. For CD133 staining, the cells were incubated with CD133/1 (AC133)-PE antibody (Miltenyi Biotec). One microgram per milliliter of 7AAD (Sigma-Aldrich, St. Louis, MO) was added to the samples before flow analysis to facilitate dead cell discrimination. Samples were analyzed on MoFlo XDP flow cytometer (Beckman Coulter, Luton, UK).
AR79 treatment in vivo
All animal studies were approved by the University of Michigan Animal Use and Care Committe. AR79 (AstraZeneca, UK) was used at 15 mg/kg and 50 mg/kg orally once daily based on in vivo studies in mice that demonstrated it inhibited GSK-3 activity and increased bone mass in femurs (11). AR 79 was formulated fresh daily in 0.5% hydroxypropylmethyl cellulose (HPMC)(Sigma-H7509) with 0.1% Tween 80 (Sigma-P8074) in water. Vehicle consisted of the carrier (0.5% HPMC/0.1% Tween 80 in water).
Statistical Analyses
All in vitro experiments were performed at least three times. Numerical data are expressed as mean ± SD. Statistical analysis was performed by analysis of One-way ANOVA and/or Student's t-test for independent analysis. The value p <0.05 was considered statistically significant.
Results
To determine if Wnt activation promotes PCa cell growth in soft tissue in vivo, we injected C4-2B cells into the subcutaneous tissue of mice and treated the mice with either vehicle or AR79 using low (15mg/kg) and high (50mg/kg) doses. Both doses of AR79 promoted PCa growth over a 42 day period (Fig. 1A and 1B). We also evaluated Du145 PCa cells using AR79 (50mg/kg). Similar to the C4-2B cells, AR79 induced Du145 cell growth (Fig. 1C–1F). In agreement with the BLI findings, individual tumor weights were greater in the Du145 tumors from mice that received AR79 (Fig. 1E and F). Mouse body weights were not impacted by treatment (Fig. S1).
Figure 1.
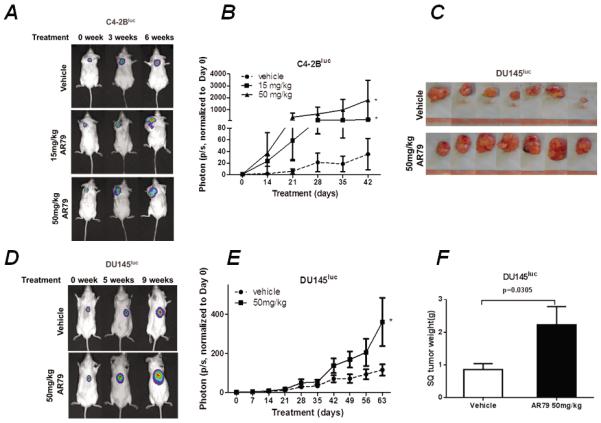
AR79 promotes prostate cancer cell growth in soft tissue in vivo. NOD/SCID mice were injected subcutaneously (SQ) with C4-2Bluc (1×106, in 100μl T medium) and DU145luc (3×105 with Matrigel, in 100μl RPMI). After establishment of tumor growth, treatment with either AR79 or vehicle was initiated and continued 6 weeks for C4-2Bluc and 9 weeks for DU145luc. Mice were imaged weekly using bioluminescent imaging (BLI) after tumor injection to monitor tumor development. There were 15 NOD/SCID mice in the C4-2Bluc injection group (50mg/kg of AR79, n=5; 15mg/kg of AR79, n=5; vehicle, n=5); 20 NOD/SCID mice in DU145luc injection group (50mg/kg of AR79, n=10; vehicle, n=10). A and C. Representativeimages of BLI. Note the increased signals in the the AR79-treated SQ implanted compared with vehicle-treated mice. B and D. Quantification of tumor volume as measured using BLI. * p<0.05 versus Vehicle. E. Pictures of the SQ DU145luc tumors. F. Tumor weight of DU145luc injection group.
In the basal state, GSK3 promotes phosphorylation and degradation of β-catenin. When Wnts are present, β-catenin dissociates from the GSK3 complex and is not phosphorylated, thus avoiding degradation resulting in increased β-catenin levels. AR79 mediates its activity through inhibiting GSK3 which results in decreased β-catenin phosphorylation and increased nuclear β-catenin. Consequently, we next assessed if AR79 impacted β-catenin expression in tumor tissues by measuring total and phospho-β-catenin. AR79 increased β-catenin in both C4-2B (Fig. 2A and B) and Du145 cells (Fig. 2D and E) and decreased total phospho-β-catenin expression in both C4-2B (Fig. 2A and C) and Du145 (Fig. 2D and F) cells. We evaluated additional PCa cell lines (PC-3, ARCaP and LNCaP) and in all instances, AR79 reduced phospho-β-catenin and increased total β-catenin (Fig. S2).
Figure 2.
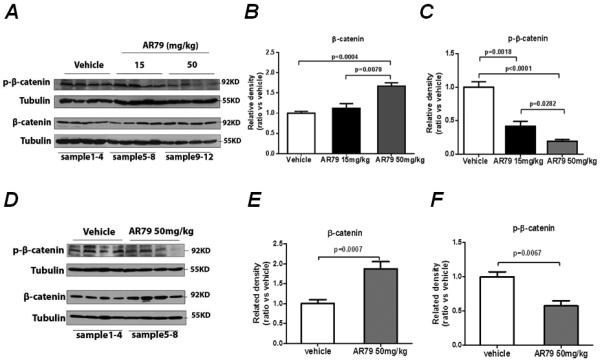
AR79 down-regulates phosphorylation of β-catenin and up-regulates total β-catenin in soft tissue prostate cancer tumors. Tumor tissues from the mice described in Figure 1 were harvested, total protein extracted and subjected to western blot for phospho-β-catenin and total β-catenin. A. C4-2Bluc western blot with 4 different samples for each treatment shown. B Phospo-β-catenin and C total β-catenin band densities from the C4-2Bluc cells were quantified using Image J software. Results were normalized to tubulin band density. D. Du145luc western blot with 4 different samples for each treatment shown. E Phospo-β-catenin and F total β-catenin band densities from the Du145luc cells were quantified using Image J software. Results were normalized to tubulin band density.
In addition to impacting Wnt signaling, in vitro chemical pharmacology screening studies with AR79 have shown that this compound also inhibits homeodomain-interacting protein kinase 2 (HIPK2) with equipotency compared with GSK3β (11). To determine if AR79 impacted HIPK2 in PCa, we exposed multiple PCa cell lines to AR79 and measured changes in phospho-HIPK2 expression. AR79 had no impact on phospho-HIPK2 expression, while it decreased GSK3 and phospho-β-catenin and increased total β-catenin (Fig. S2).
To determine if the observed effects of Wnt activation on tumor in soft tissue also extended to bone, the most common site of PCa metastases, we injected C4-2B cells into the tibiae of mice and treated the mice with either vehicle or AR79. Similar to tumors in soft tissue sites, AR79 at both 15mg/kg and 50mg/kg induced C4-2B tumor growth in bone (Fig. 3A and B). The increased tumor growth was accompanied by increased tumor-induced radiographic changes in the tibiae (Fig. 3C). AR79 in non-tumor bearing tibiae had no impact on radiographic appearance of the tibiae (Fig. S3A). To better define the impact of AR79 on tumor-induced bone changes, we subjected the tibiae to dual energy x-ray absorptiometry (DEXA) scan to measure bone mineral changes. Administration of AR79 had no impact on bone mineral density (BMD) but was associated with decreased bone mineral content (BMC) in the tibiae containing tumors (Fig. 3D and E) indicating an overall loss of bone. In contrast, AR79 (50 mg/kg) induced an increase in both BMD and BMC in tibiae without tumor (Fig S3B and C) indicating an increase in bone mineral within the bone itself. The AR79-induced tumor-mediated bone loss was associated with an overall increase of serum TRACP 5b, a marker of osteoclast activity (Fig. 3F) and no impact on serum osteocalcin, a marker of bone turnover (Fig 3G).
Figure 3.
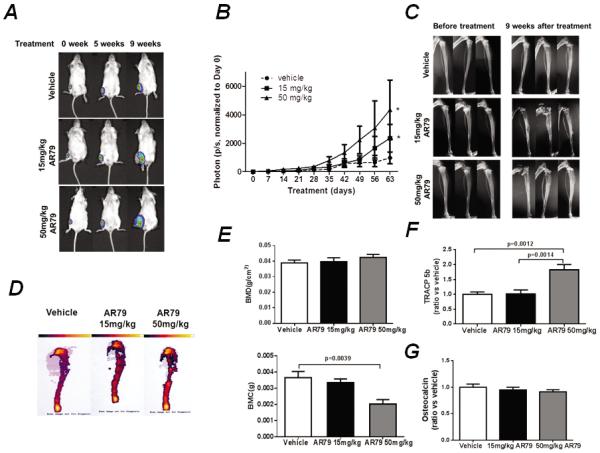
AR79 promotes the development of prostate cancer cell growth in bone. C4-2Bluc cells (5×105, in 20μl T medium) were injected into the tibiae of NOD/SCID mice (n=40). After establishment of tumors, treatment with either AR79 (50mg/kg of AR79, n=14; 15mg/kg of AR79, n=14) or vehicle (n=12) was initiated and continued for 9 weeks. Mice were euthanized, bones were subjected to Faxitron X-ray analysis and dual energy x-ray absorptiometry (DEXA), blood was collected and separated into serum that was subjected to bone marker analysis (TRAP 5b and osteocalcin). A. Representative BLI pictures. Note the increased signals in the tibiae of the AR79-treated mice compared with vehicle-treated mice. B. Total photons were measured using BLI weekly after tumor injection to monitor tumor development. * p<0.05 AR79 vs Vehicle, P value was determined by t test. C. Microradiographs revealed increased presence of tumor-induced bone changes in the tibiae from mice treated with AR79. D. Representative DEXA scans. Note decreased bone mineral in the AR79-treated mice. E. Bone mineral density (BMD) and bone mineral content (BMC) of tibiae with tumors. F Serum TRACP 5b, and G. serum osteocalcin were measured using ELISA.
To explore cellular mechanisms that could account for AR79-mediated promotion of tumor growth we assessed the tumor tissues for expression of Ki67, a marker of cellular proliferation; caspase 3, a marker of apoptosis; and nuclear β-catenin. In both the C4-2B and Du145 subcutaneous tumors and the C4-2B intratibial tumors administration of AR79 increased Ki67 and decreased caspase 3 indicating that it overall promoted tumor growth through both increased proliferation and decreased apoptosis (Fig 4). Additionally, AR79 induced nuclear β-catenin expression in all of these tumors indicating that the Wnt pathway was activated (Fig. 4).
Figure 4.
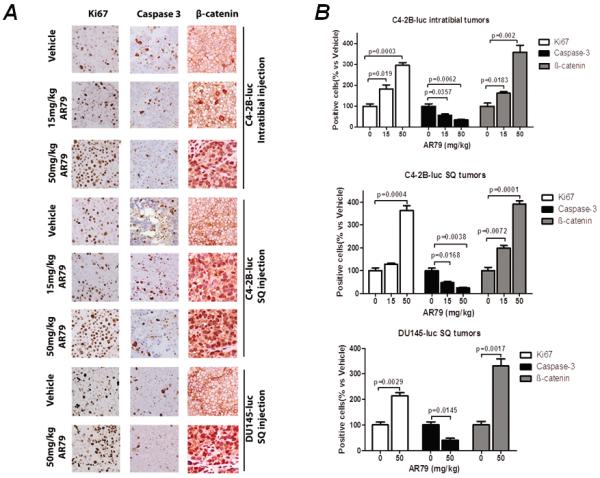
AR79 increases Ki67, decreases caspase 3 and induces nuclear beta-catenin expression in C4-2B and DU145 subcutaneous tumors and C4-2B intratibial tumors. A. Immunhistochemical analysis of paraffin sections from the SQ and intratibial tumors was performed using antibodies to Ki67, caspase 3 and β-catenin. B. Positive staining cells were counted in 4 random 40X fields from each section. Results are reported as percent of positive cells compared to control.
The in vivo results suggested that AR79 stimulated proliferation of PCa cells. To determine if there was a direct effect of AR79 on PCa or if bone stroma was required for the proliferative effect we exposed PCa cells alone or in co-culture with ST-2 stromal cells. AR79 at 0.03 μ-g/ml and 0.3 μ-g/ml had no impact; whereas, at 3.0 μ-g/ml induced C4-2B cell growth nearly 100% in vitro at both 48 and 72 hours (Fig 5); but had no effect at any dose at 24 hours (not shown). The addition of ST-2 cells at ratios of 3:1 or 6:1 (ST2:C4-2B) in the culture had no impact on cell number or AR79 effects (Fig. 5). To ensure that the effect of AR79 was not specific to C4-2B cells, the study was repeated with Du145 cells. Similar to the results observed with C4-2B cells, AR79 promoted Du145 cell growth (albeit only at 0.3 μ-g/ml) after 48 and 72 hours (Fig S4), but had no impact at 24 hours (not shown). These results confirm that AR79 directly induces PCa growth.
Figure 5.
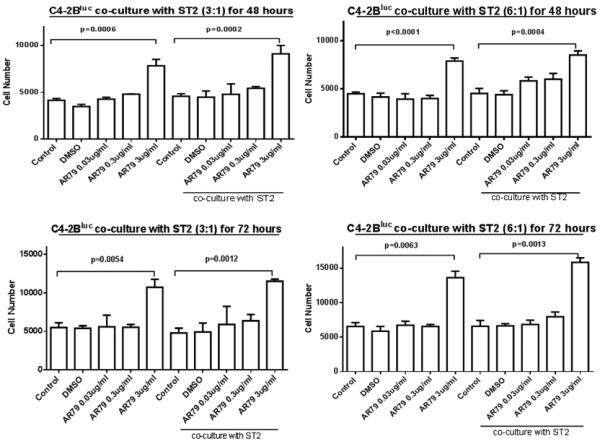
AR79 increases numbers of C4-2Bluc cells in the presence or absence of ST-2 co-culture. C4-2Bluc cells were grown alone or co-cultured with ST2 at 6:1 and 3:1. Then cells were treated with different concentrations (0.03μg/ml, 0.3μg/ml and 3μg/ml) of AR79 or vehicle (DMSO). Luciferase activity was measured after 24 (not shown), 48 and 72 hours by BLI. Data are presented as mean±SD from triplicate determinations.
We next determined if the impact of AR79 on the Wnt pathway observed in vivo extended to C4-2B PCa cells and the ST2 and MC3T3-E1 bone stromal cell lines in vitro. AR79 (3μg/ml, but not lower doses) decreased phospho-β-catenin and increased total β-catenin in all cell lines evaluated, including the colon cancer HCT116 cell line which was used as a positive control (Fig.6A). This effect was not observed until 60 minutes post-treatment and continued through at least 120 minutes post-treatment (Fig. 6B). A corresponding increase in nuclear β-catenin was observed in all cell lines evaluated which was similar to the increase observed when the cells were treated with Wnt3a as a positive control (Fig. 6C and D; see NE for nuclear extracts).
Figure 6.
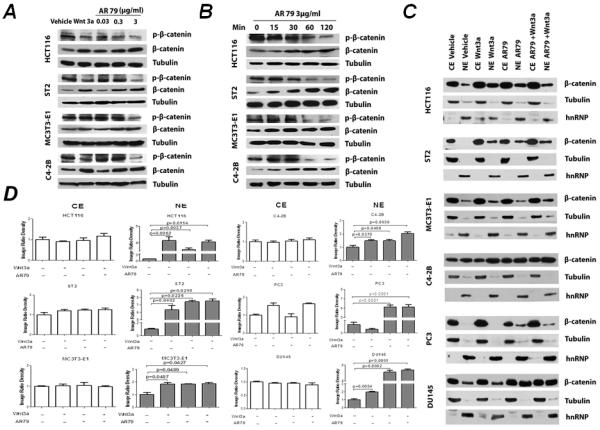
AR79 down-regulates phosphorylation of β-catenin and upregulates total β-catenin nuclear translocation in prostate cancer and bone stromal cell lines. A. Cells were treated with the indicated concentration of AR79 or Wnt3a (60ng/mL). After 3 hours, total protein was isolated and subjected to western blot analyses. B. Cells were treated with 3μg/ml AR79 for the indicated times at which time total protein was isolated and subjected to western blot. C. Cells (5×106 cells) were serum starved for 12 hours then treated with AR79 at 3μg/ml. Nuclear protein was extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents and analyzed for β-catenin bywestern blot. CE: Cytoplasmic Extract NE: Nuclear Extract D. The densities of Western blot bands were quantified with Image J software (version 1.40; National Institutes of Health, Bethesda, MD).
As kinase inhibitors can have off target effects, we further evaluated if the proliferation-inducing activity of AR79 was dependent on upregulation of β-catenin. We used two different siRNA constructs to knockdown β-catenin expression in Du145 and C4-2B cells. Both constructs mediated over >80% decreased in β-catenin protein expression in both cell lines compared to scrambled control (Fig. S5). To ensure that knockdown was functional during administration of AR79 we assessed the expression of nuclear β-catenin in the siRNA transfected cells. Administration of Wnt3a or AR79 increased nuclear β-catenin in the scrambled siRNA control cells lines compared to vehicle-treated cells; whereas, neither Wnt3a nor AR79 increased nuclear β-catenin in the β-catenin siRNA cells compared to vehicle (Supplemental Fig. 3B). These results demonstrate that knockdown of β-catenin abrogates the ability of AR79 to increase nuclear β-catenin. To next assess if blocking the ability of AR79 to induce β-catenin expression impacts its ability to promote prostate cancer proliferation, we evaluated the effect of AR79 on the cells with knockdown of β-catenin. At 24 hours, neither Wnt3a nor AR79 affected cell growth, as previously observed; however, at 48 and 72 hours, both Wnt3a and AR79 induced cell growth in the parental and scrambled siRNA cell lines and this effect was completely inhibited by knockdown of β-catenin (Fig. 7).
Figure 7.
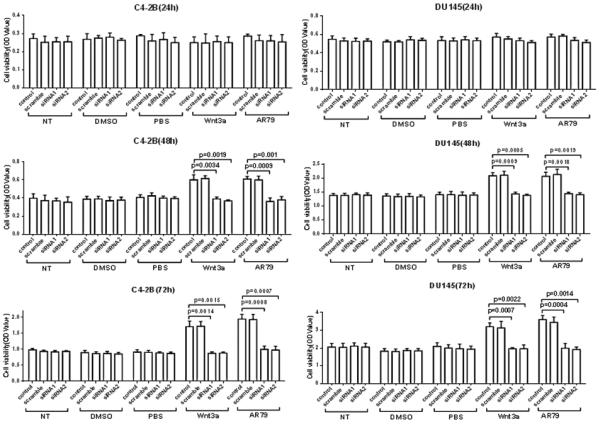
AR79 increases cell growth of DU145 and C4-2B cell lines via activating canonical Wnt signaling. DU145 and C4-2B parental cells (control) and cells transfected with scrambled or β-catenin siRNAs were grown in 96-well plates. Cells were then treated with AR79 (3μg/ml) and rhWnt3a (60ng/ml) or vehicle (DMSO or PBS) for 24hr, 48hr and 72hr. Cell numbers were measured using WST-1. Data are presented as mean±SD from triplicate determinations.
Wnts have been shown to promote maintenance of cancer stem cell-like cells (CSC) (15–17) (also called tumor initiating cells-TICs) indicating that modulation of Wnt activity may impact CSC. The definition of PCa CSC is constantly being refined; however, the presence of positive staining for aldehyde dehydrogenase (ALDH) (18, 19) and CD133 (20, 21) have been associated with CSC-like phenotype. Therefore, in order to determine if AR79 modulated stemness of the PCa cell population we assessed for the impact of AR79 on the proportion of ALDH+/CD133+ cells in the C4-2B and Du145 populations. AR79 increased the proportion of ALDH+/CD133+ cells by approximately 100% and 200% in C4-2b and Du145, respectively (Fig. 8).
Figure 8.
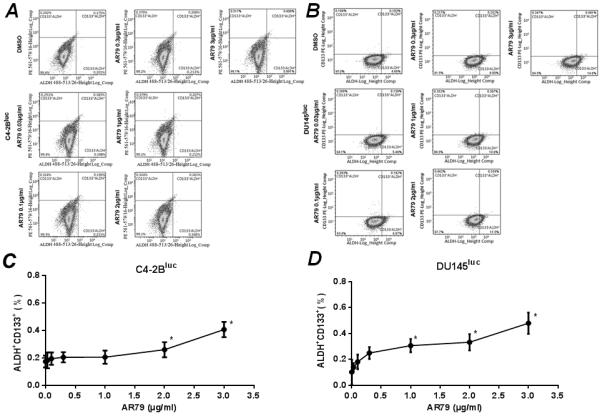
AR79 increases the proportion of ALDH+CD133+ cells in C4-2Bluc and DU145luc cell lines. C4-2Bluc and DU145luc (5×106) were seeded into 10mm plates and treated with indicated concentrations of AR79 or vehicle (DMSO) for 48 hours. Cells were dissociated with Accutase and flow cytometric analysis was performed for ALDH and CD133. A. Flow cytometric analysis showing ALDH and CD133 expression in C4-2Bluc. B. Flow cytometric analysis showing ALDH and CD133 expression in DU145luc. C and D. Proportion of ALDH+;CD133+ in C4-2Bluc and DU145luc, respectively. Data are shown as mean±SD from two independent experiments. *P<0.05 versus vehicle.
Discussion
Induction of bone anabolic activity is a major therapeutic goal to diminish bone mineral loss. Towards that end, systemic administration of PTH (22, 23) and local administration of bone morphogenetic proteins (BMPs) with bone implants (24) have been used for their bone anabolic properties. However, because these therapies have limitations including label warnings of cancer, there is a need to identify additional bone anabolic agents. In this light, manipulation of the Wnt pathway is being explored as a potential therapy due to the ability of Wnts to induce bone formation.
Wnt signaling plays a central role in osteoblast development and bone formation (25–27). Wnts promote the lineage commitment of mesenchymal precursor cells and the differentiation of progenitor cell lines into osteoblasts. They also stimulate direct effects on the formation and turnover of the mature skeleton. Mutations in components of the Wnt pathway are associated with human skeletal diseases and variation of bone mineral density (28–31), demonstrating the functional relevance of Wnt pathway for human bone biology.
Canonical Wnt signaling is mediated through increasing nuclear β-catenin levels (3). GSK3β is a key regulator of Wnt signaling through its ability to phosphorylate and target β-catenin for ubiquitination and degradation. Thus, inhibition of GSK3β is considered to be a viable and promising strategy to increase Wnt pathway activity through promotion of nuclear β-catenin expression. In agreement with the role of Wnts on bone production and inhibition of GSK3β on Wnt signaling, an orally available GSK3β inhibitor was shown to increase bone mass in rats (32). AR79 was developed in order to block GSK3β activity and hence induce bone anabolic activity as demonstrated by the stimulation of osteoblastogenesis with AR79 in vitro and a bone anabolic phenotype in rodents in vivo (11). However, prior to clinical evaluation of its bone anabolic potential, it was deemed important to determine its impact on PCa, which is known to be influenced by Wnt signaling (33, 34).
Wnts may play a role in promotion of PCa progression through several mechanisms. For example, Wnts, through β-catenin activation have been shown to influence expression of the androgen receptor, a key regulator of PCa (35). Furthermore, GSK3β has been shown to regulate the progression of PCa to the advanced castration resistant form (36). Additionally, it has been suggested that activation of GSK3β may inhibit the proliferative effects of Wnt and β-catenin in PCa (36). We have observed that as PCa progresses to the bone metastatic phenotype, expression of the endogenous Wnt inhibitor, dickkopf-1 (DKK1) is decreased, allowing for enhanced Wnt activity (37). This in turn can account for the osteoblastic phenotype of PCa as demonstrated by the observation that the PCa mediates bone production through activation of the Wnt receptor LRP5 (38). Taken together, these findings indicate that any therapy that modulates Wnt signaling could have an impact on PCa progression.
Our results clearly demonstrate that AR79 promoted PCa growth in soft tissue and bone in a murine model of PCa. Furthermore, the increase of PCa growth was associated with decreased phospho-β-catenin expression and increased total β-catenin expression. Additionally, in vitro exposure of PCa cells to AR79 decreased phospho-β-catenin and increased nuclear β-catenin providing further evidence that AR79 effectively activates the Wnt pathway. Taken together, these results suggest that AR79 promotes PCa growth through activation of the canonical Wnt pathway due to its ability to block GSK3β activity. Our findings appear to conflict with an earlier report in which the GSK-3 inhibitor CHIR99021 decreased growth of the 22Rv1 human PCa cell line in vitro (39). However, differences in cell lines and compounds could account for differences. Additionally, that report did not evaluate the in vivo impact of CHIR99021 which is critical for a thorough assessment due to the importance of microenvironment on cancer growth.
To determine cellular mechanisms through which AR79 promoted PCa growth we evaluated Ki67expression, a marker of proliferation, and caspase 3/7, markers of apoptosis. That AR79 administration was associated with both an increase of Ki67 and a decrease of caspase 3/7 expression in the tumor tissue indicated that it promoted overall tumor growth through both induction of cellular proliferation and inhibition of apoptosis. This finding is consistent with activation of the Wnt pathway as Wnts are known to induce proliferation and inhibit apoptosis in a variety of cell systems (40). It should be pointed out that we measured these parameters only at the time of sacrifice, thus this may not have been the optimal to time to see the maximal changes. To further identify whetherAR79 modified PCa cell proliferation and apoptosis directly; we evaluated its impact on PCa cells in vitro. However, we only identified an increase in cell proliferation, and no impact on apoptosis. This finding suggests that AR79 modulate apoptosis in vivo through an indirect mechanism in the tumors. However, it may be that the basal apoptosis rate in a two-dimensional culture in vitro, which is well oxygenated, is much lower than that in a three-dimensional tumor in vivo, which will have areas of hypoxia and presumably more endogenous apoptosis. Thus, it is possible we could not identify an anti-apoptotic effect in vitro due to differences in the growth environment of the PCa cells.
Since in vitro pharmacology studies with AR79 indicated that this compound also inhibits homeodomain-interacting protein kinase 2 (HIPK2) with equipotency compared with GSK3β (11) we wanted to determine if off target effects of AR79 on HIPK2 could account for the impact on growth. HIPK2 is known to act as a tumor suppressor through activation of p53 which both (1) mediates induction of apoptosis (41) and (2) inhibits the wnt/β-catenin pathway through miRNA-34 (42). Therefore, HIPK2 may have contributed to some of the responses observed in PCa studies with AR79. However, our results indicated that AR79 had no impact on HIPK2 activation in PCa cells. These data indicate that the response observed was not due to AR79 activity on HIPK2. Furthermore, the studies demonstrating that AR79 lost its ability to induce prostate cancer cell growth in vitro upon knockdown of β-catenin provides strong evidence that AR79 mediates its proliferative effect through activation of canonical Wnt signaling. The mechanism through which Wnt pathway activation mediates prostate cancer cell growth is not clear at this time; however, it has been previously demonstrated that there is crosstalk between the Wnt pathway and the androgen receptor axis in prostate cancer (43, 44) which could contribute to the growth response.
In addition to a pro-proliferative affect, AR79 increased the proportion of cancer stem cell-like cells in two of the PCa cell lines. This is consistent with the ability of Wnts to promote normal and cancer stem cell self-renewal and pluripotency (15–17, 45). This finding underscores that therapies which promote Wnt signaling could result in enhancing cancer stem cell-like activity. As cancer stem cells are considered gain resistance to immunosurveillance (46), gain resistance to apoptosis and chemotherapy (47, 48) and have diminished induction of senescence (49) they may establish a nidus of cancer recurrence. Thus, therapies that result in overall promotion of Wnt pathway activity should be carefully considered prior to their use. A caveat to this concern is that it is not absolutely clear that the ALDH+/CD133+ cell population that was increased are definitively cancer stem cells. Although there are reports that indicate that cells with these markers contribute to the cancer stem cell-like cell population (18–21), there are also indications that these markers may indicate the ability for these cells to have a greater proliferative potential as opposed to clear stem cell properties (50, 51). Even if these markers represent more of a pro-proliferative capability as opposed to a clear cancer stem cell-like phenotype, the ability of AR79 to increase this component of the population would still be a major concern as they could contribute to increased tumor growth.
A key reason for development of AR79 was to promote bone anabolic activity through activation of the canonical Wnt pathway. Along these lines, AR79 increased both BMD and BMC in normal bone indicating an overall increase in bone production. Furthermore, AR79 activated the Wnt signaling pathway in osteoblast-like cells based on its ability to decrease phospho-β-catenin and increase total and nuclear β-catenin. Taken together, these results indicate that AR79 is an effective bone anabolic agent. However, in apparent contrast to its ability to induce bone production, administration of AR79 decreased BMC in the bone containing tumor. The likely reason for this is that is that as AR79 induced PCa cell growth, tumor-associated bone destruction occurred. This was evident on radiographs and paralleled by the measureable decrease in BMC.
A limitation of these experiments was the use of subcutaneous injection to model soft tissue growth. Ideally orthotopic injection may have provided a model that more closely recapitulates human PCa than subcutaneous injection. A challenge to orthotopic injection in the murine model is the presence of four biologically different and anatomically separated lobes of the prostate. Another limitation to this model is that the cell lines used do not completely recapitulate the osteoblastic bone response that is typical of clinical PCa. Specifically, C4-2B cells induce mixed osteoblastic/osteolytic lesions and Du145 cells produce osteolytic lesions. It is unknown at this time if the type of bone response induced by PCa could influence the activity of AR79.
In summary, this research identified that inhibition of GSK3β using AR79 had a bone anabolic effect, but also stimulated PCa growth in vivo a murine model. These findings suggest that use of therapeutic agents that modulate the Wnt pathway should be used with caution, particularly in the presence of PCa. It is likely these effects would extend to other cancers as Wnts play a role in multiple cancers.
Supplementary Material
Acknowledgments
Grant support: This work was supported by the National Institutes of Health Grant P01 CA093900 and funding by AstraZeneca.
Conflicts of interest: Karen J. Escott and Hitesh J. Sanganee are employed as AstraZeneca. Evan T. Keller received research funding from AstraZeneca or the studies in this report.
Footnotes
Implications: These data suggest that clinical application of pharmaceuticals that promote Wnt pathway activation should be used with caution as they may enhance tumor growth.
References
- 1.Sibai T, Morgan EF, Einhorn TA. Anabolic agents and bone quality. Clin Orthop Relat Res. 2011;469:2215–24. doi: 10.1007/s11999-010-1722-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–9. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 4.Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. 2012;33:747–83. doi: 10.1210/er.2011-1060. [DOI] [PubMed] [Google Scholar]
- 5.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 6.Camilli TC, Weeraratna AT. Striking the target in Wnt-y conditions: intervening in Wnt signaling during cancer progression. Biochem Pharmacol. 2010;80:702–11. doi: 10.1016/j.bcp.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao H, Ashihara E, Maekawa T. Targeting the Wnt/beta-catenin signaling pathway in human cancers. Expert Opin Ther Targets. 2011;15:873–87. doi: 10.1517/14728222.2011.577418. [DOI] [PubMed] [Google Scholar]
- 8.Shah GV, Muralidharan A, Gokulgandhi M, Soan K, Thomas S. Cadherin switching and activation of beta-catenin signaling underlie proinvasive actions of calcitonin-calcitonin receptor axis in prostate cancer. J Biol Chem. 2009;284:1018–30. doi: 10.1074/jbc.M807823200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu D, Carson DA. Inhibition of Wnt signaling and cancer stem cells. Oncotarget. 2011;2:587. doi: 10.18632/oncotarget.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall CL, Bafico A, Dai J, Aaronson SA, Keller ET. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 2005;65:7554–60. doi: 10.1158/0008-5472.CAN-05-1317. [DOI] [PubMed] [Google Scholar]
- 11.Gilmour PS, O'Shea PJ, Fagura M, Pilling JE, Sanganee H, Wada H, et al. Human stem cell osteoblastogenesis mediated by novel glycogen synthase kinase 3 inhibitors induces bone formation and a unique bone turnover biomarker profile in rats. Toxicol Appl Pharmacol. 2013 doi: 10.1016/j.taap.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Comstock KE, Hall CL, Daignault S, Mandlebaum SA, Yu C, Keller ET. A bioluminescent orthotopic mouse model of human osteosarcoma that allows sensitive and rapid evaluation of new therapeutic agents In vivo. In Vivo. 2009;23:661–8. [PubMed] [Google Scholar]
- 13.Dai J, Keller J, Zhang J, Lu Y, Yao Z, Keller ET. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. 2005;65:8274–85. doi: 10.1158/0008-5472.CAN-05-1891. [DOI] [PubMed] [Google Scholar]
- 14.Dai J, Lu Y, Yu C, Keller JM, Mizokami A, Zhang J, et al. Reversal of chemotherapy-induced leukopenia using granulocyte macrophage colony-stimulating factor promotes bone metastasis that can be blocked with osteoclast inhibitors. Cancer research. 2010;70:5014–23. doi: 10.1158/0008-5472.CAN-10-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curtin JC, Lorenzi MV. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget. 2010;1:563–77. doi: 10.18632/oncotarget.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–8. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Wend P, Holland JD, Ziebold U, Birchmeier W. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol. 2010;21:855–63. doi: 10.1016/j.semcdb.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Doherty RE, Haywood-Small SL, Sisley K, Cross NA. Aldehyde dehydrogenase activity selects for the holoclone phenotype in prostate cancer cells. Biochem Biophys Res Commun. 2011;414:801–7. doi: 10.1016/j.bbrc.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 19.van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010;70:5163–73. doi: 10.1158/0008-5472.CAN-09-3806. [DOI] [PubMed] [Google Scholar]
- 20.Castellon EA, Valenzuela R, Lillo J, Castillo V, Contreras HR, Gallegos I, et al. Molecular signature of cancer stem cells isolated from prostate carcinoma and expression of stem markers in different Gleason grades and metastasis. Biol Res. 2012;45:297–305. doi: 10.4067/S0716-97602012000300011. [DOI] [PubMed] [Google Scholar]
- 21.Trerotola M, Rathore S, Goel HL, Li J, Alberti S, Piantelli M, et al. CD133, Trop-2 and alpha2beta1 integrin surface receptors as markers of putative human prostate cancer stem cells. Am J Transl Res. 2010;2:135–44. [PMC free article] [PubMed] [Google Scholar]
- 22.Miao D, He B, Jiang Y, Kobayashi T, Soroceanu MA, Zhao J, et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J Clin Invest. 2005;115:2402–11. doi: 10.1172/JCI24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettway GJ, Meganck JA, Koh AJ, Keller ET, Goldstein SA, McCauley LK. Parathyroid hormone mediates bone growth through the regulation of osteoblast proliferation and differentiation. Bone. 2008;42:806–18. doi: 10.1016/j.bone.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Even J, Eskander M, Kang J. Bone morphogenetic protein in spine surgery: current and future uses. J Am Acad Orthop Surg. 2012;20:547–52. doi: 10.5435/JAAOS-20-09-547. [DOI] [PubMed] [Google Scholar]
- 25.Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19–39. doi: 10.1016/j.gene.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 26.Baron R, Rawadi G, Roman-Roman S. Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol. 2006;76:103–27. doi: 10.1016/S0070-2153(06)76004-5. [DOI] [PubMed] [Google Scholar]
- 27.Mason JJ, Williams BO. SOST and DKK: Antagonists of LRP Family Signaling as Targets for Treating Bone Disease. J Osteoporos. 2010;2010 doi: 10.4061/2010/460120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–21. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 29.Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–35. doi: 10.1101/gr.3437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, et al. New sequence variants associated with bone mineral density. Nat Genet. 2009;41:15–7. doi: 10.1038/ng.284. [DOI] [PubMed] [Google Scholar]
- 31.Kumar J, Swanberg M, McGuigan F, Callreus M, Gerdhem P, Akesson K. LRP4 association to bone properties and fracture and interaction with genes in the Wnt- and BMP signaling pathways. Bone. 2011;49:343–8. doi: 10.1016/j.bone.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 32.Kulkarni NH, Onyia JE, Zeng Q, Tian X, Liu M, Halladay DL, et al. Orally bioavailable GSK-3alpha/beta dual inhibitor increases markers of cellular differentiation in vitro and bone mass in vivo. J Bone Miner Res. 2006;21:910–20. doi: 10.1359/jbmr.060316. [DOI] [PubMed] [Google Scholar]
- 33.Robinson DR, Zylstra CR, Williams BO. Wnt signaling and prostate cancer. Curr Drug Targets. 2008;9:571–80. doi: 10.2174/138945008784911831. [DOI] [PubMed] [Google Scholar]
- 34.Hall CL, Kang S, MacDougald OA, Keller ET. Role of Wnts in prostate cancer bone metastases. J Cell Biochem. 2006;97:661–72. doi: 10.1002/jcb.20735. [DOI] [PubMed] [Google Scholar]
- 35.Yang X, Chen MW, Terry S, Vacherot F, Bemis DL, Capodice J, et al. Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene. 2006 doi: 10.1038/sj.onc.1209366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3beta: key regulators of progression to androgen-independent prostate cancer. Oncogene. 2006;25:329–37. doi: 10.1038/sj.onc.1209020. [DOI] [PubMed] [Google Scholar]
- 37.Hall CL, Daignault SD, Shah RB, Pienta KJ, Keller ET. Dickkopf-1 expression increases early in prostate cancer development and decreases during progression from primary tumor to metastasis. Prostate. 2008;68:1396–404. doi: 10.1002/pros.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li ZG, Yang J, Vazquez ES, Rose D, Vakar-Lopez F, Mathew P, et al. Low-density lipoprotein receptor-related protein 5 (LRP5) mediates the prostate cancer-induced formation of new bone. Oncogene. 2008;27:596–603. doi: 10.1038/sj.onc.1210694. [DOI] [PubMed] [Google Scholar]
- 39.Darrington RS, Campa VM, Walker MM, Bengoa-Vergniory N, Gorrono-Etxebarria I, Uysal-Onganer P, et al. Distinct expression and activity of GSK-3alpha and GSK-3beta in prostate cancer. Int J Cancer. 2012;131:E872–83. doi: 10.1002/ijc.27620. [DOI] [PubMed] [Google Scholar]
- 40.Pecina-Slaus N. Wnt signal transduction pathway and apoptosis: a review. Cancer Cell Int. 2010;10:22. doi: 10.1186/1475-2867-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puca R, Nardinocchi L, Givol D, D'Orazi G. Regulation of p53 activity by HIPK2: molecular mechanisms and therapeutical implications in human cancer cells. Oncogene. 2010;29:4378–87. doi: 10.1038/onc.2010.183. [DOI] [PubMed] [Google Scholar]
- 42.Cha YH, Kim NH, Park C, Lee I, Kim HS, Yook JI. MiRNA-34 intrinsically links p53 tumor suppressor and Wnt signaling. Cell Cycle. 2012;11:1273–81. doi: 10.4161/cc.19618. [DOI] [PubMed] [Google Scholar]
- 43.Schweizer L, Rizzo CA, Spires TE, Platero JS, Wu Q, Lin TA, et al. The androgen receptor can signal through Wnt/beta-Catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol. 2008;9:4. doi: 10.1186/1471-2121-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verras M, Brown J, Li X, Nusse R, Sun Z. Wnt3a growth factor induces androgen receptor-mediated transcription and enhances cell growth in human prostate cancer cells. Cancer Res. 2004;64:8860–6. doi: 10.1158/0008-5472.CAN-04-2370. [DOI] [PubMed] [Google Scholar]
- 45.Kuhl SJ, Kuhl M. On the role of Wnt/beta-catenin signaling in stem cells. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbagen.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 46.Qi Y, Li RM, Kong FM, Li H, Yu JP, Ren XB. How do tumor stem cells actively escape from host immunosurveillance? Biochem Biophys Res Commun. 2012;420:699–703. doi: 10.1016/j.bbrc.2012.03.086. [DOI] [PubMed] [Google Scholar]
- 47.Crea F, Danesi R, Farrar WL. Cancer stem cell epigenetics and chemoresistance. Epigenomics. 2009;1:63–79. doi: 10.2217/epi.09.4. [DOI] [PubMed] [Google Scholar]
- 48.Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012;30:372–8. doi: 10.1002/stem.1035. [DOI] [PubMed] [Google Scholar]
- 49.Rajaraman R, Guernsey DL, Rajaraman MM, Rajaraman SR. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006;6:25. doi: 10.1186/1475-2867-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reyes EE, Kunovac SK, Duggan R, Kregel S, Griend DJ. Growth kinetics of CD133-positive prostate cancer cells. Prostate. 2012 doi: 10.1002/pros.22616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu C, Yao Z, Dai J, Zhang H, Escara-Wilke J, Zhang X, et al. ALDH Activity Indicates Increased Tumorigenic Cells, But Not Cancer Stem Cells, in Prostate Cancer Cell Lines. In Vivo. 2011;25:69–76. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.