

Summary
Eosinophilia is a hallmark characteristic of TH2-associated diseases and is critically regulated by the central eosinophil growth factor interleukin 5 (IL-5). Here we demonstrate that IL-5 activity in eosinophils was regulated by paired immunoglobulin-like receptor (PIR)-A and PIR-B. Upon self-recognition of β2M molecules, PIR-B served as a permissive checkpoint for IL-5-induced eosinophil development by suppressing the pro-apoptotic activities of PIR-A, which were mediated by the Grb2-Erk-Bim pathway. PIR-B-deficient bone marrow (BM) eosinophils underwent compartmentalized apoptosis, resulting in decreased blood eosinophilia in naïve, IL-5- and aeroallergen-challenged mice. Subsequently, Pirb−/− mice displayed impaired aeroallergen-induced lung eosinophilia and induction of lung TH2 responses. Collectively, these data uncovers an intrinsic, self-limiting pathway regulating IL-5-induced eosinophil expansion, which has broad implications for eosinophil-associated diseases.
Eosinophils are bone marrow (BM)-derived myeloid cells that differentiate under the control of the transcription factors GATA-1, PU.1, c/EBP, and the common β chain (βc)-signaling cytokines interleukin 5 (IL-5), IL-3 and granulocyte macrophage-colony stimulating factor (GM-CSF)1. IL-5 is the most potent and specific cytokine for the eosinophil lineage and is responsible for cellular expansion2, release from the bone marrow (BM) into the peripheral blood (PB)3, and survival4 following a variety of triggers, typically TH2 stimuli. At baseline, eosinophils mainly reside in the gastrointestinal tract, fat pads, spleen, lymph nodes and thymus5, 6 where they function to maintain homeostasis6-8. Yet, in settings of allergic inflammation such as those found in asthma, eosinophils expand in the BM and infiltrate the lung where their accumulation is a characteristic disease hallmark5, 7. While the extrinsic pathways (e.g. IL-5) that regulate eosinophil expansion are relatively well characterized, the presence of intrinsic, self-limiting, molecular checkpoints regulating IL-5-induced eosinophilia is largely unknown and remains to be defined.
A major checkpoint in curtailing cytotoxic T cells and natural killer (NK) cells is inhibitory signaling driven by major histocompatibility complex (MHC) restricted self-recognition, mediated by receptors that are capable of recognizing self via MHC-I-binding. The T cell receptor (TCR) enables positive and negative selection, which is critical for adaptive immunity while avoiding autoreactivity9. NK cell inhibitory receptors (KIR) govern cytotoxicity towards virally infected or transformed cells10.
An additional receptor system that mediates self-recognition by binding to classical and non-classical MHC-I molecules has drawn considerable less attention. This receptor system is comprised of the paired immunoglobulin-like receptors (PIR)-A and PIR-B11. PIRs are predominantly expressed by myeloid cells endowing them with the potential to discriminate self from non-self12. Comprehensive structural and biochemical analyses have shown that the amino acid sequences of the PIR-A and PIR-B ectodomains are over 92% identical and bind the same MHC-I ligands12,13. The deduced structure of PIR-B is a type I transmembrane glycoprotein with six extracellular immunoglobulin-like domains, a hydrophobic transmembrane segment, and an intracellular polypeptide with four immunoreceptor tyrosine-based inhibitory motifs (ITIM) or ITIM-like sequences12. In contrast, PIR-A lacks the extended intracellular motif and requires assistance of adaptor molecules, such as the FcRγ chain for efficient expression and function12, 14. Subsequent binding of MHC-I molecules by PIRs leads to the recruitment of cytosolic phosphatases by PIR-B, which ultimately results in inhibition of yet undefined signaling cascades triggered by PIR-A14, 15. Of specific interest, PIR-B is capable of suppressing cellular responses elicited by the βc-signaling cytokines, IL-3 and GM-CSF16, 17 as well as by Flt-3L18, thus suggesting a role for PIR-B in myeloid cell hematopoiesis. The recent demonstration that eosinophils express PIRs19 together with the suggested hematopoietic functions of PIR-B raised the hypothesis that PIR-B may regulate IL-5-induced responses in eosinophils.
In this study, we demonstrate a critical role for PIR-B and PIR-A in eosinophil development. PIR-B suppressed PIR-A-induced eosinophil apoptosis, thereby counterbalancing survival and growth signals selectively driven by IL-5. PIR expression was regulated by IL-5 and increased during distinct eosinophil maturation stages. Eosinophils maintained a dominant expression of PIR-B over PIR-A under homeostatic conditions. Thus, despite abundant MHC-I ligand availability, which may trigger PIR-A-induced apoptosis, eosinophil development was intact due to inhibitory signals driven by PIR-B. Furthermore, we demonstrate that PIR-A bound the adaptor protein Grb2 and that apoptotic eosinophils displayed increased Erk activation and Bim expression. These data open a new paradigm in our understanding of the intrinsic molecular pathways regulating IL-5-induced eosinophilia and highlight self-recognition via PIRs as a key molecular checkpoint in eosinophil expansion.
Results
PIR-B is required for eosinophil differentiation in vitro
To begin to assess the function of PIR-B in eosinophils, we used the recently described BM-derived eosinophil cell culture system20. Cultures obtained from the low-density (LD) fraction of wild-type BM displayed by day fourteen >95% eosinophils of uniform morphological appearance, including stereotypical bilobed nuclei (Fig. 1a, day 14). In contrast, starting from day 10, cultures of Pirb−/− LDBM cells showed the appearance of myeloid morphology (represented by the black arrows) and by day 14, the entire Pirb−/− culture was comprised of large often-multinucleated cells lacking eosinophil morphology (represented by the dotted arrows, Fig 1a). Total cell counts that were obtained from the Pirb−/− LDBM cultures were lower than those obtained from wild-type cultures (Fig. 1b). At day 14, cells retrieved from wild-type but not Pirb−/− cultures expressed CCR3 (Fig. 1c) and major basic protein (MBP) (Fig. 1d) both of which are classical eosinophil markers. Moreover, Pirb−/− cells exhibited distinct physical parameters (i.e. granularity and size, Fig. 1e). Furthermore, Pirb−/− cells expressed increased expression of myeloid-associated adhesion molecules and activation markers including CD11b, CD18, α5 integrin, CD103, CD62L, and CD69 (Supplementary Fig. 1). LDBM culture-derived Pirb−/− cells displayed markedly decreased mRNA encoding the eosinophil-associated transcription factors GATA-1 and GATA-2 (Fig. 1f,g), though c/EBP, PU.1 and Fog-1 expression were comparable to wild-type controls (data not shown). By the end of the cell cultures, total eosinophil counts in Pirb−/− LDBM cultures were reduced by 97.3 ± 0.06% as compared to wild-type cultures (Fig. 1h). Decreased eosinophil generation was not due to alterations in the numbers of eosinophil progenitors (EoPs) (defined as Sca1−CD34+Lin−c-kitintIL-5Rα+, Fig. 1i,j)21 or alteration in IL-5Rα surface expression in EoPs (Fig. 1k). The inability to generate mature eosinophils in vitro was an intrinsic defect of Pirb−/− cells since growing Pirb−/− cells in supernatants obtained from wild-type LDBM-derived eosinophil cultures did not render the Pirb−/− cells into eosinophils. Moreover, the generation of eosinophils was not impaired when cultured in supernatants harvested from Pirb−/− cultures (data not shown). Importantly, the requirement of PIR-B in eosinophil development was PIR-B and eosinophil specific since we were able to generate mature eosinophils from LDBM cells of other ITIM-bearing receptor deficient mice (e.g. CMRF35-like molecule (CLM)-122) and Pirb−/− BM yielded normal numbers of macrophages, dendritic cells (DCs), neutrophils and mast cells in the respective cytokine-driven cultures (data not shown and16, 23, 24). Collectively, these data indicate that PIR-B regulates an intrinsic pathway, which is specifically required for eosinophilopoiesis.
Figure 1. Pirb−/− low-density bone marrow-derived cells fail to differentiate into mature eosinophils in vitro.

Low-density bone marrow cells were obtained from wild-type (WT) and Pirb−/− mice and differentiated in vitro into eosinophils. Representative photomicrograph images at the indicated time points of stained cytospins (a) and day 14 total cell counts (b) are shown. Filled and broken black arrows (a) indicate single nucleated and multi-nucleated myeloid cells, respectively. CCR3 surface expression (c), eosinophil major basic protein (Mbp, d), cell size and granularity (e), Gata1 (f) and Gata2 (g) expression were assessed in LDBM WT and Pirb−/− cells. All qPCR analyses were normalized to the house keeping gene hypoxanthine-guanine phosphoribosyltransferase (Hprt). In (h), total eosinophil numbers as determined by CCR3+Siglec-F+ cells is depicted. Assessment of CD45+CD34+Lin−Sca-1−C-KitintIL-5Rα+ eosinophil progenitors (EoPs) (i-j) and IL-5 receptor (R) α expression in EoPs (k) is shown. ns-non significant, *P < 0.05, **P < 0.01, ***P < 0.001. Data are representative of at least five independent experiments conducted in triplicates (a-e); mean and s.e.m. of cultures obtained from three (f-h) or five (j-k) mice per group.
PIR-B regulates eosinophil apoptosis
Impaired eosinophil development in Pirb−/− LDBM cell cultures could be either due to a decreased proliferative ability or to increased apoptosis. Assessment of cellular proliferation in response to a combination SCF plus Flt-3L during the first two days of LDBM cell culture revealed no difference between the proliferative activity of wild-type and Pirb−/− EoPs (Supplementary Fig. 2a). Due to the loss of the distinct EoP cell surface markers (i.e. C-Kit, IL-5Rα) after 1-2 days in culture, we subsequently assessed cell proliferation in the general LDBM cultures and observed no proliferative difference between wild-type and Pirb−/− cells (Supplementary Fig. 2b).
Morphological assessment of Pirb−/− LDBM cell cultures revealed the presence of apoptotic eosinophils as identified by nuclear fragmentation and cell blebbing (Fig. 2a). Moreover, apoptotic eosinophils and/or apoptotic bodies in Pirb−/− cultures were observed to be engulfed by the abundant mononuclear myeloid cells in these cultures (Fig. 2b). In accordance with these findings, assessment of early and late apoptosis using Annexin-V staining combined with propidium iodide (PI), revealed that Pirb−/− LDBM cell cultures contained a prominent fraction of Annexin-V+PI− cells (Fig. 2c,d). Co-staining for Siglec-F, an early eosinophil development surface marker25 and Annexin-V demonstrated that starting at day 10 of the culture, nearly 20% of the Pirb−/− LDBM cells were Siglec-F+Annexin-V+ comprising approximately half of all Pirb−/−Siglec-F+ cells (Fig. 2e); in contrast, wild-type eosinophils showed good viability and exhibited only 3-5% Siglec-F+Annexin-V+ cells. Increased percentage of Siglec-F+Annexin-V+ cells in Pirb−/− cultures (Fig. 2f) was reflected by increased total dead eosinophils (Fig. 2g). Eventually, the numbers of total dead eosinophils declined due to the overall decrease in total cell counts, which was observed in the Pirb−/− LDBM culture (Fig. 1b). Analysis of the percentage and total Siglec-F+Annexin-V− cells demonstrated markedly increased percentage and total viable eosinophils in the wild-type cell cultures in comparison with the Pirb−/− cell cultures (Fig. 2h,i).
Figure 2. PIR-B regulates eosinophil apoptosis during low-density bone marrow-derived eosinophil differentiation.

Low-density bone marrow (LDBM) cells were obtained from wild-type (WT) and Pirb−/− mice and differentiated in vitro. Representative photomicrograph images at the indicated time points of apoptotic eosinophils (a) as well as engulfed eosinophils (b) are shown. Representative density plot analysis (c) and a summary of annexin-V propidium idodide (PI) stained cells (d) is depicted. Numbers in each quadrant indicates the percentage of cells corresponding with this quadrant. Representative density plots of anti-Siglec F and annexin-V stained cells (e) and analysis of the percentage and total cell counts of Siglec-F+annexin V+ cells and Siglec-F+annexin V− (f, g, h, i) is shown. LDBM cells were stained with anti-Siglec-F, annexin-V and anti-active caspase 3 (j), anti-Bim (l), or anti-GATA-1 (m) and expression was assessed using flow cytometry. Bim expression was assessed in cDNA obtained from LDBM cells by qPCR analysis (k) and normalized to the house keeping gene hypoxanthine-guanine phosphoribosyltransferase (Hprt). *P < 0.05, **P < 0.01,***P < 0.001. Data are representative of at least five independent experiments conducted in triplicates (a-m); mean and s.e.m. of data obtained from three mice per group (d,f,g-I,k) or representative histograms from the respective experiments (j, i, m).
Consistent with their apoptotic appearance, Siglec-F+Annexin-V+ cells in Pirb−/− LDBM cultures expressed increased expression of active caspase 3 (Fig. 2j). Moreover, Pirb−/− LDBM cell cultures displayed increased mRNA expression of the pro-apoptotic molecule Bcl-interacting molecule (Bim)26 in comparison to wild-type cells (Fig. 2k). No differences were observed in the expression of additional pro- or anti-apoptotic molecules (Supplementary Fig. 3a-c). Indeed, Pirb−/− Siglec-F+Annexin-V+ cells displayed substantially increased amounts of Bim compared with Siglec-F+Annexin-V− wild-type or Pirb−/− cells (Fig. 2l). Intracellular staining assessing GATA-1 abundance demonstrated that Pirb−/− Siglec-F+Annexin-V+ cells displayed less GATA-1 protein compared with Siglec-F+Annexin-V− wild-type or Pirb−/− cells (Fig. 2m). GATA-1 and Bim protein expression was similar between wild-type and mutant Siglec-F+Annexin-V− and Siglec-F−Annexin V− cells (Fig. 2l,m). Thus, Pirb−/− cells do not mature into eosinophils in vitro likely due to Bim-mediated apoptosis.
PIR-B is required for IL-5-induced colony formation
To further assess the specificity of PIR-B to IL-5-induced eosinophil differentiation wild-type and Pirb−/− LDBM cells were subjected to colony forming unit (CFU) assays using IL-5, GM-CSF or IL-3. Pirb−/− cells displayed a specific decrease in IL-5- but not IL-3-and/or GM-CSF-induced CFUs (Fig. 3). Assessment of the relative cell counts per colony demonstrated that the amount of cells per colony was lower in IL-5-induced (but not IL-3- or GM-CSF-induced) Pirb−/− CFUs (Fig. 3). Furthermore, Pirb−/− CFUs displayed a 2-fold increase in Siglec-F+Annexin-V+ cells specifically in the IL-5-driven colonies (Fig. 3c). Thus, in response to IL-5 (specifically), Pirb−/− BM produced fewer colonies, which were smaller and displayed increased apoptosis in comparison with wild-type cells.
Figure 3. PIR-B regulates IL-5- but not IL-3/GM-CSF-induced colony formation.

Low-density bone marrow cells were obtained from wild-type (WT) and Pirb−/− mice and subjected to colony forming unit (CFU) assays in response to IL-5 (a, b, c), IL-3 (d, e) and GM-CSF (f, g). Total CFU counts (a, d, f) and relative cell counts per CFU (b, e, g) were assessed. In addition, IL-5-induced CFUs from WT and Pirb−/− cultures were obtained and stained with anti-Siglec-F and annexin-V. Thereafter, Siglec-F+ cells were assessed for annexin-V expression (c); ns-non significant, **P < 0.01, ***P < 0.001. Data are mean and s.e.m. of three independent experiments of three mice per group (a-g).
PIR-B regulates eosinophil apoptosis in vivo
The requirement for PIR-B in IL-5-induced eosinophil development in vitro led us to assess eosinophil numbers in the BM of naïve Pirb−/− mice. Flow cytometric analysis of BM eosinophils revealed two distinct eosinophil populations in the BM; namely a Siglec-F+CCR3int population and a Siglec-F+CCR3hi population (likely representing immature and mature eosinophils, respectively)25, both of which displayed eosinophil physical parameters (Fig. 4a), morphology and eosinophil granule proteins (e.g. MBP, Supplementary Fig. 4). Both eosinophil populations retrieved from naïve Pirb−/− mice displayed more Annexin-V+ staining, as compared to wild-type controls (Fig. 4b,c). Analysis of other BM-resident myeloid cell populations such as monocytes (gated as Ly6G−Ly6B.2+, R1, Fig. 4d) or neutrophils (gated as Ly6G+Ly6B.2int, R2, Fig. 4d) revealed that increased baseline apoptosis in Pirb−/− mice was specific to eosinophils (Fig. 4d). Assessment of PB of naïve wild-type and mutant mice revealed that the Pirb deficiency resulted in 30% fewer blood eosinophils in the steady-state (Fig. 4e,f). Eosinophil viability was similar between wild-type and Pirb−/− PB eosinophils (data not shown), suggesting that reduction is due to a compartmentalized apoptotic fate in the BM.
Figure 4. PIR-B regulates eosinophil apoptosis in-vivo even in the presence of increased IL-5.

Bone marrow cells were obtained from naïve wild-type (WT) and Pirb−/− mice. Siglec-F+CCR3int and Siglec-F+CCR3hi cells (a) were gated (R1 and R2, respectively) and assessed for annexin-V expression (b, c). BM neutrophils and monocytes were gated (d) and assessed for annexin-V expression (d). PB was obtained from naïve WT and Pirb−/− mice and assessed for circulating eosinophil levels using anti-Siglec-F and anti-CCR3 staining (e, f). WT and Pirb−/− mice were treated with anti-IL-5 (TRFK5) or isotype control antibodies (Iso). Thereafter, bone marrow cells were stained with anti-Siglec-F and anti-CCR3 and the levels of Siglec-F+CCR3int (g) and Siglec-F+CCR3hi (h) cells as well as PB eosinophils (i) were assessed. In addition, the expression of PIRs was assessed on the surface of blood eosinophils (j). IL-5 was administered daily for 5 consecutive days and PB and BM were obtained. BM cells were stained with anti-Siglec-F, anti-CCR3 and bone marrow Annexin-VSiglec-F+CCR3int and Annexin-VSiglec-F+CCR3hi cells were gated (R1 and R2, respectively) and assessed for annexin-V expression (k, l). The levels of PB eosinophils were assessed (m). Expression of PIRs in BM (n, o) and PB (p) eosinophils is shown; ns-non significant, *P < 0.05, **P < 0.01, ***P < 0.001. Data are representative histograms, dot-plots and mean and s.e.m. of at least fifteen mice (a-f); or mean and s.e.m of two independent experiments with five to seven mice per group (g-p).
Previous studies utilizing Il5−/− mice as well as anti-IL-5 neutralization in vivo revealed that under homeostatic conditions, BM eosinophils are only partially regulated by an IL-5-dependent pathway27, 28. Rather, IL-5 is critical in eosinophil expansion under various disease settings29. To define whether PIR-B regulates in vivo steady-state eosinophilia in an IL-5-dependent fashion, IL-5 was neutralized in naïve wild-type and Pirb−/− mice using the TRFK5 antibody30. IL-5 neutralization caused an evident (but not complete) decrease in BM and PB eosinophils of wild-type but not Pirb−/− mice (Fig. 4g–i). Moreover, anti-IL-5-treatment caused a specific reduction in the expression of PIR-B but not PIR-A in mature BM eosinophils (i.e. Siglec-F+CCR3hi) but not PB eosinophils (Fig. 4j). Collectively, these data demonstrate that PIR-B regulates the IL-5-dependent but not IL-5-independent baseline frequencies of eosinophils.
Consistently, increased apoptosis of Pirb−/− BM eosinophils persisted even when IL-5 availability was artificially elevated by exogeneous delivery of recombinant IL-5. Thus, while this regimen was capable to decrease the apoptotic fate of some Pirb−/− Siglec-F+CCR3int and Siglec-F+CCR3hi cells (Fig. 4k,l), the frequencies of apoptotic eosinophils were still higher in Pirb−/− mice in comparison to wild-type controls (Fig. 4k,l). The frequency of eosinophils in the blood of IL-5-treated wild-type animals increased 1.76 ± 0.17-fold whereas; IL-5-treated Pirb−/− mice displayed only a 1.2 ± 0.025-fold increase (Fig. 4m). Moreover, IL-5-treatment increased the expression of PIR-B in BM but not PB eosinophils (Fig. 4n–p). These results establish an intricate relationship between IL-5 and PIR expression.
Self-recognition regulates eosinophil expanison
The β2 microglobulin (β2M) subunit of MHC-I molecules serves as a ligand for PIR-B11. Thus, we hypothesized that loss of β2M will result in similar eosinophil apoptosis as observed in Pirb−/− cell cultures and mice. Surprisingly however, CFU counts obtained from B2m−/− LDBM cells demonstrated increased numbers of colonies in response to IL-5 (Fig. 5a). Furthermore, assessment of relative cell counts per colony revealed more cells in the B2m−/− CFUs (Fig. 5b) and less Siglec-F+Annexin-V+ cells compared with wild-type cells (Fig. 5c). Consistently, both Siglec-F+CCR3int and Siglec-F+CCR3hi BM eosinophils obtained from naïve B2m−/− mice were less apoptotic than naïve wild-type BM eosinophils (Fig. 5d,e). The finding that loss of MHC-I recognition resulted in decreased eosinophil apoptosis suggested the involvement of an additional receptor, which can bind MHC-1 and is suppressed by PIR-B, such as PIR-A13.
Figure 5. Increased eosinophil development and decreased apoptosis in the absence of MHC-I expression.
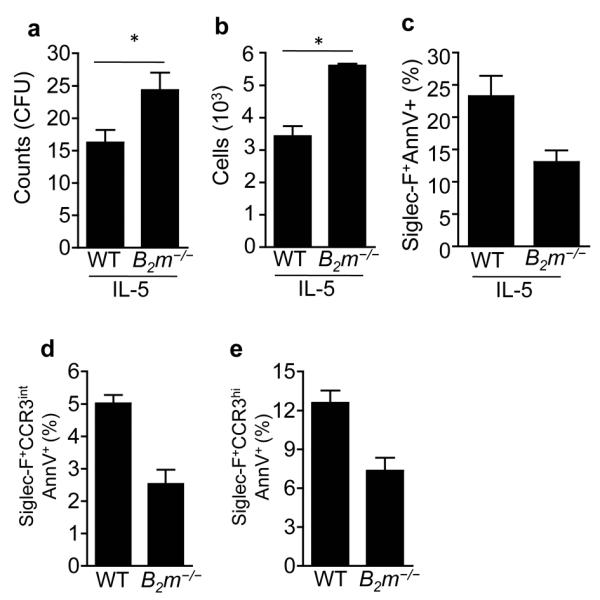
Low-density bone marrow cells were obtained from wild-type (WT) and B2m−/− mice and subjected to colony forming unit (CFU) assays in response to IL-5 (a-c). Thereafter, total CFU counts (a) and relative cell counts per CFU (b) were assessed. In addition, IL-5-induced CFUs from WT and B2m−/− cell cultures were stained with anti-Siglec-F and annexin-V for assessment of apoptosis (c). Bone marrow cells were obtained from naïve WT and B2m−/− mice. Cells were stained with anti-Siglec-F, anti-CCR3 and annexin-V. Thereafter, Siglec-F+CCR3int and Siglec-F+CCR3hi cells were gated and assessed for annexin-V expression (d, e); ns-non significant, *P < 0.05. Data are mean and s.e.m of three independent experiments using three (a-c) and seven (d, e) mice.
Absence of PIR-B enables pro-apoptotic signals by PIR-A
To address the hypothesis that PIR-A delivers pro-apoptotic signaling in eosinophils, we first analyzed the expression of PIRs throughout the LDBM culture system. Both PIRs were progressively upregulated on the cultured cells (Fig. 6a). Expression of PIR-A preceded that of PIR-B by 2 days, with PIR-A increasing at day 5 (Fig. 6a). The induction of PIR-A (but not of PIR-B) expression thus correlated with the induction of the pro-apoptotic factor Bim (Fig. 6b and Supplementary Fig. 5). To assess whether PIR-A is responsible for the apoptotic phenotype that was observed in Pirb−/− LDBM cultures, PIR-A was neutralized in the Pirb−/− LDBM eosinophil cell cultures using the anti-PIR-A/B antibody 6C1. This antibody was specifically chosen since it recognizes ectodomain 1 and 2 of PIR molecules, which are the major MHC-I recognizing ectodomains in PIRs31.
Figure 6. Neutralization of PIR-A in Pirb−/− eosinophil cultures, attenuates eosinophil apoptosis.

The expression of PIR-A throughout the low-density bone marrow (LDBM)-derived eosinophil culture was determined at the indicated time points (a). In (b) Pearson correlation between PIR-A and Bim expression is shown (r=0.943, P = 0.0004). LDBM cells from wild type (WT) and Pirb−/− mice were obtained and eosinophils were grown untreated (UT), in the presence of anti-PIR-A/B, or isotype control antibodies (Iso). Representative dot plots (c) and a summary of the flow cytometric analysis of anti-Siglec-F and annexin-V stained LDBM cells is shown (d, e). Cells were stained with modified Wright Giemsa stain and cellular morphology was assessed (f). Viable (i.e. Siglec-F+Annexin-V−) Pirb−/− cells were sorted and PIR-A immunoprecipitated. Thereafter, antibody coated membranes were treated with anti-phospho-tyrosine antibodies conjugated to horseradish peroxidase and PIR-A:protein interactions were assessed (g). Representative flow cytometric histogram plot (h) and quantitative analysis of phopsho-Erk 1/2 expression in Siglec-F+Annexin-V− and Siglec-F+Annexin-V+ cells from WT and Pirb−/− cells is shown (i). Expression of PIR-A and PIR-B on the surface of eosinophils from various in vivo sources is depicted (j); *P < 0.05, **P < 0.01, ***P < 0.001. Data are representative dot-plots and mean and s.e.m of five experiments conducted in duplicates (a-f); representative blots of one out of three independent experiments (g); representative histogram plot and pooled mean and s.e.m from five mice; and mean and s.e.m of at least five mice.
Neutralization of PIR-A markedly attenuated the apoptotic phenotype, which was observed in Pirb−/− eosinophils (Fig. 6c,d) and the numbers of mature eosinophils were noticeably increased (Fig. 6e,f). Pirb−/− cultures, which received isotype control antibodies, displayed a similar number of apoptotic cells and total eosinophils as untreated Pirb−/− cultures (Fig. 6e). Morphologically, PIR-A neutralization partially restored the typical eosinophil granular appearance in Pirb−/− cell cultures (Fig. 6f). Collectively, these data demonstrate that unless counteracted by PIR-B, PIR-A induces eosinophil apoptosis, an activity that is revealed in the absence of PIR-B.
To gain mechanistic insight into the functional activities driven by PIR-A, we have utilized a mini-phosphoproteomic approach where PIR-A was immunoprecipitated from the surface of sorted viable Pirb−/− LDBM cells and incubated with a membrane that was pre-coated with antibodies against various signaling molecules (Supplementary Fig. 6a,b). After incubation, the membrane was blotted with anti-phospho-tyrosine, thus detecting interactions between PIR-A and functional downstream signaling intermediates. Given the abundant availability of MHC-I ligands to PIR-A, we hypothesized that the kinases and/or adaptor molecules interacting with PIR-A will be active as well. We found that PIR-A was strongly associated with the adaptor Grb2 (Fig. 6g). Given the association between Grb2 and Erk activation32, we hypothesized that apoptotic Pirb−/− eosinophils will display increased Erk activation in comparison with viable cells. Assessment of phopho-Erk-1/2, in Siglec-F+Annexin-V− and Siglec-F+Annexin-V+ cells revealed an evident increase in Erk-1/2 phosphorylation in Siglec-F+Annexin-V+ in comparison with Siglec-F+Annexin-V− cells (Fig. 6h,i). Notably, this effect was specific to ERK1/2 activation since activation of Jnk and p38 were not changed between viable and dead eosinophils (Supplementary Fig. 6).
Distinct expression for PIRs during eosinophil maturation
Given the pro-apoptotic role of PIR-A, we sought to understand why do peripheral eosinophils survive in the presence of abundant MHC-I expression. To this end, we analyzed the relative expression of PIR-A and PIR-B in various eosinophil populations in the BM (i.e. EoPs, Sigec-F+CCR3int, Sigec-F+CCR3hi cells), PB and tissue (i.e. peritoneal cavity). EoPs expressed very low and equivalent amounts of PIR-A and PIR-B (1.11 ± 0.02 and 1.14 ± 0.04, fold-increase over isotype control, P = 0.26, respectively). The expression of PIR-A and PIR-B gradually increased as eosinophils developed (Fig. 6j) and eosinophils displayed higher amounts of PIR-B in comparison with PIR-A. Therefore, although PIRs compete and bind the same ligands13, pro-apoptotic signaling by PIR-A in eosinophils is constitutively balanced by inhibitory signals mediated by PIR-B, due to its relatively higher expression.
PIR-B is required for allergic airway inflammation
Regulation of eosinophil development by PIRs suggested a role for PIRs in allergic eosinophilic airway inflammation. Therefore, wild-type and Pirb−/− mice were sensitized and exposed to extracts of the aeroallergens Aspergillus fumigatus (Asp) or house dust mite (HDM). These models were specifically chosen since mucosal aeroallergen-challenged Pirb−/− mice displayed similar induction of IgE compared to wild-type mice (Fig. 7a). In contrast, systemic OVA and Alum sensitization of Pirb−/− mice results in increased total IgE production, which is likely due to the dysregulation of DC functions16 (Supplementary Fig. 7a). Mucosal aeroallergen-challenge had no effect on the apoptotic frequency of wild-type BM eosinophils (Fig. 7b,c). However, it was not sufficient to fully rescue BM Pirb−/− eosinophils from apoptosis (Fig. 7b,c). Subsequently, aeroallergen-challenged Pirb−/− mice displayed a significant reduction in aeroallergen-induced PB eosinophilia (Fig. 7d) and had less eosinophilic BAL infiltrates (Fig. 6e,f). This phenotype was not due to increased local eosinophil cell death in the lungs as Pirb−/− mice displayed similar frequencies of Annexin-V+Siglec-F+CCR3hi cells as wild-type mice (Fig. 7g). Decreased lung eosinophilia in aeroallergen-challenged Pirb−/− mice was also associated with a minor but statistically significant decrease in CD4+ T cell accumulation (P < 0.01) and decreased expression of TH2 cytokines and chemokines, including IL-4, IL-13 and CCL17 (Supplementary Fig. 7b–e). Decreased eosinophilia and TH2-associated allergic airway disease were not an allergen-specific phenomenon since also HDM-challenged Pirb−/− mice exhibited less lung eosinophil infiltration as well (Supplementary Fig. 7f,g).
Figure 7. Pirb−/− mice display increased bone marrow eosinophil apoptosis as well as decreased PB and tissue eosinophilia following mucosal aeroallergen challenge.
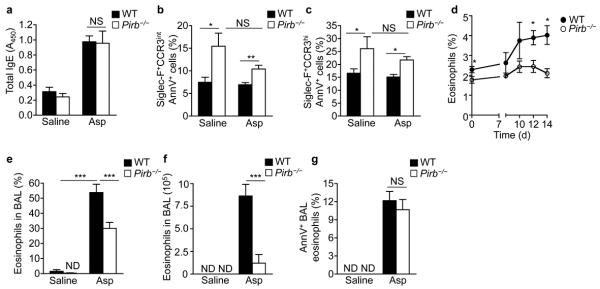
Wild-type (WT) and Pirb−/− mice were challenged with saline or Aspergillus fumigatus (Asp). Following six challenges, mice were bled and assessed for total serum IgE (a). Bone marrow was obtained and stained with anti-Siglec-F, anti-CCR3 antibodies and annexin-V. Siglec-F+CCR3int and Siglec-F+CCR3hi cells were gated and assessed for the frequencies of annexin-V positive cells (b, c). Saline- and Asp-challenged WT and Pirb−/− mice were assessed for allergen-induced PB eosinophils (d), bronchoalveolar lavage fluid (BALF) eosinophil percentages (e) and total BAL eosinophils (f). Eosinophils from saline and Asp-challenged BALF were assessed for annexin-V positivity (g). *P < 0.05, **P < 0.01, ***P < 0.001. Data are representative of five experiments each containing six to eight mice per group (a-g).
Since PIR-B is also expressed in myeloid cells other than eosinophils, impaired lung eosinophilia in aeroallergen-challenged Pirb−/− mice could result from deficiency in these cells. To specifically probe for the involvement of CD11c+ myeloid cells in the impaired response of the Pirb−/− mice to the aeroallergen-challenge, we generated animals that harbor a specific PIR-B deficiency in their CD11c+ cell compartment (including lung DCs and alveolar macrophages), using a mixed BM chimera approach33,34. Diphtheria toxin (DTx) treatment of mixed BM chimeras generated with CD11c-DTR BM and Pirb−/− BM result in animals that retain a Pirb-deficient DC compartment, while other hematopoietic cell populations retain their mixed wild-type:Pirb−/− make up (Supplementary Fig. 7h–j). Restricted absence of PIR-B from CD11c+ myeloid cells did not affect the ability of the mice to respond to Asp as CD11c-DTR Pirb−/− chimeric mice displayed similar expression of IL-4 and CCL17, as CD11c-DTR/wild-type control chimeras (Supplementary Fig. 7k,l). Taken together, these data demonstrate that decreased eosinophil accumulation and TH2-associated inflammation is not due to the expression of PIR-B in CD11c+ cells, rather it is likely an eosinophil-dependent phenomenon.
Discussion
The ability to discriminate “self” from “non-self” via binding of MHC-I molecules is a major immunological concept leading to tolerance of cytotoxic T cells and NK cells35. However, myeloid cells are also capable of self-recognition through PIR-A and PIR-B, which elicit activating and inhibitory signaling, respectively11. In this study, we dissected the function of the MHC-I binding receptors PIR-A and PIR-B in eosinophil maturation and subsequent allergic airway responses and demonstrated key roles for PIRs in IL-5-induced eosinophil expansion.
IL-5 has an exclusive requirement for the rapid expansion of eosinophils especially in disease settings such as asthma27. Our findings demonstrate that IL-5-regulated eosinophilia is governed by a constant “tugging war” with PIR-A. TH2 settings (such as those observed in asthma) substantially increase the ability of IL-5 to induce eosinophilia by strengthening the “positive” signals for eosinophil development. Eosinophils likely escape their apoptotic fate, which is regulated by PIR-A, exit the BM and enter the PB, consequently migrating into the inflamed tissue. PIR-B, which is hierarchically dominant to PIR-A, serves as a “permissive” checkpoint for IL-5-induced eosinophil development by suppressing the activities of PIR-A. Thus, a constant crosstalk between PIR-A (acting as a pro-apoptotic “vector”) and IL-5 and PIR-B (acting as anti-apoptotic “vectors”) exists in eosinophils. Collectively, our model demonstrates that upon self-recognition, PIR-B is a negative regulator of PIR-A-induced apoptosis, which counter balances IL-5-mediated expansion signals (Figure S8).
Various candidate “self”-recognizing molecules are widely (and sometime exclusively) expressed by myeloid cells including eosinophils. These include PIRs19, 36, CD200 molecules; Siglec-family receptors and CD47, which interacts with signal regulatory protein-α (SIRP-α, also known as CD172)37. Despite this, the requirement for self-recognition in eosinophil activities is unknown. Our data demonstrates that inhibitory signaling by PIR-B is dominant over PIR-A induced eosinophil apoptosis and raises the question on how can this occur if they both bind similar ligands? One evident explanation for the relative dominance of PIR-B over PIR-A is the finding that eosinophils express higher amounts of PIR-B than PIR-A and therefore MHC-I binding will lead to more inhibitory signals than pro-apoptotic signaling, consequently maintaining high eosinophil survival rate. Interestingly, IL-5 neutralization caused a specific decrease in PIR-B expression and shifted the balance towards dominant expression of PIR-A relative to PIR-B. Conversely, IL-5 administration resulted in a compartmentalized increase in PIR-B expression (and to lesser extent PIR-A) in BM eosinophils. These data suggest that PIRs constitute an additional “fine-tuning” mechanism regulating IL-5-driven eosinophilia. Hence, increased IL-5 abundance will result in decreased apoptosis due to increased pro-survival signals that are directly mediated by IL-5 receptor; and decreased apoptosis due to increased PIR-B expression, suppressing PIR-A-induced apoptosis. Similarly, the absence of IL-5 renders eosinophils apoptotic due to lack of survival signals directly mediated by IL-5 and increased apoptotic signaling by PIR-A -due to decreased PIR-B expression and hence lack of inhibition.
We have recently established that Pirb−/− mice display increased tissue eosinophilia in the gastrointestinal tract19 and adipose tissue (data not shown). Thus, despite increased BM eosinophil apoptosis in Pirb−/−, steady-state eosinophil numbers in the gastrointestinal compartment are elevated. This is likely due to the fact that the regulation of eosinophil expansion by PIRs is restricted to the BM compartment. Once eosinophils “escape” the developmental regulation by PIRs and enter the blood, PIR-B is capable of suppressing eosinophil CCR3 signaling and subsequent eosinophil migration19. Therefore, tissues that display a constant eotaxin-gradient such as the gastrointestinal tract and adipose tissue will display eosinophilia in the absence of PIR-B6, 38. Indeed, intratracheal administration of IL-13 that generates a strong chemotactic gradient for eosinophil recruitment (but not IL-5-dependent generation of eosinophils in the BM) results in increased lung eosinophil accumulation in Pirb−/− mice19. In contrast to IL-13-administration, under typical TH2 settings, rapid IL-5-induced eosinophil generation in the BM occurs, and PIR-B is required for eosinophil generation and subsequent lung infiltration. While we cannot entirely exclude the involvement of additional mechanisms governed by PIR-B in TH2 settings, the finding that the expression of PIR-B in the CD11c+ myeloid cell compartment is dispensable for decreased TH2-associated disease in Pirb−/− mice and the finding that mucosal sensitization was similar between wild-type and Pirb−/− mice strongly suggests that decreased CD4+ T cell accumulation and expression of TH2-associated cytokines and chemokines is eosinophil dependent. In support of this notion, numerous studies demonstrate a key contribution for eosinophils in the development of allergic asthma partially by their ability to regulate local lung TH2 immunity5, 39-41.
Mechanistically, our data suggest that PIR-B does not regulate IL-5-induced eosinophil expansion by direct interactions with IL-5 receptor signaling components (similar to the way PIR-B regulates GM-CSF and/or IL-3-induced responses16, 17). This finding is of particular interest since it is different than the current reported interactions of other ITIM-bearing receptors with IL-5 receptor. For example, IL-5 primes the pro-apoptotic effects of Siglec-842, and increased IL-5 expression potentiates the suppression of IL-5-induced survival by CD300a43. Collectively suggesting direct interactions of these inhibitory receptors with IL-5 receptor. In addition, we demonstrate that PIR-A associated with the adaptor molecule Grb2, an upstream regulator of the Mek-Erk pathway32. This interaction is likely mediated via the FcRγ chain, which is required for PIR-A’s surface expression12, has been shown to interact with Grb244, and can mediate pro-apoptotic signaling45. Interestingly, and similar to our findings suggesting a role for Erk in PIR-A-mediated eosinophil apoptosis, Erk activation has been identified as critical in delivering pro-apoptotic signals via Siglec-8 in IL-5-activated eosinophils42.
In summary, we provide evidence that self-recognition of MHC-I molecules by PIRs has a critical role in eosinophil development and consequent expansion in settings of allergic airway disease. These data open a new paradigm in understanding the molecular pathways regulating eosinophilia and have broad implications for eosinophilic diseases.
Methods
Mice
Male and female, 6- to 8-week-old Pirb−/− mice (backcrossed >F9 to C57BL/6)16. CC10-Il13Tg and MHC-class I-deficient (B2m−/−) mice were recently described elsewhere46, 47. C57BL/6 wild-type (WT) mice were obtained from Harlan Laboratories. 6-week-old CD11c-diphtheria toxin receptor (DTR) transgenic mice (B6.FVB-Tg(Itgax-DTR/GFP)57Lan/J) were used for generation of BM chimeras33. In all experiments age-, weight and gender-matched mice were housed under specific pathogen-free conditions according to Tel-Aviv University and/or Weizmann Institute Animal Care Committee approved protocols.
Eosinophil bone-marrow culture
Eosinophils were grown from the BM of WT and Pirb−/− mice with modifications based on a prior report20. Briefly, BM cells were harvested and loaded on a histopaque gradient (Sigma). Low-density BM cells were collected and cultured in the presence of SCF and Flt3L for 4 days. Thereafter, the medium was replaced with IL-5 for the rest of the culture (up to day 14). All cytokines were purchased from Peprotech.
Cell proliferation
Cell proliferation was assessed using the Click-iT EdU kit (Invitrogen) according to the manufacturers’ instructions.
Allergen sensitization and challenge
Allergic eosinophilic airway inflammation was induced by challenging mice intranasally with either A. fumigatus (Asp) or house dust mite (HDM)(Bayer Pharmaceuticals), three times a week for 2-3 weeks as previously described19. In brief, mice were lightly anesthetized with isoflurane inhalation, and 10 μg of total Asp protein extract in 50 μl saline was applied to the nasal cavity by using a micropipette with the mouse held in the supine position. After instillation, mice were held upright until alert. Mice were euthanized 24–48 h after the last challenge and bronchoalveolar lavage was performed and assessed for differential cell counts and mediator content as described19. The concentration of LPS in the Asp extracts was less than 2 pg/ml as detected by the Limulus assay.
IL-5 administration and neutralization
WT and Pirb−/− mice were treated with recombinant mouse IL-5 (5 μg/mouse) or anti-IL-5 (TRFK5 antibody, 50 μg/mouse) for 5 consecutive days. Thereafter, the mice were sacrificed and BM and blood were obtained for flow cytometric analyses.
Real-time quantitative (q) PCR
RNA samples from the whole lung were subjected to reverse transcription analysis using SuperScript II reverse transcriptase (Invitrogen) according to manufacturer’s instructions. qPCR analysis was performed using the CFX96 system (Bio-Rad laboratories) in conjunction with the ready-to-use fast-start SYBR Green I Master reaction kit (Roche Diagnostic Systems). Results were normalized to Hprt cDNA as previously described 24. The primers that were used in this study were as follows: Il4, Fwd- TCAACCCCCAGCTAGTTGTC, Rev- TGTTCTTCGTTGCTGTGAGG; Mbp, Fwd-CAAAGCTCAGTCAGTTTGCC, Rev- ATCGAAAGCGTTTGCATCGG; Gata1, Fwd-TCAAGCTCCATCAGGTGAACC, Rev- TCCCTTTGCCAGATGCCTT; Pu.1, Fwd-CCCGGATGTGCTTCCCTTAT, Rev- TCCAAGCCATCAGCTTCTCC, Fog1, Fwd-AGCTGTGACCCTGATGGTGG, Rev- GGCGTCATCCTTCCTGTAGATCT; Gata2, Fwd- AGACGACAACCACCACCTTA, Rev- TCCTTCTTCATGGTCAGTGG; c/Ebpa, Fwd- GTTAGCCATGTGGTAGGAGACA, Rev- CCCAGCCGTTAGTGAAGAGT; Bcl2, Fwd-GTCCCGCCTCTTCACCTTTCAG, Rev- GATTCTGGTGTTTCCCCGTTGG; Bax, Fwd- GCGTGGTTGCCCTCTTCTACTTTG, Rev-AGTCCAGTGTCCAGCCCATGATG; Bim, Fwd-CGACAGTCTCAGGAGGAACC, Rev-CCTTCTCCATACCAGACGGA; Bid, Fwd-TGGAAAGACCTTGGCCTGAAGACA, Rev-AAGGTACATGGTGTGGATGCAGGA; Bad, Fwd- GAGTATGTTCCAGATCCCAG, Rev-GTCCTCGAAAAGGGCTAAGC; Pirb, Fwd- GGTGACAGGACACTACTGGAACCC, Rev-CCACGAGAGCTTCTGTGGTCCT; Il5ra, Fwd- ACACTTTTCCAGCATTCGGCT, Rev- TGCCTTTGCTCTTGGTCAGG; Hprt, Fwd- GTAATGATCAGTCAACGGGGGAC, Rev-CCAGCAAGCTTGCAACCTTAACCA.
Flow cytometry
Flow cytometric analysis of LDBM cells and enzymatically digested lung or bronchoalveolar lavage cells was conducted using the following antibodies (all purchased from eBioscience unless stated otherwise): anti-PIR-A/B (10-1-PIR), anti-CD62L (MEL-14), anti-β7-FITC (FIB27, Biolegend), anti-α5-FITC (HMA5-1), anti-CD48 (HM-48-1), anti-CD18 (M18/2), anti-CD69 (H1.2F3), anti-CD103 (2E7), anti-F4/80 (BM8), anti-CD86 (GL1), anti-MHC-II (NIMR-4), anti-PIR-A/B (6C1,BD Bioscience) anti-CD45 (30-F11), anti-CD11c (N418), anti-CD11b (M1/70), anti-B220 (RA3-6B2), anti-Gr-1 (RB6-8C5), anti-CD3 (145-2C11), anti-CD3 (17A2), anti-CD4 (GK1.5), anti-CD8 (SK1), anti-Siglec-F (E50-2440, BD Bioscience) and anti-CCR3 (83101, R&D Systems). Events were acquired by a Gallios flow cytometer system (Beckman Coulter) or FACScalibur (BD Bioscience) and data were analyzed using the Kaluza (Beckman Coulter) or FlowJo (TreeStar) software on at least 10,000-50,000 events. Surface molecule expression was calculated by defining the delta mean fluorescent intensity between the specific antibody stain and the isotype-matched control antibody. All sorting experiments were performed using FACSAria III (BD Bioscience) in the flow cytometer core of the Tel Aviv University. PIR-A/B expression was calculated by defining the delta mean fluorescent intensity between anti-PIR-A/B stain and isotype control. The expression of PIR-A was deduced by staining WT and Pirb−/− cells with anti-PIR-A/B, which enables us to determine the relative expression of PIR-A (present on the surface of Pirb−/− cells vs. PIR-A and PIR-B on the surface of WT cells as described previously19, 24.
Immunoprecipitation and phosphorylation assays
Siglec-F+Annexin-V− cells were sorted from LDBM Pirb−/− cell cultures and lysed in MPER lysis buffer (Pierce Endogen) supplemented with protease inhibitor cocktail (Sigma-Aldrich) on ice for 30 min. The cell lysate was pre-cleared and PIR-A immunoprecipitated using Protein A/G-conjugated anti-PIR-A/B antibodies or isotype-matched control (Dynabeads, Invitrogen). Custom-designed membranes coated with various antibodies were purchased from Hypromatrix. The immunoprecipitated PIR-A:protein complex was diluted in 2 ml of PBS and incubated with the pre-coated membrane. After washing, the membrane was incubated with anti-mouse phosphotyrosine conjugated to horseradish peroxidase (pY99; Santa Cruz Biotechnology)19. The membrane was developed using enhanced chemiluminescence (ECL)-plus (GE Healthcare).
Colony formation assays
A total of 1 × 105 bone marrow-nucleated cells in a volume of 0.1 ml was plated in 0.9 ml methylcellulose media (MethoCult 3234, Stem Cell Technologies), supplemented with 20 ng/ml mouse rIL-5 (50 ng/ml), rIL-3 (20 ng/ml) or rGM-CSF (10 ng/ml) (Peprotech), and placed in a humidified incubator with 5% CO2 at 37 °C. Colonies containing at least 50 cells were counted 10 days after incubation. The number of colonies/105-nucleated bone marrow cells was calculated. Relative cell counts were determined using flow cytometry by obtaining a fixed amount of colonies and acquiring events for 120 s as described43.
Annexin-V apoptotic assay
Apotosis was detected using an Annexin V apoptosis detection kit (eBioscience), according to the manufacturer’s instructions.
ELISA
Bronchoalveolar lavage fluid CCL17, IL-4 and IL-13 were assessed using a commercial ELISA (R&D Systems Duo Set, Lower detection limits: 15.62, 3.91 and 62.5 pg/ml, respectively). Serum IgE was measured using a commercial kit purchased from BD bioscience according to the manufacturers’ instructions. Lower detection limit for IgE was 15 pg/ml.
Bone marrow-derived macrophage and dendritic cell generation
Macrophages and dendritic cells were grown from the BM of WT and Pirb−/− mice. Briefly, BM cells were harvested and cultured in the presence of M-CSF or GM-CSF (20 ng/ml, Peprotech), respectively, for 7-9 days.
Bone marrow chimera mice
Syngeneic BM chimeras were generated as described33, 48. Briefly, WT C57BL/6 mice were exposed to a single-lethal total body irradiation of 950 rad. One day after irradiation a 1:1 mixture of 5 × 106 BM cells obtained from CD11c-DTR mice (CD45.1+) and WT or Pirb−/− BM cells (CD45.2+) was injected into the irradiated mice, thus generating mixed BM chimeras. Thereafter, the mice were allowed to rest for 10 weeks and engraftment was validated in PB samples using anti-CD45.1 and anti-CD45.2 staining. Following confirmation of engraftment, the mice were challenged with Asp extract as described above. To deplete CD11c+ cells, diphtheria toxin (DTx) (8 ng/g body weight) was injected (intraperitoneally) every other day starting 24 h prior to the initial allergen challenge. CD11c+ cell depletion was monitored by flow cytometry using anti-CD45.1 and anti-CD11c staining.
Statistical analysis
Data were analyzed by ANOVA followed by Tukey post-hoc test using GraphPad Prism 4. Alternatively, several experiments were analyzed by Student’s t-test. Data are presented as mean ± s.e.m., and values of P < 0.05 were considered statistically significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We wish to thank H. Kubagawa (Univ. of Alabama) for providing Pirb−/− mice; S.P. Hogan (Cincinnati Children’s Hospital Medical Center, Cincinnati, OH), O. Mandelboim (Hebrew University), I. Bachelet (Bar-Ilan University) and N. Osherov and A. Ben-Baruch (Tel-Aviv University) for critically reviewing this manuscript and helpful discussions. This work was performed in partial fulfillment of the requirements for the PhD degree of Netali Morgenstern-Ben Baruch and Dana Shik at the Sackler Faculty of Medicine, Tel Aviv University, Ramat Aviv, Israel.
Supported by the FP7 Marie-Curie Reintegration grant (grant no. 256311), the Israel Science Foundation (grant nos. 955/11 and 1708/11) the Israel Cancer Research Foundation Research Career Development Award and the Fritz Thyssen Foundation (to AM); the US-Israel Bi-national Science Foundation (grant no. 2009222 AM and MER); NIH NIAID (R01AI083450, R37AI045898), CURED Foundation, FARE, and Buckeye Foundation (to MER).
Footnotes
Author contributions N.BB.M, D.S, I.M, M.I, D.K.A, C.B, P.F, D.R and A.M did in vitro and in vivo experiments and analyzed the data; M.E.R and S.J provided critical reagents for the experiments and analyzed the data; A.M supervised the study and wrote and edited the manuscript.
Competing financial interests The authors declare no competing financial interests
References
- 1.Nishinakamura R, et al. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor-deficient mice are normal. Immunity. 1995;2:211–22. doi: 10.1016/1074-7613(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 2.Sanderson CJ. Interleukin-5, eosinophils, and disease. Blood. 1992;79:3101–9. [PubMed] [Google Scholar]
- 3.Collins PD, Marleau S, Griffiths Johnson DA, Jose PJ, Williams TJ. Cooperation between interleukin-5 and the chemokine eotaxin to induce eosinophil accumulation in vivo. J Exp Med. 1995;182:1169–74. doi: 10.1084/jem.182.4.1169. issn: 0022-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rothenberg ME, et al. IL-5-dependent conversion of normodense human eosinophils to the hypodense phenotype uses 3T3 fibroblasts for enhanced viability, accelerated hypodensity, and sustained antibody-dependent cytotoxicity. J Immunol. 1989;143:2311–6. [PubMed] [Google Scholar]
- 5.Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol. 2006;24:147–74. doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- 6.Wu D, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–7. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 2013;13:9–22. doi: 10.1038/nri3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu VT, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. 2011;12:151–9. doi: 10.1038/ni.1981. [DOI] [PubMed] [Google Scholar]
- 9.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–76. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 10.Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–44. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 11.Takai T. Paired immunoglobulin-like receptors and their MHC class I recognition. Immunology. 2005;115:433–40. doi: 10.1111/j.1365-2567.2005.02177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubagawa H, et al. Biochemical nature and cellular distribution of the paired immunoglobulin-like receptors, PIR-A and PIR-B. J Exp Med. 1999;189:309–18. doi: 10.1084/jem.189.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura A, Kobayashi E, Takai T. Exacerbated graft-versus-host disease in Pirb-/- mice. Nat Immunol. 2004;5:623–9. doi: 10.1038/ni1074. [DOI] [PubMed] [Google Scholar]
- 14.Maeda A, Kurosaki M, Kurosaki T. Paired immunoglobulin-like receptor (PIR)-A is involved in activating mast cells through its association with Fc receptor gamma chain. J Exp Med. 1998;188:991–5. doi: 10.1084/jem.188.5.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blery M, et al. The paired Ig-like receptor PIR-B is an inhibitory receptor that recruits the protein-tyrosine phosphatase SHP-1. Proc Natl Acad Sci U S A. 1998;95:2446–51. doi: 10.1073/pnas.95.5.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ujike A, et al. Impaired dendritic cell maturation and increased T(H)2 responses in PIR-B(-/-) mice. Nat Immunol. 2002;3:542–8. doi: 10.1038/ni801. [DOI] [PubMed] [Google Scholar]
- 17.Wheadon H, Paling NR, Welham MJ. Molecular interactions of SHP1 and SHP2 in IL-3-signalling. Cell Signal. 2002;14:219–29. doi: 10.1016/s0898-6568(01)00241-8. [DOI] [PubMed] [Google Scholar]
- 18.Mitsuhashi Y, et al. Regulation of plasmacytoid dendritic cell responses by PIR-B. Blood. 2012;120:3256–9. doi: 10.1182/blood-2012-03-419093. [DOI] [PubMed] [Google Scholar]
- 19.Munitz A, McBride ML, Bernstein JS, Rothenberg ME. A dual activation and inhibition role for the paired immunoglobulin-like receptor B in eosinophils. Blood. 2008;111:5694–703. doi: 10.1182/blood-2007-12-126748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dyer KD, et al. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J Immunol. 2008;181:4004–9. doi: 10.4049/jimmunol.181.6.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwasaki H, et al. Identification of eosinophil lineage-committed progenitors in the murine bone marrow. J Exp Med. 2005;201:1891–7. doi: 10.1084/jem.20050548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moshkovits I, et al. CMRF35-like molecule 1 (CLM-1) regulates eosinophil homeostasis by suppressing cellular chemotaxis. Mucosal Immunol. 2013 doi: 10.1038/mi.2013.47. [DOI] [PubMed] [Google Scholar]
- 23.Uehara T, et al. Inhibition of IgE-mediated mast cell activation by the paired Ig-like receptor PIR-B. J Clin Invest. 2001;108:1041–50. doi: 10.1172/JCI12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karo-Atar D, Moshkovits I, Eickelberg O, Konigshoff M, Munitz A. Paired immunoglobulin-like receptor-B inhibits pulmonary fibrosis by suppressing profibrogenic properties of alveolar macrophages. Am J Respir Cell Mol Biol. 2013;48:456–64. doi: 10.1165/rcmb.2012-0329OC. [DOI] [PubMed] [Google Scholar]
- 25.Voehringer D, van Rooijen N, Locksley RM. Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages. J Leukoc Biol. 2007;81:1434–44. doi: 10.1189/jlb.1106686. [DOI] [PubMed] [Google Scholar]
- 26.Bouillet P, O’Reilly LA. CD95, BIM and T cell homeostasis. Nat Rev Immunol. 2009;9:514–9. doi: 10.1038/nri2570. [DOI] [PubMed] [Google Scholar]
- 27.Kopf M, et al. IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity. 1996;4:15–24. doi: 10.1016/s1074-7613(00)80294-0. [DOI] [PubMed] [Google Scholar]
- 28.Rennick DM, et al. In vivo administration of antibody to interleukin-5 inhibits increased generation of eosinophils and their progenitors in bone marrow of parasitized mice. Blood. 1990;76:312–6. [PubMed] [Google Scholar]
- 29.Rosenberg HF, Phipps S, Foster PS. Eosinophil trafficking in allergy and asthma. J Allergy Clin Immunol. 2007;119:1303–10. doi: 10.1016/j.jaci.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 30.Tomaki M, et al. Eosinophilopoiesis in a murine model of allergic airway eosinophilia: involvement of bone marrow IL-5 and IL-5 receptor alpha. J Immunol. 2000;165:4040–50. doi: 10.4049/jimmunol.165.7.4040. [DOI] [PubMed] [Google Scholar]
- 31.Matsushita H, et al. Differential but competitive binding of Nogo protein and class i major histocompatibility complex (MHCI) to the PIR-B ectodomain provides an inhibition of cells. J Biol Chem. 2011;286:25739–47. doi: 10.1074/jbc.M110.157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kessels HW, Ward AC, Schumacher TN. Specificity and affinity motifs for Grb2 SH2-ligand interactions. Proc Natl Acad Sci U S A. 2002;99:8524–9. doi: 10.1073/pnas.142224499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varol C, Landsman L, Jung S. Probing in vivo origins of mononuclear phagocytes by conditional ablation and reconstitution. Methods Mol Biol. 2009;531:71–87. doi: 10.1007/978-1-59745-396-7_6. [DOI] [PubMed] [Google Scholar]
- 34.van Rijt LS, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med. 2005;201:981–91. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 36.Ho LH, Uehara T, Chen CC, Kubagawa H, Cooper MD. Constitutive tyrosine phosphorylation of the inhibitory paired Ig-like receptor PIR-B. Proc Natl Acad Sci U S A. 1999;96:15086–90. doi: 10.1073/pnas.96.26.15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van den Berg TK, van der Schoot CE. Innate immune ‘self’ recognition: a role for CD47-SIRPalpha interactions in hematopoietic stem cell transplantation. Trends Immunol. 2008;29:203–6. doi: 10.1016/j.it.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Ahrens R, et al. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol. 2008;181:7390–9. doi: 10.4049/jimmunol.181.10.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang D, et al. Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2-MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J Exp Med. 2008;205:79–90. doi: 10.1084/jem.20062027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JJ, Jacobsen EA, McGarry MP, Schleimer RP, Lee NA. Eosinophils in health and disease: the LIAR hypothesis. Clin Exp Allergy. 2010;40:563–75. doi: 10.1111/j.1365-2222.2010.03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bochner BS. Verdict in the case of therapies versus eosinophils: the jury is still out. J Allergy Clin Immunol. 2004;113:3–9. doi: 10.1016/j.jaci.2003.09.046. quiz 10. [DOI] [PubMed] [Google Scholar]
- 42.Kano G, Almanan M, Bochner BS, Zimmermann N. Mechanism of Siglec-8-mediated cell death in IL-5-activated eosinophils: Role for reactive oxygen species-enhanced MEK/ERK activation. J Allergy Clin Immunol. 2013;132:437–45. doi: 10.1016/j.jaci.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munitz A, et al. The inhibitory receptor IRp60 (CD300a) suppresses the effects of IL-5, GM-CSF, and eotaxin on human peripheral blood eosinophils. Blood. 2006;107:1996–2003. doi: 10.1182/blood-2005-07-2926. [DOI] [PubMed] [Google Scholar]
- 44.Chu J, Liu Y, Koretzky GA, Durden DL. SLP-76-Cbl-Grb2-Shc interactions in FcgammaRI signaling. Blood. 1998;92:1697–706. [PubMed] [Google Scholar]
- 45.Kanamaru Y, et al. IgA Fc receptor I signals apoptosis through the FcRgamma ITAM and affects tumor growth. Blood. 2007;109:203–11. doi: 10.1182/blood-2006-06-025882. [DOI] [PubMed] [Google Scholar]
- 46.Zuo L, et al. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13R alpha 2-inhibited pathway. J Immunol. 2010;185:660–9. doi: 10.4049/jimmunol.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells. Science. 1990;248:1227–30. doi: 10.1126/science.2112266. [DOI] [PubMed] [Google Scholar]
- 48.Sapoznikov A, Jung S. Probing in vivo dendritic cell functions by conditional cell ablation. Immunol Cell Biol. 2008;86:409–15. doi: 10.1038/icb.2008.23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.