

Abstract
Ursolic acid (UA), present in apples, rosemary, and other sources, is known to inhibit tumor formation and tumor cell viability in multiple systems, including skin. However, various cancers are resistant to UA treatment. Herein, skin carcinoma cells (Ca3/7) as compared to skin papilloma cells (MT1/2) displayed more resistance to UA-induced cytotoxicity. Interestingly, Ca3/7 cells had elevated levels of P-glycoprotein (P-gp), an ATP-dependent efflux pump that mediates resistance to chemotherapy in pre-clinical and clinical settings, and not only accumulated less but also more rapidly expelled the P-gp substrate Rhodamine 123 (Rh123) indicating UA is transported by P-gp. To determine if P-gp inhibition can enhance UA-mediated cytotoxicity, cells were challenged with P-gp inhibitors verapamil (VRP) or cyclosporin A (CsA). Alternatively, cells were pre-treated with the natural compound resveratrol (RES), a known chemotherapy sensitizer. VRP and RES enhanced the effects of UA in both cell lines, while CsA only did so in Ca3/7 cells. Similarly, VRP inhibited Rh123 efflux in both lines, while CsA only inhibited Rh123 efflux in Ca3/7 cells. RES did not inhibit Rh123 efflux in either line, indicating the synergistic effects of RES and UA are not manifest by inhibition of P-gp-mediated efflux of UA. These results indicate that the anti-skin cancer effects of UA are enhanced with P-gp inhibitors. In addition, RES and UA interact synergistically, but not through inhibition of P-gp.
Implications
Resveratrol and/or p-glycoprotein inhibitors in combination with ursolic acid are an effective anti-skin cancer regimen.
Keywords: skin cancer, ursolic acid, p-glycoprotein, resveratrol, cytotoxicity
Introduction
In many cancer therapies, tumors have devised ways to resist treatment. Multidrug resistance (MDR) in tumors can be mediated by a large number of factors, depending on tumor location or tumor type as well as the etiology of mutations. MDR can be mediated by factors including alterations in p53 or related pathways and decreased susceptibility to apoptosis via upregulation of antiapoptotic factors like bcl-2 (1, 2). Increased activity of Nuclear Factor-kappaB (NFκB) may also induce the MDR phenotype by decreasing p53 activity as well as transcribing various inhibitors of apoptosis (3). The tumor microenvironment, specifically the disorganized vascular system surrounding the tumor, can promote chemoresistance by creating high interstitial pressure in the tumor to limit drug influx, or by inducing hypoxia or low pH (4). MDR is also associated with increased levels of efflux pumps, which have normal physiological functions but also mediate resistance to chemotherapy by actively removing anti-cancer compounds from cells. Efflux pumps which can mediate MDR in different tumor types include breast cancer resistance protein (BCRP), multidrug resistance protein (MRP1), and the highly-characterized P-glycoprotein (P-gp) (5). P-gp, encoded by the gene ABCB1 is composed of two subunits, each containing six transmembrane regions and an ATP-binding domain (6, 7). P-gp is transcribed by a number of factors implicated in MDR, including NFκB (8). P-gp is upregulated in many cancer forms and can efflux a variety of drugs to mediate resistance to chemotherapy. P-gp is highly expressed in both normal and cancerous liver, kidney, and colon tissues, making these tumors intrinsically chemoresistant (9). In addition, other cancer types, including hematological malignancies and breast cancers increase P-gp as a result of tumor progression and in response to chemotherapeutic challenge, leading to decreased therapeutic responses (10, 11). Studies of P-gp inhibitors, in vitro, in vivo, and in some clinical trials have shown they can enhance the effects of traditional chemotherapeutics (11-14).
Chemotherapy of existing skin tumors may be a less scarring alternative to surgery and provide benefit for those with higher risk of metastasis (15). However, a number of studies have shown that metastatic squamous cell carcinoma (SCC) can resist certain chemotherapies, but become sensitized upon inhibition of specific pathways (15, 16). In addition, P-gp was found to be upregulated in human SCC tissue and SCC cell lines as compared to epidermis and normal keratinocytes (17). Therefore, P-gp inhibition may enhance the effects of SCC chemotherapies that are also P-gp substrates.
Natural phytochemical ursolic acid (UA) has been shown to have anti-cancer effects in different tumor models, including those of the breast (18), lung (19), and skin (20), and likely targets many different processes in tumorigenesis (21). However, a number of studies have revealed a variety of resistance mechanisms to UA. In one report, UA treatment of HT-29 human colorectal cancer cells induced increases in pro-inflammatory cyclooxygenase-2 (COX-2) and activation of upstream COX-2 regulator p38 MAPK in association with apoptosis. COX-2 siRNA or inhibition of p38 MAPK enhanced UA-mediated apoptosis (22). A similar study in HT-29 cells and DU145 human prostate carcinoma cells showed UA-mediated apoptosis is enhanced by inhibition of upstream inducers of p38 such as Src (23). Also, while one report showed a number of chemoresistant cancer cell types were sensitive to UA, UA-mediated cytotoxicity was still stronger in the parental cell types (24), which have lower levels of P-gp (25, 26). In addition, another study showed that UA inhibits substrate efflux through P-gp at high concentrations. However, UA also enhanced the ATPase function of P-gp, indicating it is a substrate for P-gp and likely inhibits efflux of other substrates through a competitive interaction (7). These results show UA may be a substrate for P-gp. Therefore, in addition to sensitization by inhibition of other resistance mechanisms, the anti-cancer effects of UA may be enhanced by P-gp inhibition.
As UA has been shown to have anti-tumor effects in chemically-mediated skin cancer (20), we conducted experiments to determine if modulation of P-gp could enhance the effects of UA in skin cancer-relevant systems. This was done using verapamil (VRP) and cyclosporin A (CsA), which both function as P-gp substrates and competitive P-gp inhibitors (7, 14, 27). We also tested UA in combination with the phytochemical resveratrol (RES), which has been shown to sensitize cancer cells to chemotherapeutics by multiple mechanisms, potentially including enhancement of apoptotic pathways and inhibition of P-gp-mediated efflux (28, 29). UA with or without P-gp inhibitors may provide another treatment avenue besides surgery, and may also provide therapeutic leads for related epithelial cancers, such as lung cancer.
Materials and Methods
Reagents
UA, VRP, CsA, RES, doxorubicin hydrochloride (DOX), thiazolyl blue tetrazolium bromide (MTT reagent), rhodamine 123 (Rh123), and a kit for Annexin V/propidium iodide staining were obtained from Sigma (St. Louis, MO). Stock solutions of UA (up to 40 mM), RES (up to 400 mM), and Rh123 (100 mM) were prepared in DMSO. Stock solutions of CsA (100 mM in EtOH), VRP (100 mM in H2O), and DOX (40 mM in H2O) were also prepared. For Western blotting, primary antibodies used were for phosphorylated (Ser536) p65 and total p65 (Cell Signaling Technology, Danvers, MA), P-gp (Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin (Abcam, Cambridge, MA).
Cell culture
MT1/2 mouse skin papilloma cells and Ca3/7 mouse skin carcinoma cells were maintained in Joklik MEM containing 8% FBS, 50 U/ml penicillin, 50 ng/ml streptomycin, 10 μg/ml transferrin, 50 μg/ml gentamicin sulfate, 5 μg/ml insulin, 5 ng/ml EGF, 10 μM o-phosphorylethanolamine, and 10 μM 2-aminoethanol. B16F10 metastatic mouse melanoma cells were maintained in DMEM containing 10% FBS, 50 U/ml penicillin, and 50 ng/ml streptomycin. A549 human lung adenocarcinoma cells were grown in DMEM/F12 containing 10% FBS, 50 U/ml penicillin, and 50 ng/ml streptomycin. All cells were grown in an incubator at 5% CO2 and 37°C. MT1/2 are papilloma-producing cells derived from SENCAR mouse skin initiated with N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and treated with 12-O-tetradecanoylphorbol-13-acetate (TPA) (30). Ca3/7 were obtained from carcinomas grown using the 7,12-dimethylbenz[α]anthracene (DMBA)/TPA skin carcinogenesis protocol in SENCAR mice, and reform metastatic carcinomas when implanted into nude mice (31). B16F10 cells were provided by the lab of Dr. Tyler Curiel (UTHSCSA). A549 cells were provided by the lab of Dr. Sung-Jen Wei (UTHSCSA).
MTT assay
Cells were treated with various doses of UA, DOX, or 0.1% vehicle for 24 hr. In combination experiments, cells were pretreated with VRP, CsA, RES, or 0.1% vehicle for 1 hr followed by various doses of UA or 0.1% vehicle for 24 hr. In the study with A549 cells, cells were treated for 48 hr. MTT reagent (0.5 mg/ml) was added, and cells were incubated for an additional 2.5 hr. Media was removed and formazan crystals were dissolved with DMSO. Samples were measured at 570 nm with background subtraction at 650 nm, using a Biotek Synergy HT spectrophotometer (Biotek, Winooski, VT).
Annexin V/Propidium Iodide staining
Ca3/7 cells were treated with indicated doses of RES, UA, or 0.1% DMSO vehicle for 12 hr. Media and trypsinized cells were collected and spun at 150×g for 3 min, rinsed and spun again, and resuspended in 1× binding buffer containing Annexin V and propidium iodide according to the manufacturer's instructions. Cells were read on an LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ) with dead cells gated out.
Western blotting
MT1/2 and Ca3/7 cells were treated with indicated doses of VRP, CsA, RES, or 0.1% vehicle for 24 hr. Media was aspirated and cells were rinsed twice with cold PBS before lysis in buffer containing 1% Triton X-100, 0.5% IGEPAL, 0.05 M TrisHCl and 0.1M NaCl as well as protease/phosphatase inhibitors and 5 mM EDTA. Proteins were extracted by centrifugation and quantified by using the Bradford reagent method. Proteins were denatured in XT Sample Buffer containing XT reducing agent. Proteins were separated on 4-12% gradient gels, transferred onto PVDF, blocked in 5% bovine serum albumin or 5% milk in TBST for 1 hr before incubation overnight in primary antibody solution. Blots were rinsed, incubated in secondary antibody for 1 hr and developed using ECL2 Western blotting substrate (Pierce, Rockford, IL). Digital images of films were acquired using EPSON Scan (EPSON, Long Beach, CA). Densitometry was measured using UN-SCAN-IT (Silk Scientific, Orem, UT).
Rh123 assay
Cells suspended in Hank's Balanced Salt Solution (HBSS) were incubated with 10 μM Rh123 for 1 hr. Cells were centrifuged and rinsed twice, followed by efflux for the desired timepoints at 37°C. Cells were rinsed and kept on ice in the dark in HBSS. Cells were rinsed and resuspended in PBS before analysis. For flow cytometry, live cells were gated and FITC channel mean fluorescence was measured. At least 2500 live cells per group per experiment were analyzed. Results were normalized to the Rh123 initial (“accumulation”) level in the MT1/2 cells.
Alternatively, in experiments with P-gp inhibitors or RES, Rh123 levels were measured using a plate reader. MT1/2 and Ca3/7 cells were treated with vehicle or 100 μM VRP, CsA, or RES for 24 hr. 10 μM Rh123 was added for 1 hr. Plates were rinsed twice with cold PBS and resupplied with the compounds in warm media for 30 min. Plates were rinsed twice with cold PBS and lysed with lysis buffer. Proteins were extracted by centrifugation and quantified by Bradford method. 50 μL extracted lysate per sample was transferred to a black plate and read at 485 nm/530 nm using a Biotek Synergy HT spectrophotometer. Rh123 levels for each sample were normalized to the protein concentration.
Statistical Analyses
Differences between individual groups were determined by ANOVA. IC50 values were calculated with GraphPad Prism. Standard deviation is given except where otherwise noted.
Results
Effect of UA and DOX on skin cancer cell viability
Preliminary cell viability experiments showed Ca3/7 cells are more resistant to the effects UA than are MT1/2 cells (Figure 1A). UA had an IC50 of 8.17±0.49 in MT1/2 cells versus an IC50 of 19.66±3.69 in Ca3/7 cells (p<0.001). In addition, the Ca3/7 cells are also chemoresistant to DOX, a P-gp substrate chemotherapeutic, relative to MT1/2 cell lines (Figure 1B). DOX reduced viability of MT1/2 cells with an IC50 of 0.95±0.29 versus an IC50 of 45.2±3.47 in Ca3/7 cells (p<0.005). While UA is less potent than DOX in MT1/2 cells, UA achieves greater cell viability decreases than does DOX in Ca3/7 carcinoma cells (p<0.005).
Figure 1.

A) Ca3/7 cells are less sensitive to UA than are MT1/2 cells (Average of 4 independent experiments, n=3/exp, * p<0.05, ** p<0.01, *** p<0.005, and **** p<0.001 between cell lines). B) Ca3/7 cells are less sensitive to DOX than are MT1/2 cells (Average of 2 independent experiments, n=3/exp, * p<0.05, ** p<0.01, *** p<0.005, and **** p<0.001 between cell lines).
Ca3/7 cells have higher basal P-gp levels and activity
Factors typically involved in chemoresistance were measured by Western blotting. We found that Ca3/7 cells expressed higher levels of P-gp, relative to MT1/2 cells (Figures 2A and 2C). We also found Ca3/7 cells have higher levels of activated NFκB, indicated by phosphorylation status of the active subunit p65 (Figures 2B and 2C). NFκB contributes to transcription of P-gp, which may partially explain the chemoresistant effects of NFκB activation (8). In addition, we analyzed cells for basal levels of cyclin D1 and bcl-2, but found no difference between cell lines (data not shown).
Figure 2.

A) Ca3/7 cells have higher basal p-glycoprotein levels than MT1/2 cells (Average of 3 experiments, n=3/experiment). B) Ca3/7 cells have higher basal NFκB activity (measured by phosphorylation of p65) than MT1/2 cells (Average of 3 experiments, n=3/experiment). C) Representative Western blot for the data in Figures 2A and 2B. D) Ca3/7 cells have higher basal p-glycoprotein activity than MT1/2 cells. This was indicated by decreased accumulation and increased efflux of Rh123, a p-glycoprotein substrate, measured by flow cytometry (Average of 4 experiments, n=2/exp). For these experiments, significance between cell lines is noted by * p<0.05, ** p<0.01, *** p<0.005, and **** p<0.001.
We also used P-gp substrate Rh123 to determine if the increase in P-gp levels led to an increase in P-gp activity. Flow cytometry experiments indicated Ca3/7 cells accumulated less Rh123 than did MT1/2 cells (Figure 2D). Similar results were obtained in preliminary studies measuring Rh123 with a plate reader (data not shown). The decreased intracellular levels of Rh123 were also evident 15 min, 30 min, and 60 min after rinsing (Figure 2D). Data normalized to the accumulation level of Rh123 (data not shown) indicated that Ca3/7 cells also had a higher rate of efflux at the 15 and 30 min timepoints. Preliminary experiments using 30 min, 60 min, and 120 min timepoints indicated overall Rh123 levels had equalized by 120 min, but Ca3/7 cells still had a significantly higher efflux rate than MT1/2 cells at 30 min (data not shown). Similar correlations between efflux of Rh123 and relative protein or mRNA levels of P-gp have been shown for different cancer cell lines (32).
P-gp inhibitors enhance the cytotoxic effects of UA
The differences in sensitivity to UA between cell lines, combined with differences in P-gp expression and activity, indicate that UA may be a P-gp substrate in these skin cancer cells. We next examined pharmacological inhibitors of P-gp to see if they could enhance the cytotoxic effects of UA.
VRP and CsA have been shown to inhibit P-gp in multiple systems, leading to enhanced chemotherapeutic effects (12-14, 33). In addition, RES has been shown to sensitize cancer cells to traditional chemotherapeutics, perhaps by inhibiting P-gp expression and/or activity (28, 29). We pretreated MT1/2 and Ca3/7 cells with a range of doses of VRP, CsA, or RES for 1 hr before incubation with different doses of UA.
For these studies, the viable cells in each treatment group were normalized to vehicle control, and IC50 values for UA in the presence of vehicle or 5-100 μM of VRP, CsA, or RES were calculated. In our experiments, we found that VRP significantly and dose-dependently enhanced UA-mediated decreases in viability of both MT1/2 and Ca3/7 tumor cells. This effect was significant with as low as 50 μM VRP in MT1/2 cells (Table 1) and as little as 5 μM VRP in Ca3/7 cells (Table 2). Similarly, CsA dose-dependently and significantly enhanced UA-mediated decreases in viability of Ca3/7 cells. This effect was significant with as little as 5 μM CsA (Table 2). However, the enhancing effects of CsA were not evident in MT1/2 cells, even up to 100 μM CsA (Table 1). Finally, RES starting at 25 μM enhanced the anti-proliferative effect of UA in the Ca3/7 cell line in a dose-dependent manner (Table 2). RES also enhanced the effects of UA in MT1/2 cells, starting at 25 μM, however this effect did not increase at higher doses (Table 1). Also, the synergistic effects of RES in Ca3/7 cells were significantly stronger (p<0.05) than in MT1/2 cells at 100 μM. Representative experiments for the data in Tables 1 and 2 are shown in Supplementary Figure S1.
Table 1. VRP and RES enhanced UA-mediated cytotoxicity in MT1/2 cells, indicated by a decrease in the IC50 for UA (average of 3-4 independent experiments, n=3/exp).
| compound | dose | fold decrease of IC50 | p-value |
|---|---|---|---|
| +veh | 1 | ||
| +5 μM | 1.14±0.16 | ||
| VRP | +10 μM | 1.17±0.19 | |
| +25 μM | 1.53±0.31 | ||
| +50 μM | 1.99±0.41 | p<0.01 | |
| +100 μM | 2.52±0.53 | p<0.05 | |
| +veh | 1 | ||
| +5 μM | 1.00±0.02 | ||
| CsA | +10 μM | 1.03±0.11 | |
| +25 μM | 1.13±0.11 | ||
| +50 μM | 1.25±0.27 | ||
| +100 μM | 1.24±0.23 | ||
| +veh | 1 | ||
| +5 μM | 1.01±0.06 | ||
| RES | +10 μM | 1.01±0.07 | |
| +25 μM | 1.38±0.2 | p<0.05 | |
| +50 μM | 1.35±0.17 | p<0.05 | |
| +100 μM | 1.3±0.11 | p<0.005 |
Table 2. VRP, CsA, and RES enhanced UA-mediated cytotoxicity in Ca3/7 cells, indicated by a decrease in the IC50 for UA (average of 3-4 independent experiments, n=3/exp).
| compound | dose | fold decrease of IC50 | p-value |
|---|---|---|---|
| +veh | 1 | ||
| +5 μM | 1.22±0.1 | p<0.01 | |
| VRP | +10 μM | 1.22±0.13 | p<0.05 |
| +25 μM | 1.86±0.32 | p<0.05 | |
| +50 μM | 1.85±0.15 | p<0.0001 | |
| +100 μM | 2.85±0.41 | p<0.005 | |
| +veh | 1 | ||
| +5 μM | 1.34±0.06 | p<0.005 | |
| CsA | +10 μM | 1.53±0.2 | p<0.05 |
| +25 μM | 1.82±0.27 | p<0.05 | |
| +50 μM | 1.96±0.43 | p<0.05 | |
| +100 μM | 1.93±0.39 | p<0.05 | |
| +veh | 1 | ||
| +5 μM | 0.91±0.16 | ||
| RES | +10 μM | 1.0±0.29 | |
| +25 μM | 1.35±0.15 | p<0.01 | |
| +50 μM | 1.42±0.17 | p<0.005 | |
| +100 μM | 2.31±0.52 | p<0.01 |
Stronger synergistic effects, indicated by an average fold IC50 decrease of 3.26 ± 0.45, were observed in the Ca3/7 cell line pre-treated with 200 μM RES (Supplementary Figure S2). The synergistic effects of RES and UA in Ca3/7 cells were confirmed by flow cytometry, which showed these compounds induced a mixed apoptotic/necrotic cell death (Supplementary Figure S3). A synergistic cytotoxic effect of RES and UA was also manifest in the metastatic mouse melanoma cell line B16F10 (average fold IC50 decrease of 1.84 ± 0.13, Supplementary Figure S4), as well as in the transformed mouse epidermal line JB6 P+ (John DiGiovanni lab, personal communication, data not shown) and in the human lung cancer cell line A549 (Supplementary Figure S5).
The discrepancies for the sensitization effects of VRP, CsA, and RES in UA-treated cells may be explained by potential differences in substrate dependence of P-gp inhibitors. Studies have shown that different tea catechins can affect the transport of some P-gp substrates but not others, indicating the potential of P-gp modulation via allosteric sites (34). Others have also found P-gp has multiple distinct sites for interaction with compounds (6, 35, 36). In addition, these cell lines may express variants of P-gp, which could differentially affect the ability of inhibitors to interact with P-gp at these multiple sites (37).
VRP, CsA, and RES do not decrease P-gp levels
Western blotting was performed to determine if the effects of these compounds on potentiating UA-induced cytotoxicity were due to a decrease in P-gp expression. Levels of phosphorylated p65, indicative of NFκB activity, were also measured in order to explain potential changes in P-gp levels. However, none of the treatments significantly decreased P-gp levels or NFκB activity. Some treatments, including CsA in both cell lines and RES in MT1/2 cells significantly increased NFκB activity, while 100 μM RES increased P-gp levels in Ca3/7 cells (Figure 3). In preliminary experiments, 200 μM RES also significantly enhanced P-gp levels in Ca3/7 cells (p<0.05) by a factor of 2.11±0.29 (average of 2 independent experiments).
Figure 3.

None of the compounds used decreased p65 phosphorylation or p-glycoprotein expression (all data normalized to β-actin and vehicle control with S.E.M. shown and doses indicated on the x axis, Figures 3A-3D are the average of 4 independent experiments, Figures 3E and 3F are the average of 6 independent experiments, and a representative Western blot is shown in Figure 3G. Statistical significance is noted by * p<0.05, ** p<0.01, *** p<0.005, and **** p<0.001 relative to vehicle control).
Other studies have shown cell line-specific effects of VRP, CsA, and RES on P-gp protein and mRNA levels. RES has been shown to decrease ABCB1 mRNA expression in HeLa, HepG2, and KBv200 cells but not MCF-7 cells (28, 29). Interestingly, cytotoxic doses of VRP increased ABCB1 mRNA levels in leukemia cells, while non-cytotoxic doses decreased ABCB1 mRNA levels (38). Similarly, studies in human colon carcinoma showed VRP and CsA, at slightly growth-suppressing doses, increased ABCB1 mRNA levels (39). These inhibitors as well as RES may increase P-gp expression as part of a cell stress response to efflux harmful chemicals, as cytotoxic non-P-gp substrates have been shown to increase P-gp expression (40).
VRP and CsA, but not RES, inhibit Rh123 efflux
In order to confirm that VRP, CsA, and RES are functioning via inhibition of P-gp, levels of Rh123 in MT1/2 and Ca3/7 cells after 30 minutes efflux were measured in the presence of these chemicals. The effect of each individual chemical was normalized to their vehicle (H2O, EtOH, or DMSO). Each individual vehicle did not affect the relative Rh123 efflux differences between cell lines (Supplementary Figure S6). As expected, VRP significantly decreased efflux of Rh123 in both cell lines. CsA significantly decreased Rh123 efflux in Ca3/7 cells while showing a trend towards this effect in MT1/2 cells. Interestingly, RES significantly increased Rh123 efflux in MT1/2 cells, but had no effect in Ca3/7 cells (Figure 4).
Figure 4.
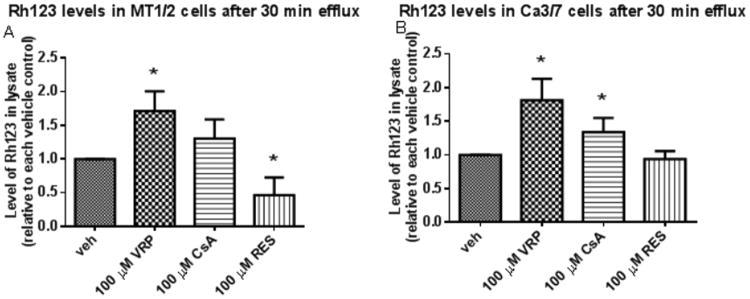
A) Rh123 efflux is inhibited by VRP, unchanged by CsA, and enhanced by RES in MT1/2 cells. B) Rh123 efflux is inhibited by VRP and CsA, but unchanged by RES in Ca3/7 cells (average of 3 independent experiments, n=3/exp, * indicates p<0.05 relative to vehicle control for each group).
These results mirror those found with sensitization of UA-mediated cytotoxicity, which revealed VRP sensitized cells of both lines while CsA only significantly sensitized Ca3/7 cells to UA (Tables 1 and 2, Supplementary Figure S1). However, these results show that RES did not increase Rh123 levels. This, combined with the fact RES does not decrease P-gp levels (Figure 3) indicates RES does not inhibit P-gp to enhance the effects of UA in these skin-relevant systems.
Discussion
In this study we attempted to determine if the cytotoxic effects of the phytochemical UA in skin cancer cell lines could be enhanced with various chemosensitizing compounds. These included VRP and CsA, inhibitors of the efflux pump P-gp, and RES, a phytochemical which can sensitize cells to chemotherapeutics through various mechanisms. In addition, we used DOX, a chemotherapeutic compound effective in a multitude of tumor types including aggressive skin carcinoma (41), as a positive control P-gp substrate. The cell lines used were MT1/2, a cell line derived from chemically-induced mouse skin papilloma (30), and Ca3/7, derived from chemically-induced mouse skin carcinoma (31).
Our results indicated that the Ca3/7 carcinoma line was significantly more chemoresistant to both UA and DOX, relative to the MT1/2 papilloma cell line. In addition, UA more strongly inhibited viability of the skin cancer cell line than DOX. In order to determine the mechanism of relative resistance, pathways associated with chemoresistance were analyzed. We found Ca3/7 cells exhibit higher basal P-gp expression and NFκB activity than MT1/2 cells. As described earlier, the increased P-gp levels may be driven by higher NFκB activity (8), which contributes to transcription of a wide variety of genes involved in progression to carcinomas (42). In addition, Rh123 accumulation and efflux assays showed Ca3/7 cells have higher P-gp activity than MT1/2 cells.
Because P-gp appeared to be a contributing factor to resistance to UA, we combined UA with well-characterized P-gp inhibitors VRP and CsA as well as the chemosensitizing phytochemical RES. While VRP, CsA, and RES all enhanced the cytotoxic effects of UA in Ca3/7 cells, only VRP and RES synergized with UA in MT1/2 cells. One explanation for these discrepancies is the potential that VRP and CsA bind and inhibit P-gp at different sites (43). In addition, studies of different ABCB1 variants have shown no differences in basal activity but differences in the ability of inhibitors to block P-gp activity (37). The differing sensitivities of MT1/2 and Ca3/7 to DOX or UA may be mediated by P-gp amounts, while different interactions between P-gp and inhibitor across cell lines may be explained by ABCB1 polymorphisms as well as separate inhibitor binding sites.
In order to determine if VRP, CsA, and RES are inhibiting P-gp function in these cells, Western blotting for total P-gp levels as well as Rh123 efflux assays for P-gp activity was performed. None of the chemosensitizers significantly decreased P-gp levels or NFκB activity. However, the Rh123 efflux assay indicated VRP increased Rh123 levels in both cell lines, while CsA only increased Rh123 levels in Ca3/7 cells. These results parallel that seen with sensitization to UA-mediated cytotoxicity in these cell lines. Interestingly, the Rh123 efflux assay also showed RES actually decreased Rh123 levels in MT1/2 cells, while having no effect in Ca3/7 cells. Regardless, these results indicate that while VRP and CsA can sensitize skin cancer cells to UA via P-gp inhibition, the synergistic effects of RES and UA occur through a different mechanism. Because RES synergizes with UA through a separate mechanism than P-gp inhibitors, we also propose future studies examining combinations of RES, UA, and P-gp inhibitors in skin cancer-relevant systems.
Other studies have shown RES may sensitize cells by blocking chemotherapy-induced NFκB activity (44). However, in our cancer cell lines, RES did not inhibit NFκB activity (data not shown). Our other preliminary studies showed that RES and UA actually synergistically increased NFκB activity in Ca3/7 cells, measured by Western blotting for phosphorylated p65 (data not shown). Another potential explanation for the synergistic activities of RES and UA is through RES-mediated potentiation of apoptotic pathways or inhibition of metabolizing enzymes (44). While the metabolism of UA is not well-characterized, related compounds such as glycyrrhetinic acid (45) and protopanaxatriol ginsenosides (46) are metabolized by the cytochrome p450 isoform CYP3A4. In addition, a number of studies have shown RES inhibits the activities of this isoform (47). This indicates RES may enhance the effects of UA by preventing its metabolism and subsequently maintaining its intracellular concentration.
Contrary to some positive results, a number of other clinical trials have shown that P-gp inhibitors do not enhance response to chemotherapy. However, many of these studies suffered from various design flaws, including failure to enrich the patient population for those whose tumors overexpress P-gp, show increased P-gp activity through the use of radioimaging, or have demonstrated resistance to previous chemotherapy. In addition, some clinical trials have also combined P-gp inhibitors with a chemotherapy that was not a P-gp substrate (5). As a consequence, recent research and subsequent clinical trials have focused on specific pathway inhibitors and moved away from chemotherapy plus P-gp inhibitors as a treatment modality. However, many of these targeted therapies (gefitinib, gleevec etc) are substrates for P-gp themselves (48). We suggest clinical trials using P-gp inhibitors, perhaps in combination with tolerated natural compounds like UA, need to be explored in chemoresistant tumor types.
UA has been shown to inhibit tumor formation in a variety of animal models, including cancers of the breast (18), lung (19), and skin (20) among others. In addition, UA is reasonably tolerated in humans, and can achieve concentrations as high as 7.57 μM in the serum when supplied intravenously in nanoliposomes (49). We suggest use of UA in addition to approved P-gp inhibitors may be a valuable method to prevent or reverse tumor formation in P-gp-expressing human tumors. Also, topically applied UA plus P-gp inhibitors may provide an easily-administered, non-scarring alternative to traditional surgery for resectable SCC. In addition to enhancing UA accumulation in P-gp-expressing SCC, P-gp inhibition will help limit topical applications of UA or other substrate chemotherapeutics to the epidermis. Studies have shown that knocking out P-gp in mouse skin kept P-gp substrates Rh123 and itraconzaole localized to the epidermis. Topical application of P-gp inhibitors propranolol and VRP had a similar effect on itraconazole distribution (50). A treatment strategy of UA combined with P-gp inhibitors would allow higher and longer-lasting doses of UA in the epidermis and in SCC in particular, while limiting systemic exposure and potential side effects or other interactions.
Supplementary Material
Acknowledgments
We thank Dr. Tyler Curiel for supplying us with B16F10 cells and Dr. Sung-Jen Wei for the A549 cells. Flow cytometry data was generated in the Flow Cytometry Shared Resource Facility which is supported by UTHSCSA, NIH-NCI P30 CA54174 (CTRC at UTHSCSA) and UL1RR025767 (CTSA grant).
Grant Support: Financial Support was provided by NIH (R01 CA164159, P30 CA54174), CTSA (UL1RR025767), an Oppenheimer Multi-Investigator Research Award, and a PhRMA Pre Doctoral Fellowship.
Financial support: Financial Support was provided by NIH (R01 CA164159, P30 CA54174), CTSA (UL1RR025767), an Oppenheimer Multi-Investigator Research Award, and a PhRMA Pre Doctoral Fellowship.
Footnotes
Conflicts of interest: None
References
- 1.Martinez-Rivera M, Siddik ZH. Resistance and gain-of-resistance phenotypes in cancers harboring wild-type p53. Biochem Pharmacol. 2012;83(8):1049–62. doi: 10.1016/j.bcp.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reed JC. Bcl-2 family proteins: regulators of chemoresistance in cancer. Toxicol Lett. 1995;82-83:155–8. doi: 10.1016/0378-4274(95)03551-6. [DOI] [PubMed] [Google Scholar]
- 3.Dey A, Tergaonkar V, Lane DP. Double-edged swords as cancer therapeutics: simultaneously targeting p53 and NF-kappaB pathways. Nat Rev Drug Discov. 2008;7(12):1031–40. doi: 10.1038/nrd2759. [DOI] [PubMed] [Google Scholar]
- 4.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74(2-3):72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robey RW, Massey PR, Amiri-Kordestani L, Bates SE. ABC transporters: unvalidated therapeutic targets in cancer and the CNS. Anticancer Agents Med Chem. 2010;10(8):625–33. doi: 10.2174/187152010794473957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc Natl Acad Sci U S A. 1997;94(20):10594–9. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nabekura T, Yamaki T, Hiroi T, Ueno K, Kitagawa S. Inhibition of anticancer drug efflux transporter P-glycoprotein by rosemary phytochemicals. Pharmacol Res. 2010;61(3):259–63. doi: 10.1016/j.phrs.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Bentires-Alj M, Barbu V, Fillet M, Chariot A, Relic B, Jacobs N, et al. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene. 2003;22(1):90–7. doi: 10.1038/sj.onc.1206056. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein LJ, Galski H, Fojo A, Willingham M, Lai SL, Gazdar A, et al. Expression of a multidrug resistance gene in human cancers. J Natl Cancer Inst. 1989;81(2):116–24. doi: 10.1093/jnci/81.2.116. [DOI] [PubMed] [Google Scholar]
- 10.Clarke R, Leonessa F, Trock B. Multidrug resistance/P-glycoprotein and breast cancer: review and meta-analysis. Semin Oncol. 2005;32(6 Suppl 7):S9–15. doi: 10.1053/j.seminoncol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Ross DD. Modulation of drug resistance transporters as a strategy for treating myelodysplastic syndrome. Best Pract Res Clin Haematol. 2004;17(4):641–51. doi: 10.1016/j.beha.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 12.Belpomme D, Gauthier S, Pujade-Lauraine E, Facchini T, Goudier MJ, Krakowski I, et al. Verapamil increases the survival of patients with anthracycline-resistant metastatic breast carcinoma. Ann Oncol. 2000;11(11):1471–6. doi: 10.1023/a:1026556119020. [DOI] [PubMed] [Google Scholar]
- 13.List AF, Kopecky KJ, Willman CL, Head DR, Persons DL, Slovak ML, et al. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood. 2001;98(12):3212–20. doi: 10.1182/blood.v98.12.3212. [DOI] [PubMed] [Google Scholar]
- 14.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41(5):1967–72. [PubMed] [Google Scholar]
- 15.Franco R, Nicoletti G, Lombardi A, Di Domenico M, Botti G, Zito Marino F, et al. Current treatment of cutaneous squamous cancer and molecular strategies for its sensitization to new target-based drugs. Expert Opin Biol Ther. 2013;13(1):51–66. doi: 10.1517/14712598.2012.725720. [DOI] [PubMed] [Google Scholar]
- 16.Claerhout S, Verschooten L, Van Kelst S, De Vos R, Proby C, Agostinis P, et al. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer. 2010;127(12):2790–803. doi: 10.1002/ijc.25300. [DOI] [PubMed] [Google Scholar]
- 17.Skazik C, Wenzel J, Marquardt Y, Kim A, Merk HF, Bickers DR, et al. P-glycoprotein (ABCB1) expression in human skin is mainly restricted to dermal components. Exp Dermatol. 2011;20(5):450–2. doi: 10.1111/j.1600-0625.2010.01237.x. [DOI] [PubMed] [Google Scholar]
- 18.De Angel RE, Smith SM, Glickman RD, Perkins SN, Hursting SD. Antitumor effects of ursolic acid in a mouse model of postmenopausal breast cancer. Nutr Cancer. 2010;62(8):1074–86. doi: 10.1080/01635581.2010.492092. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Tan X, Shu L, Sun H, Song J, Jin P, et al. Ursolic acid inhibits cigarette smoke extract-induced human bronchial epithelial cell injury and prevents development of lung cancer. Molecules. 2012;17(8):9104–15. doi: 10.3390/molecules17089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang MT, Ho CT, Wang ZY, Ferraro T, Lou YR, Stauber K, et al. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994;54(3):701–8. [PubMed] [Google Scholar]
- 21.Shanmugam MK, Dai X, Kumar AP, Tan BK, Sethi G, Bishayee A. Ursolic acid in cancer prevention and treatment: molecular targets, pharmacokinetics and clinical studies. Biochem Pharmacol. 2013;85(11):1579–87. doi: 10.1016/j.bcp.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Limami Y, Pinon A, Leger DY, Mousseau Y, Cook-Moreau J, Beneytout JL, et al. HT-29 colorectal cancer cells undergoing apoptosis overexpress COX-2 to delay ursolic acid-induced cell death. Biochimie. 2011;93(4):749–57. doi: 10.1016/j.biochi.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Limami Y, Pinon A, Leger DY, Pinault E, Delage C, Beneytout JL, et al. The P2Y2/Src/p38/COX-2 pathway is involved in the resistance to ursolic acid-induced apoptosis in colorectal and prostate cancer cells. Biochimie. 2012;94(8):1754–63. doi: 10.1016/j.biochi.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Shan JZ, Xuan YY, Ruan SQ, Sun M. Proliferation-inhibiting and apoptosis-inducing effects of ursolic acid and oleanolic acid on multi-drug resistance cancer cells in vitro. Chin J Integr Med. 2011;17(8):607–11. doi: 10.1007/s11655-011-0815-y. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Zhao Y, Jiang L, Miao X, Zhou H, Jia L. Glycomic alterations are associated with multidrug resistance in human leukemia. Int J Biochem Cell Biol. 2012;44(8):1244–53. doi: 10.1016/j.biocel.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 26.Shi R, Li W, Zhang X, Zhang Y, Peng H, Xie Y, et al. A novel indirubin derivative PHII-7 potentiates adriamycin cytotoxicity via inhibiting P-glycoprotein expression in human breast cancer MCF-7/ADR cells. Eur J Pharmacol. 2011;669(1-3):38–44. doi: 10.1016/j.ejphar.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 27.Goldberg H, Ling V, Wong PY, Skorecki K. Reduced cyclosporin accumulation in multidrug-resistant cells. Biochem Biophys Res Commun. 1988;152(2):552–8. doi: 10.1016/s0006-291x(88)80073-1. [DOI] [PubMed] [Google Scholar]
- 28.Al-Abd AM, Mahmoud AM, El-Sherbiny GA, El-Moselhy MA, Nofal SM, El-Latif HA, et al. Resveratrol enhances the cytotoxic profile of docetaxel and doxorubicin in solid tumour cell lines in vitro. Cell Prolif. 2011;44(6):591–601. doi: 10.1111/j.1365-2184.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quan F, Pan C, Ma Q, Zhang S, Yan L. Reversal effect of resveratrol on multidrug resistance in KBv200 cell line. Biomed Pharmacother. 2008;62(9):622–9. doi: 10.1016/j.biopha.2008.07.089. [DOI] [PubMed] [Google Scholar]
- 30.Conti CJ, Fries JW, Viaje A, Miller DR, Morris R, Slaga TJ. In vivo behavior of murine epidermal cell lines derived from initiated and noninitiated skin. Cancer Res. 1988;48(2):435–9. [PubMed] [Google Scholar]
- 31.Klann RC, Fitzgerald DJ, Piccoli C, Slaga TJ, Yamasaki H. Gap-junctional intercellular communication in epidermal cell lines from selected stages of SENCAR mouse skin carcinogenesis. Cancer Res. 1989;49(3):699–705. [PubMed] [Google Scholar]
- 32.Lee JS, Paull K, Alvarez M, Hose C, Monks A, Grever M, et al. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol Pharmacol. 1994;46(4):627–38. [PubMed] [Google Scholar]
- 33.Ross DD, Wooten PJ, Sridhara R, Ordonez JV, Lee EJ, Schiffer CA. Enhancement of daunorubicin accumulation, retention, and cytotoxicity by verapamil or cyclosporin A in blast cells from patients with previously untreated acute myeloid leukemia. Blood. 1993;82(4):1288–99. [PubMed] [Google Scholar]
- 34.Wang EJ, Barecki-Roach M, Johnson WW. Elevation of P-glycoprotein function by a catechin in green tea. Biochem Biophys Res Commun. 2002;297(2):412–8. doi: 10.1016/s0006-291x(02)02219-2. [DOI] [PubMed] [Google Scholar]
- 35.Pascaud C, Garrigos M, Orlowski S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J. 1998;333(Pt 2):351–8. doi: 10.1042/bj3330351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol. 2000;58(3):624–32. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 37.Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–8. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 38.Muller C, Goubin F, Ferrandis E, Cornil-Scharwtz I, Bailly JD, Bordier C, et al. Evidence for transcriptional control of human mdr1 gene expression by verapamil in multidrug-resistant leukemic cells. Mol Pharmacol. 1995;47(1):51–6. [PubMed] [Google Scholar]
- 39.Herzog CE, Tsokos M, Bates SE, Fojo AT. Increased mdr-1/P-glycoprotein expression after treatment of human colon carcinoma cells with P-glycoprotein antagonists. J Biol Chem. 1993;268(4):2946–52. [PubMed] [Google Scholar]
- 40.Chaudhary PM, Roninson IB. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J Natl Cancer Inst. 1993;85(8):632–9. doi: 10.1093/jnci/85.8.632. [DOI] [PubMed] [Google Scholar]
- 41.Cranmer LD, Engelhardt C, Morgan SS. Treatment of unresectable and metastatic cutaneous squamous cell carcinoma. Oncologist. 2010;15(12):1320–8. doi: 10.1634/theoncologist.2009-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loercher A, Lee TL, Ricker JL, Howard A, Geoghegen J, Chen Z, et al. Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Res. 2004;64(18):6511–23. doi: 10.1158/0008-5472.CAN-04-0852. [DOI] [PubMed] [Google Scholar]
- 43.Litman T, Skovsgaard T, Stein WD. Pumping of drugs by P-glycoprotein: a two-step process? J Pharmacol Exp Ther. 2003;307(3):846–53. doi: 10.1124/jpet.103.056960. [DOI] [PubMed] [Google Scholar]
- 44.Kaminski BM, Steinhilber D, Stein JM, Ulrich S. Phytochemicals resveratrol and sulforaphane as potential agents for enhancing the anti-tumor activities of conventional cancer therapies. Curr Pharm Biotechnol. 2012;13(1):137–46. doi: 10.2174/138920112798868746. [DOI] [PubMed] [Google Scholar]
- 45.Liu L, Xiao J, Peng ZH, Chen Y. In vitro metabolism of glycyrrhetic acid by human cytochrome P450. Yao Xue Xue Bao. 2011;46(1):81–7. [PubMed] [Google Scholar]
- 46.Hao H, Lai L, Zheng C, Wang Q, Yu G, Zhou X, et al. Microsomal cytochrome p450-mediated metabolism of protopanaxatriol ginsenosides: metabolite profile, reaction phenotyping, and structure-metabolism relationship. Drug Metab Dispos. 2010;38(10):1731–9. doi: 10.1124/dmd.110.033845. [DOI] [PubMed] [Google Scholar]
- 47.Chow HH, Garland LL, Hsu CH, Vining DR, Chew WM, Miller JA, et al. Resveratrol modulates drug- and carcinogen-metabolizing enzymes in a healthy volunteer study. Cancer Prev Res (Phila) 2010;3(9):1168–75. doi: 10.1158/1940-6207.CAPR-09-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Modok S, Mellor HR, Callaghan R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr Opin Pharmacol. 2006;6(4):350–4. doi: 10.1016/j.coph.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Z, Qian Z, Yan Z, Zhao C, Wang H, Ying G. A phase I pharmacokinetic study of ursolic acid nanoliposomes in healthy volunteers and patients with advanced solid tumors. Int J Nanomedicine. 2013;8:129–36. doi: 10.2147/IJN.S38271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito K, Nguyen HT, Kato Y, Wakayama T, Kubo Y, Iseki S, et al. P-glycoprotein (Abcb1) is involved in absorptive drug transport in skin. J Control Release. 2008;131(3):198–204. doi: 10.1016/j.jconrel.2008.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.