

Abstract
Aims
The importance of transforming growth factor beta (TGFβ) as an immune regulatory cytokine in atherosclerosis has been established. However, the role of TGFβ signalling in dendritic cells (DCs) and in DC-mediated T cell proliferation and differentiation in atherosclerosis is unknown.
Methods and results
Here, we investigated the effect of disrupted TGFβ signalling in DCs on atherosclerosis by using mice carrying a transgene resulting in functional inactivation of TGFβ receptor II (TGFβRII) signalling in CD11c+ cells (Apoe−/−CD11cDNR). Apoe−/−CD11cDNR mice exhibited an over two-fold increase in the plaque area compared with Apoe−/− mice. Plaques of Apoe−/−CD11cDNR mice showed an increase in CD45+ leucocyte content, and specifically in CD3+, CD4+ and CD8+ cells, whereas macrophage content was not affected. In lymphoid organs, Apoe−/−CD11cDNR mice had equal amounts of CD11c+ cells, and CD11c+CD8+ and CD11c+CD8− subsets, but showed a subtle shift in the CD11c+CD8− population towards the more inflammatory CD11c+CD8−CD4− DC subset. In addition, the number of plasmacytoid-DCs decreased. Maturation markers such as MHCII, CD86 and CD40 on CD11chi cells did not change, but the CD11cDNR DCs produced more TNFα and IL-12. CD11c+ cells from CD11cDNR mice strongly induced T-cell proliferation and activation, resulting in increased amounts of effector T cells producing high amounts of Th1 (IFN-γ), Th2 (IL-4, IL-10), Th17 (IL-17), and Treg (IL-10) cytokines.
Conclusion
Here, we show that loss of TGFβRII signalling in CD11c+ cells induces subtle changes in DC subsets, which provoke uncontrolled T cell activation and maturation. This results in increased atherosclerosis and an inflammatory plaque phenotype during hypercholesterolaemia.
Keywords: Atherosclerosis, Inflammation, TGFβ, Dendritic cell
See page 3684 for the editorial comment on this article (doi:10.1093/eurheartj/ehs228)
Introduction
The adaptive immune system plays a crucial role in atherosclerosis. Different T cell subsets, B cells, and natural killer (NK) T cells modulate atherosclerotic plaque progression, plaque inflammation, and athero-thrombosis.1 The polarization of the immune response determines whether a response is pro-atherogenic or anti-atherogenic: differentiation of T cells along the Th1-pathway are pro-atherogenic, along the Treg pathway are anti-atherogenic, and along the Th2 and Th17 pathway either pro- or anti-atherogenic.1
Dendritic cells (DCs) have a central role in initiating and regulating immune responses and especially T cell responses,2 both in lymphoid and peripheral tissue.
It has become evident that many distinctive DC subtypes exist, each with a particular location, a specialized function in the immune system and expressing a unique set of cell surface markers.3,4 Commonly, DCs are classified in the CD11chi MHCII+ conventional DCs (cDCs) and the non-conventional DCs. cDCs are subdivided into migratory and lymphoid-tissue-resident DCs (which can be further divided into two subsets; the CD8+ cDC and the CD8− cDC).5 The non-conventional DCs consist of the plasmacytoid (p)DCs and monocyte-derived DCs. Plasmacytoid DCs are found in many tissues including blood, thymus, bone marrow, liver, and lymphoid organs. Plasmacytoid DCs are known to secrete large amounts of type I interferons (IFNs) and have been associated with the preservation of peripheral tolerance and with the initiation of autoimmune responses.3 However, the specific role of these distinct DC subsets in atherosclerosis remains to be elucidated.6
In the vasculature, DCs are present in the arterial intima7 and were recently ascribed an immune-modulatory role in atherosclerosis.8 Intimal DCs are able to accumulate lipid.9 In more advanced stages of atherosclerosis, DCs were found to affect plaque growth as well as cholesterol homeostasis. Hypercholesterolaemic mice containing DCs with an enhanced lifespan and immunogenicity (DC-hBcl2Apoe−/− DC-hBcl2Ldlr−/− mice) had increased Th1 and Th17 cytokine expression levels, but decreased plasma cholesterol levels, and no change in the atherosclerotic plaque area.6 In contrast, Apoe−/− mice in which DCs were depleted (CD11c-DTRApoe−/−) mice had increased cholesterol levels.10 Vaccination using mature DCs pulsed with oxidized low-density lipoprotein (oxLDL) reduced lesion size,11 whereas vaccination with malondialdehyde (MDA)-LDL pulsed DCs aggravated atherosclerosis,12 illustrating the tight and complex modulation of atherosclerosis by DCs in different conditions.
Other studies stressing the importance of DC-T cell interactions in atherosclerosis are interventions in co-stimulatory molecules such as CD80/86, CD40/CD40L, OX40L/OX40, CD137 and ICOS, which are all pivotal for DC-mediated T cell proliferation and polarization.13–18 For example, we have shown that inhibition of CD40L, CD40, or CD40-TNF-Receptor Associated Factor (TRAF) 6 interactions reduces atherosclerosis by affecting T cell activation, polarization, and monocyte activation, and by upregulating the anti-inflammatory cytokine transforming growth factor beta (TGFβ).19–21
Transforming growth factor beta is a powerful modulator of immune responses. It not only regulates immune responses by inhibiting the proliferation of naïve and activated T cells, and by mediating T cell polarization, but also has a crucial role in mediating DC functions.22,23 Transforming growth factor beta can immobilize DCs, induce DC apoptosis, and can decrease CD40, CD80, and CD86 expression on the cell surface. Moreover, TGFβ can dampen tumour necrosis factor (TNF)α, IL12, and CCL5 production in DCs, thereby rendering a more tolerogenic DC that promotes Treg development.24
In the present study, we investigated the effects of TGFβRII signalling in DCs on atherosclerosis in Apoe−/− mice that contained a dysfunctional TGFβ Receptor II in CD11c+ cells (Apoe−/−CD11cDNR). We found that impairment of TGFβRII signalling in CD11c+ cells accelerated atherosclerosis, enhanced the influx of both CD4+ and CD8+ T cells into the plaque, and decreased plaque fibrosis, while lowering cholesterol levels. This aggravated atherosclerosis was accompanied by augmented pro-inflammatory T cell and DC responses.
Methods
Apoe−/−CD11c-dnTGFβRII transgenic mice
CD11c-dnTGFβRII (CD11cDNR) mice were generated as described previously and were backcrossed to Apoe−/− mice.25 The transgene was composed of the human TGFβ type II receptor sequence (–7 and +573) that encodes for the extracellular and transmembrane regions, but not the intracellular region of the TGFβ type II receptor, thereby preventing TGFβ signalling.25 Genotypes were verified by polymerase chain reaction (PCR) as described before and hemizygous transgenes and their littermate wild types (both Apoe−/−) were used.
The animals were fed a normal chow diet and at the age of 20 weeks, mice (n = 34) were euthanized. Blood was obtained from the retro-orbital plexus and spleen, liver and lymph nodes were harvested after in situ perfusion using PBS followed by 1% paraformaldehyde. Hearts were isolated and frozen in Tissue-Tek (Shandon, Veldhoven, The Netherlands). Other organs collected during autopsy were fixed in 4% paraformaldehyde. All animal experiments were performed under approved Institutional Animal Care and Use Committee protocols of the respective universities.
Histology and morphometry
The plaque area was analysed in the aortic root using serial 6 µm sections with 42 µm intervals, beginning from the onset of the aortic valves until the valves had disappeared. For histological analysis of atherosclerosis, sections were stained with haematoxylin and eosin (HE). The plaque area was measured on a Leica DM3000 Light microscope (Leica Microsystems) coupled to a computerized morphometry system (Leica Qwin 3.5.1).
Immunohistochemistry
Consecutive sections were immunolabelled with anti-CD45 rat monoclonal antibody (1:5000; BD Biosciences) to detect all inflammatory cells, anti-Moma-2 rat monoclonal antibody (1:50; Serotec) to detect macrophages, anti-αSMA monoclonal mouse antibody (1:500; DAKO) as a marker of vascular smooth muscle cells and myofibroblasts and anti-MMP9 goat polyclonal antibody (1:200; Santa Cruz) to detect matrix metalloproteinase 9. Anti-CD3 rabbit monoclonal antibody (1:200; DAKO) was used to detect T lymphocytes, and anti-CD4 and anti-CD8 rat monoclonal antibodies (undiluted, gift from W. Buurman, Department of General Surgery, Academic Hospital Maastricht) to distinguish between T-helper cells and cytotoxic T-cells, respectively. Sirius red staining was used to detect collagen content, both by brightfield- and polarization light microscopy. Morphometric analyses were performed using a Leica Quantimet with Qwin3.5.1 software (Leica Microsystems).
Fluorescent immunohistochemistry was used to determine the presence of CD11c+ cells in the aortic lesions. CD11c, CD11b, DX5, CD4, and CD8 antibodies (all BD Biosciences) were conjugated to fluorescein isothiocyanate (FITC), phycoerythrin (PE), or peridinin chlorophyll protein (PerCP). Sections were analysed with a Leica TCS SP5 multi-photon microscope (Leica).
Lipids and lipoproteins
Plasma cholesterol and triglyceride levels were measured using colorimetric assays (Roche). Size fractionation of lipoproteins was performed by fast-performance liquid chromatography (FPLC) using a 30 × 0.32 cm Superose 6B micro-FPLC column (GE Healthcare) followed by in-line cholesterol detection, as described previously.26
Antibody measurements
Antibody (Ab) titres to Cu2+-LDL and MDA-LDL were measured in the plasma as previously described.27 In brief, copper-oxidized LDL (CuOx-LDL) and malondialdehyde-modified LDL (MDA-LDL) were generated from freshly isolated human LDL. Binding of specific IgM, IgG1, and IgG2c antibodies in individual plasma samples to coated antigens were measured by chemilluminescent enzyme-linked immunosorbent assay (ELISA) at indicated dilutions. Bound antibodies were detected using alkaline phosphatase (AP)-conjugated goat-anti-mouse IgM or biotinylated goat-anti-mouse IgG1 and goat-anti-mouse IgG2c (Jackson Immuno Research) followed by AP-conjugated Neutravidin (Thermo Scientific).
Real-time polymerase chain reaction
RNA was isolated from cultured bone marrow-derived macrophages using the RNeasy Mini kit II (Qiagen). One microgram of total RNA was reverse transcribed into cDNA using the SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen). Real-time PCR reactions (7900HT Fast Real Time PCR system, Applied Biosystems) were carried out with cDNA (equivalent to 10 ng total RNA), TaqMan® Fast Advanced Master Mix, and TaqMan® Gene Expression assays (all Applied Biosystems) for CD40, CD86, TNFα, MHCII, iNOS, Mannose receptor, Arginase 1, RELMα, and, Ym-1 according to the instructions of the manufacturer. Samples were assayed in quadruplicates. The mRNA expression was normalized to that of the housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA expression.
Flow cytometry
Spleen and lymph nodes were harvested from the mice and single cell suspensions were prepared and stained with anti-TCR, -CD3, -CD4, -CD8, -CD25, -Foxp3, -CD44, -CD62L, -CD11c, -CD40, -MHCII, -CD86, -Ly6C, -CD19 (BD Biosciences), or isotype control IgG (all BD Biosciences). Antibodies conjugated to FITC, PE, allophycocyanin (APC), or PerCP were used and cells were analysed using a FACS-Canto II (BD Biosciences).
Cell culture
Spleen and lymph nodes from Apoe−/− and Apoe−/−CD11cDNR mice (n = 5) were isolated and gently mashed through a 70 µm nylon cell strainer. Splenocytes and lymph node cells were cultured in triplicate at 3 × 105 cells/well in the presence of soluble anti-CD3/CD28 (2 μg/mL) in complete RPMI [RPMI 1640 medium supplemented with 2 mM l-glutamine, 10% FCS, and 100 U/mL streptomycin/penicillin (PAA)]. After 48 h, supernatants were collected and TGFβ, IFNγ, IL-10, IL-12p, IL-4, and IL-17 production were measured by ELISA (Ebioscience).
Isolation and purification of CD4+ and CD11c+ cells
CD4+ cells were negatively selected using of a cocktail of antibody-coated magnetic beads (Miltenyi Biotech), resulting in 92% pure CD4+ T cells. CD11c+ cells were positively selected with biotin-conjugated anti-CD11c mAb (N418, PharMingen), streptavidin microbeads (Miltenyi Biotec), followed by two consecutive magnetic cell separations using LS columns (Miltenyi Biotec), resulting in 95% CD11c+ cells.
Bone marrow-derived dendritic cells/macrophages
Femurs and tibiae were removed and the ends of the bones were carefully cut off. The bone marrow was flushed out and filtered. Red blood cells were lysed and the cells were counted and adjusted to 0.5 × 106 cells/mL by adding RPMI 1640 containing 10% heat-inactivated FCS, glutamine, 100 U/mL penicillin/streptomycin, and 50 µM β-Mercaptoethanol. Granulocyte-macrophage colony-stimulating factor (20 ng/mL) was added to generate DCs. At day 10, 99% of the cells were CD11c+. A similar protocol was used to obtain macrophages where L929-conditioned medium (M-CSF) was added.
Cell culture, proliferation, and cytokine assays
Cells were cultured in RPMI 1640 supplemented with Glutamax, 10% FCS, 0.02 mM 2β-mercaptoethanol, and antibiotics. For cytokine measurements, bone marrow-derived CD11c+ cells (2 × 106 cells/well) were stimulated with LPS (10 μg/mL) and IFNγ (100 U/mL) for 48 h. TNFα and IL-12 production in the supernatants were measured using specific ELISAs (R&D Systems).
CD4+ T cells were cultured at 1 × 105 cells/well for 48h in anti-CD3-coated microplates (10 μg/mL) either alone or with CD11c+ cells (2 × 104 cells/well). Cytokine production in the supernatants was measured using specific ELISAs (R&D Systems). For proliferation, cells were cultured at 37°C for 72 h and pulsed with 1 μCi of [3H] thymidine (Amersham) for the last 18 h of culture. Thymidine incorporation was assessed using a TopCount NXT scintillation counter (Perkin Elmer).
Statistical analysis
Values are expressed as mean ± SEM. A non-parametric Mann–Whitney U test (two-tailed), or, when appropriate, a student's t-test was performed (two-tailed). Statistical analysis was performed using Prism (GraphPad). Probability values of P < 0.05 were considered significant.
Results
Localization and subtype of CD11c+ cells in atherosclerotic plaques
Previously, Laouar et al.28 found specific expression of the DNR transgene in CD11c expressing DCs and NK cells but not in T cells, NKT cells, or B cells. To assess which cells express CD11c (and thus the construct) in atherosclerotic lesions, plaques of both Apoe−/−CD11cDNR and Apoe−/− mice were double stained to determine the presence, localization, and identity of CD11c+ cells (Figure 1). CD11c+ cells were found in the shoulder region of the plaque and in the adventitia (Figure 1A–C). Interestingly, only a small percentage of CD11c+ cells in the plaque expressed CD11b (monocytes/macrophages), indicating that the majority of CD11c+ cells in the plaque were either DCs, or macrophage-derived foam cells (CD11c+ CD11b−).29 Interestingly, CD11c+CD8+ DCs, which are well known for the production of IL-12 and their capacity for cross presentation, were only occasionally observed in the atherosclerotic lesions (see Supplementary material online, Figure S1A–C). CD11c+ DX5+ NK cells were not identified (data not shown).
Figure 1.
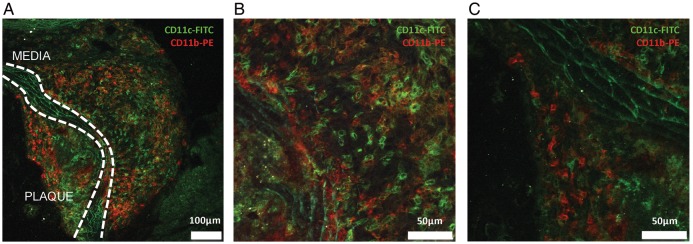
Localization and characterization of CD11c+ cells in atherosclerotic plaques of the aortic root of Apoe−/−CD11cDNR mice fed a normal chow diet for 20 weeks. (A–C) Multiphoton microscopy. CD11c+ (FITC, green) and CD11b+ (PE, red) cells are present in the plaque and lymphoid aggregates in the adventitia. White lines indicate the media. (B) Detail of the adventitia demonstrating infiltration of CD11c+ cells (FITC). (C) Detail of the atherosclerotic plaque. CD11c+ cells (FITC) and CD11b+ cells (PE) are present in the shoulder region of the plaque.
Disruption of TGFβRII signalling in CD11c+ cells affects DC subset distribution and DC function in atherosclerosis
Apoe−/−CD11cDNR mice and their Apoe−/− littermates had equal amounts of CD11c+ cells in spleen and lymph nodes (Figure 2A), with similar CD11c+CD8+ (Figure 2B) and CD11c+CD8− (Figure 2C) DC fractions (for gating strategies see Supplementary material online, Figure S2). Interestingly, within the CD8− DC subset, the CD8−CD4− subset, generally thought to have a more pro-inflammatory capacity, prevailed in the Apoe−/−CD11cDNR mice (Figure 2D).30 Indeed, TNFα and IL12 production were increased in in vitro matured and stimulated CD11cDNR DCs (Figure 2E and F). Plasmacytoid DC (CD3−CD19−CD11clowB220highLy6chigh) levels were significantly decreased in the Apoe-/-CD11c animals (Figure 2G). Remarkably, maturation markers such as MHCII, CD86, and CD40 were similar (see Supplementary material online, Figure S3A–C).
Figure 2.

Inhibition of transforming growth factor beta signalling in CD11c+ cells leads to a shift in dendritic cell subsets. Flow cytometric analysis of splenic cells from Apoe−/− and Apoe−/−CD11cDNR mice (n = 5, *P < 0.05). (A) Percentage of CD11chigh cells (% of non-CD3+CD19+ cells). (B) Percentage of CD8+ dendritic cells (% of CD11chigh cells). (C) Percentage of CD8− dendritic cells (% of CD11chigh cells). (D) Percentage of CD8−CD4− dendritic cells (% of CD11chigh cells, n = 5, *P < 0.05). (E and F) TNFα and IL-12 cytokine production by CD11c+ cells purified from spleen, stimulated by LPS, measured by ELISA (pg/mL). Disruption of transforming growth factor beta signalling in CD11c+ cells does not alter dendritic cell maturation in vivo. (G) MHCII, (H) CD86, (I) CD40 of Apoe−/− and Apoe−/−CD11cDNR splenocytes.
Bone marrow-derived CD11c+ cells from CD11cDNR mice strongly induced T cell proliferation when co-cultured with T cells from wild-type mice upon stimulation with CD3/CD28 antibodies (Figure 3A). In addition, CD11cDNR splenocytes (Figure 3B–E) or splenic DCs co-cultured with T cells (Figure 3F and G) stimulated with CD3/CD28 resulted in strong activation of T cells that produced high levels of Th1 (increased IFN-γ), Th2 (increased IL-4), Th17 (increased IL-17), and Treg (increased IL-10) associated cytokines, compared with splenocytes and DCs from Apoe−/− littermates.
Figure 3.

Disruption of transforming growth factor beta signalling in CD11c+ cells increases T cell proliferation and cytokine expression. (A) T cell proliferation (×104 cpm, n = 3, *P < 0.05). Bone marrow-derived CD11c+ cells from CD11cDNR mice co-cultured with wild-type CD4+ T cells (magnetic bead-isolated). (B–E) Cytokine expression, concentrations (pg/mL) of (B) interferon-γ, (C) IL-4, (D) IL-10, and (E) IL-17 in supernatants of cultured splenocytes from Apoe−/− and Apoe−/−CD11cDNR mice (n = 5, *P < 0.05), stimulated with anti-CD3/CD28 antibodies. Disruption of transforming growth factor beta signalling in bone marrow-derived CD11c+ cells co-cultured with wild type T cells increases (F) interferon-γ and (G) IL-4 cytokine expression (n = 8, *P < 0.05).
In Apoe−/−CD11cDNR mice, lymphoid organs showed a slightly increased CD4+/CD8+ ratio (Figure 4A) and an increased percentage of CD25+Foxp3+ Tregs within the CD4+ population (Figure 4B). Remarkably, the Apoe−/−CD11cDNR had less naïve T cells (CD44lowCD62Lhigh cells), and more effector memory T cells (CD44highCD62Llow) within both the CD4+ and CD8+ subset, suggesting increased mobilization of mature, activated T cells in the absence of TGFβRII-signalling (Figure 4C–F).
Figure 4.

Inhibition of transforming growth factor beta signalling in CD11c+ cells disturbs T cell homeostasis. Flow cytometric analysis of splenic cells from Apoe−/− and Apoe−/−CD11cDNR mice. (A) CD4/CD8 ratio T cells (n = 5, *P < 0.05). (B) Percentage CD25+Foxp3+ of CD4+ T cells (n = 5, *P < 0.05). (C) Percentage of naive (CD44lowCD62Lhigh) CD4+ T cells. (D) Percentage of naive (CD44lowCD62Lhigh) CD8+ T cells (n = 5, *P < 0.05). (E) Percentage of effector (CD44lowCD62Lhigh) CD4+ T cells (n = 5, *P < 0.05). (F) Percentage of effector (CD44high CD62Llow) CD8+ T cells (n = 5, *P < 0.05). (G) Plasma titres of antibodies against Cu2+-oxLDL and MDA-LDL, relative light units (RLU).
Anti-malondialdehyde-LDL or oxLDL antibodies
We next tested whether deficiency of TGFβRII signalling in DCs would change antibody titres or subtypes of IgG antibodies directed against modified LDL in the plasma of Apoe−/−Cd11cDNR mice. However, no significant differences in plasma titres of IgG1, IgG2b, IgG2c, or IgM antibodies against Cu2+-oxLDL and MDA-LDL could be detected between the two groups of mice (Figure 4G).
Deficient transforming growth factor beta receptor II signalling in dendritic cells accelerates atherosclerosis and promotes T cell influx in atherosclerotic plaques
At sacrifice, body weight of Apoe−/− and Apoe−/−CD11cDNR mice was similar. Surprisingly, Apoe−/−CD11cDNR mice had decreased total plasma cholesterol levels, particularly in the chylomicron remnant/VLDL fraction, while plasma triglycerides did not differ (see Supplementary material online, Figure S4).
Despite the decrease in total cholesterol levels, Apoe−/−CD11cDNR mice exhibited a two-fold increase in atherosclerotic plaque area in the aortic root (Figure 5A, C, D) and a two-fold increase in the percentage of stenosis (Figure 5B) compared with the Apoe−/− littermates. Plaque CD45+ leucocyte content was strongly increased in Apoe−/−CD11cDNR animals compared with controls (Figure 6A–D), consistent with the increase in effector memory T cells (Figure 4). To more precisely define the actual leucocyte subsets that were increased in Apoe−/−CD11cDNR mice, we analysed the lesions for the presence of different T cell (CD3, CD4, CD8, Foxp3), and macrophage markers (Moma-2). Plaques of Apoe−/−CD11cDNR mice contained significantly more CD3+ T cells then the controls (Figure 6E–H). Interestingly, increased levels of both CD4+ T-helper cells and CD8+ cytotoxic T cells were found in plaques of Apoe−/−CD11cDNR mice (Figure 6I–P). No differences in the number of Foxp3+ cells could be observed, which was not surprising considering the low numbers of Foxp3+ cells present in plaques (Apoe−/− 1.37 × 10–5 ± 5.51 × 10−6 vs. Apoe−/−CD11cDNR 5.04 × 10−6 ± 2.30 × 10−6 FoxP3+cells/μm2).
Figure 5.

Defective transforming growth factor beta signalling in CD11c+ cells accelerates atherosclerosis. (A) The plaque area (μm2) in the aortic root of Apoe−/− and Apoe−/−CD11cDNR animals (n = 8, *P < 0.05). (B) The fractional area of the lesion (% stenosis) of Apoe−/− and Apoe−/−CD11cDNR mice (n = 8, *P < 0.05). (C and D) Representative HE staining of atherosclerotic lesions in the aortic root.
Figure 6.

Disruption of transforming growth factor beta signalling in CD11c+ cells enhances plaque inflammation. (A) CD45+ cells per total cell number in the plaque and (B) CD45+ cells per μm2 plaque area (n = 8, *P < 0.05). (C and D) Representative CD45 immunostaining of Apoe−/− and Apoe−/−CD11cDNR aortic root lesions. (E) CD3+ cells per total cell number in the plaque and (F) number of CD3+ cells per μm2 plaque area (n = 8, *P < 0.05). (G and H) Representative CD3 immunostaining of Apoe−/− and Apoe−/−CD11cDNR aortic root lesions. (I) CD4+ cells per total cell number in the atherosclerotic plaque not and (J) CD4+ cells per μm2 plaque area (n = 8, *P < 0.05). (K and L) Representative CD4 immunostaining of Apoe−/− and Apoe−/−CD11cDNR aortic root lesions. (M) CD8+ cells per total cell number in the lesions and (N) CD8+ cells per μm2 plaque area (n = 8, *P < 0.05). (O and P) Representative CD8 immunostaining of Apoe−/− and Apoe−/−CD11cDNR aortic root lesions.
Absence of transforming growth factor beta receptor II signalling in CD11c+ cells does not significantly alter macrophage phenotype
Plaque macrophage (Moma-2) content did not differ between both groups (Apoe−/− 21.08 ± 2.41 vs. Apoe−/−CD11cDNR 19.10 ± 4.42; % Moma-2+ positive area in the lesion), and, only a slight alteration in macrophage phenotype was detected. Bone marrow-derived macrophages from Apoe−/−CD11cDNR mice stimulated with oxLDL showed increased expression of CD40 and MHCII, but not CD80 or CD86 by flow cytometry (see Supplementary material online, Figure S3D–G). Using real-time PCR, we could not detect any significant changes in expression of TNFα, CD206, Arginase-1, or YM1 (see Supplementary material online, Figure S3H). These data suggest that the absence of TGFβ signalling in CD11c+ cells does not affect macrophage phenotype or alters the M1/M2 balance, but rather affects DC and T cell populations.
Atherosclerotic lesions of Apoe−/−CD11cDNR mice exhibit increased matrix turnover
In atherosclerotic lesions of Apoe−/−CD11cDNR mice, the sirius red positive area had significantly decreased compared with controls (Figure 7A–C). Polarization microscopy confirmed this decrease in collagen content, and revealed a shift towards the red spectrum, specifying thicker and more mature collagen fibres (Figure 7A, D, E), probably reflecting the more advanced stage of the plaques in Apoe−/−CD11cDNR mice. This was accompanied by a decrease in α-smooth muscle cell actin+ (ASMA) cell content in the lesion (Figure 7F–H). MMP9 expression significantly increased in lesions of Apoe−/−CD11cDNR mice (Figure 7I–K).
Figure 7.

Defective transforming growth factor beta signalling in CD11c+ cells leads to an unstable plaque phenotype. (A) Percentage of Sirius red positive staining per aortic root lesions area of Apoe−/− and Apoe−/−CD11cDNR aortic root lesions (n = 8, *P < 0.05). The total of Sirius red positive staining is further divided in percentages of red, yellow, and green fibres as indicated in the graph bars. (B and C) Representative sirius red stainings. (D and E) Representative polarization microscopy images of sirius red stained sections. (F) Amount of SMC content in the atherosclerotic plaques of Apoe−/− and Apoe−/−CD11cDNR aortic roots (n = 8, *P < 0.05). (G and H) Representative ASMA+ stainings. (I) Percentage of MMP9+ area in the plaques (n = 8, *P < 0.05). (J and K) Representative MMP9 immunostainings (magnification ×20).
Discussion
The present study highlights the importance of TGFβRII signalling in DCs for the progression of atherosclerosis. The current experiments clearly show that the absence of TGFβRII signalling in DCs caused an increase in atherosclerotic plaque size, despite a 50% reduction in plasma cholesterol levels. Moreover, plaques contained more CD4+ and CD8+ T cells and less collagen and an increased matrix turnover, and generally were reminiscent of a vulnerable, rupture–prone plaque phenotype in humans.
Several studies have provided evidence for an important role of TGFβ as an immune modulating cytokine in atherosclerosis. Systemic inhibition of TGFβ signalling in Apoe−/− mice by using a recombinant soluble TGFβ type II receptor31 or a blocking TGFβ1 antibody32 resulted in accelerated atherosclerosis. Lesions exhibited an unstable phenotype that contained low amounts of fibrosis, an increased amount of inflammatory cells, and even intraplaque hemorrhages.31,32 Cardiac overexpression of TGFβ1, resulting in increased plasma levels of TGFβ, limited plaque growth and induced plaque stabilization.33 These effects have predominantly been attributed to TGFβ signalling in T cells. Indeed, mice with abrogated TGFβ signalling in T cells (Apoe−/−CD4-dnTGFβRII) also showed accelerated lesion progression, with plaques containing abundant inflammatory cells paralleled by a decrease in plaque fibrosis.34,35 The defect in TGFβRII signalling in T cells increased systemically the amount of Th1 and Th2 cytokines, with profound increases in plasma levels of IFNγ, but also of IL-10 and IL-4. Gojova et al.36 who transplanted bone marrow from CD2-dnTGFβRII mice into Ldlr−/− recipients obtained similar results in plaque phenotype, although plaque area decreased.
The immune modulatory effects of TGFβ are not restricted to T cells. Although TGFβ producing Tregs are well known to control T cell activation and differentiation and play a crucial role in atherosclerosis,22 the immune-regulatory capacity of other cell types is also controlled via TGFβ.24 Phagocytosis of apoptotic cells leads to TGFβ secretion, which inhibits the production of inflammatory cytokines and chemokines (including IL-1β and TNFα) in macrophages.37 Moreover, TGFβ also inhibits macrophage activation and may be involved in polarization towards the M2 phenotype.24 However, in our hands, disruption of the TGFβ signalling in CD11c+ cell did not affect macrophage polarization significantly, although M1 markers like CD40 showed a slight decrease, and expression of M2 markers such as Arginase-1 and the mannose receptor a slight increase.
The exact role of TGFβ in DC biology remains to be elucidated. What is known is that TGFβ can immobilize tumour Langerhans cells,38 and reduces their expression of MHCII, CD86, and CD40 as well as the production of IL-12, TNFα, and CCL5, thereby reducing its antigen-presenting capacity and DC-mediated T cell responses.39–41 In DC development, TGFβ accelerates the differentiation of common DC progenitors (CDP) towards conventional CD8+ and CD8− DCs (cDC) and blocks the differentiation into pDCs.42 Although the majority of these studies focused on Langerhans cells and tumour-associated DCs, these data corroborate our findings. We also observed increased DC cytokine production, which had massive effects on T cell activation and augmented T cell proliferation when TGFβ signalling was abrogated in DCs in an atherosclerosis setting. However, in contrast to earlier findings,42 inhibition of TGFβ-signalling decreased the number of pDCs significantly. A recent report by Daissormont et al.43 showed that inhibition of pDCs is associated with plaque progression and inflammation. Therefore, the observed decrease in pDCs in our mouse model is likely secondary to the increase in plaque area and plaque inflammation.
TGFβ signalling in CD11c+ cells seems also crucial for the recruitment of inflammatory cells into the lesion. A lack of TGFβ signalling in CD11c+ cells results in substantial infiltration of CD4+ and CD8+ cells in atherosclerotic plaques and systemic activation of the T cell compartment as evidenced by an expansion of CD44highCD62Llow effector memory T cells. These activated T cells expressed elevated levels of IFN γ, TNFα, IL-4, IL-17, IL-10, and TGFβ. This broad array of expressed cytokines refers to strong activation of the Th1, Th2, and Th17 T cell subsets. Especially, the Th1 subset is crucial for atherosclerosis, while the Th2 subset can be either pro- or anti-atherogenic.1 The increase in Th17 cells is rather surprising, since TGFβ was considered to be crucial for a Th17 response. However, Th17 cells can develop independently of TGFβ44 and the role of Th17 cells in atherosclerosis is still controversially discussed with some groups reporting a pro-atherogenic,45–47 and others an anti-atherogenic role for Th17 cytokines.48 Interestingly, Apoe−/−CD11cDNR mice develop a compensatory Treg response, which was insufficient to prevent the increase in atherosclerosis.
Both CD11c+ cells, as well as TGFβ, play a crucial, but yet unidentified role in cholesterol homeostasis. During expansion of CD11c+ cells, cholesterol levels and the amount of atherosclerosis decreases, while a depletion of CD11c+ cells results in hypercholesterolaemia, suggesting a tight regulation between DC homeostasis and lipid metabolism.10 Frutkin et al.33 described that hypercholesterolaemia induces TGFβ expression, and Robertson et al.34 observed decreased cholesterol values in the plasma of Apoe−/−CD4DNR mice. Surprisingly, decreased levels of cholesterol, especially in the chylomicron remnant/VLDL fraction, were observed in the plasma of Apoe−/−CD11cDNR mice, suggesting an interaction between TGFβ, DCs, and cholesterol metabolism, which requires further investigation.
In conclusion, we observed that TGFβRII signalling in CD11c expressing cells plays a key role in the regulation of the activation of T cells during atherogenesis. More specifically, our data show that TGFβ signalling in DCs modulates the interaction between the innate and adaptive immune response by affecting DC and T cell activation, thus dampening inflammation in atherosclerosis.
Our data substantially contribute to the understanding of TGFβ signalling in atherosclerosis and underline the important role of TGF-β in regulating immune responses in atherogenesis. Many TGFβ agonists have been developed and their efficacy was tested in numerous animal models.49 However, to apply activation of TGFβ as a therapy for atherosclerosis, a number of obstacles need to be overcome. TGFβ signalling is highly dependent on the environmental milieu and can be either beneficial or detrimental. In neoplastic disease for example, TGFβ suppresses the progression of early tumours, but at more advanced stages of the disease cancer cells start producing TGFβ, which then promotes metastasis.37,50 In addition, chronic administration of TGFβ has been reported to induce interstitial fibrosis and hepatic fibrosis.51 Targeted induction of TGF-β signalling in specific cells such as DCs and T cells might circumvent these adverse side effects. The results of this study will aid the development of immunotherapies to combat a broad range of inflammatory diseases.
Translational aspects/clinical perspectives
In the present paper, we discovered that TGFβ expressed by DCs plays an important role in the modulation of the immune system during atherosclerosis, and consequently in mediating the progression of atherosclerosis. Dendritic cell-specific TGFβ not only mediates DC polarization, but is even more important in keeping the different T-lymphocytes in control, both within the plaque and in blood and lymphoid organs. This renders TGFβ as one of the most powerful regulators of the immune system in atherosclerosis. Dampening of the immune responses by stimulation of TGFβ would therefore slow down the progression of atherosclerosis and induce atherosclerotic plaques with a low inflammatory burden, thereby reducing the incidence of atherosclerotic plaque rupture and subsequent clinical symptoms such as myocardial infarction or stroke.
Until now, different TGFβ agonists have been developed and their efficacy was tested in numerous animal models.49 However, besides an immune-modulatory cytokine, TGFβ is also a pro-fibrotic factor, and systemic therapy consequently induces interstitial and liver fibrosis.51
Based on our present data, stimulation of TGFβ in DCs only would still be beneficial for atherosclerosis by dampening immune responses, but may prevent the adverse effects of global TGFβ stimulation.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This work was supported by the Humboldt Foundation (Sofja Kovalevskaja grant to E.L.), the Netherlands Organization for Scientific Research (VIDI grant to E.L.), and the Netherlands Heart Foundation (established investigator grant to E.L. and E.B.). R.A.F. is an Investigator of the Howard Hughes Medical Institute and A.K.R. was a Wenner-Gren fellow.
Conflict of interest: none declared.
Supplementary Material
References
- 1.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. doi:10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. doi:10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 3.Kushwah R, Hu J. Complexity of dendritic cell subsets and their function in the host immune system. Immunology. 2011;133:409–419. doi: 10.1111/j.1365-2567.2011.03457.x. doi:10.1111/j.1365-2567.2011.03457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. doi:10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naik SH. Demystifying the development of dendritic cell subtypes, a little. Immunol Cell Biol. 2008;86:439–452. doi: 10.1038/icb.2008.28. doi:10.1038/icb.2008.28. [DOI] [PubMed] [Google Scholar]
- 6.Manthey H, Zernecke A. Dendritic cells in atherosclerosis: functions in immune regulation and beyond. Thromb Haemost. 2011;106:772–778. doi: 10.1160/TH11-05-0296. [DOI] [PubMed] [Google Scholar]
- 7.Bobryshev YV, Lord RS. Ultrastructural recognition of cells with dendritic cell morphology in human aortic intima. Contacting interactions of Vascular Dendritic Cells in athero-resistant and athero-prone areas of the normal aorta. Arch Histol Cytol. 1995;58:307–322. doi: 10.1679/aohc.58.307. doi:10.1679/aohc.58.307. [DOI] [PubMed] [Google Scholar]
- 8.Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005;26:1700–1704. doi: 10.1093/eurheartj/ehi282. doi:10.1093/eurheartj/ehi282. [DOI] [PubMed] [Google Scholar]
- 9.Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390. doi: 10.1161/CIRCRESAHA.109.210781. doi:10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- 10.Gautier EL, Huby T, Saint-Charles F, Ouzilleau B, Pirault J, Deswaerte V, Ginhoux F, Miller ER, Witztum JL, Chapman MJ, Lesnik P. Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis. Circulation. 2009;119:2367–2375. doi: 10.1161/CIRCULATIONAHA.108.807537. doi:10.1161/CIRCULATIONAHA.108.807537. [DOI] [PubMed] [Google Scholar]
- 11.Habets KL, van Puijvelde GH, van Duivenvoorde LM, van Wanrooij EJ, de Vos P, Tervaert JW, van Berkel TJ, Toes RE, Kuiper J. Vaccination using oxidized low-density lipoprotein-pulsed dendritic cells reduces atherosclerosis in LDL receptor-deficient mice. Cardiovasc Res. 2010;85:622–630. doi: 10.1093/cvr/cvp338. doi:10.1093/cvr/cvp338. [DOI] [PubMed] [Google Scholar]
- 12.Hjerpe C, Johansson D, Hermansson A, Hansson GK, Zhou X. Dendritic cells pulsed with malondialdehyde modified low density lipoprotein aggravate atherosclerosis in Apoe(−/−) mice. Atherosclerosis. 2010;209:436–441. doi: 10.1016/j.atherosclerosis.2009.10.003. doi:10.1016/j.atherosclerosis.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Buono C, Pang H, Uchida Y, Libby P, Sharpe AH, Lichtman AH. B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109:2009–2015. doi: 10.1161/01.CIR.0000127121.16815.F1. doi:10.1161/01.CIR.0000127121.16815.F1. [DOI] [PubMed] [Google Scholar]
- 14.Olofsson PS, Soderstrom LA, Wagsater D, Sheikine Y, Ocaya P, Lang F, Rabu C, Chen L, Rudling M, Aukrust P, Hedin U, Paulsson-Berne G, Sirsjo A, Hansson GK. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117:1292–1301. doi: 10.1161/CIRCULATIONAHA.107.699173. doi:10.1161/CIRCULATIONAHA.107.699173. [DOI] [PubMed] [Google Scholar]
- 15.van Wanrooij EJ, van Puijvelde GH, de Vos P, Yagita H, van Berkel TJ, Kuiper J. Interruption of the Tnfrsf4/Tnfsf4 (OX40/OX40L) pathway attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:204–210. doi: 10.1161/01.ATV.0000251007.07648.81. doi:10.1161/01.ATV.0000251007.07648.81. [DOI] [PubMed] [Google Scholar]
- 16.Afek A, Harats D, Roth A, Keren G, George J. A functional role for inducible costimulator (ICOS) in atherosclerosis. Atherosclerosis. 2005;183:57–63. doi: 10.1016/j.atherosclerosis.2005.03.040. doi:10.1016/j.atherosclerosis.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 17.Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394:200–203. doi: 10.1038/28204. doi:10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 18.Lievens D, Eijgelaar WJ, Biessen EA, Daemen MJ, Lutgens E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb Haemost. 2009;102:206–214. doi: 10.1160/TH09-01-0029. [DOI] [PubMed] [Google Scholar]
- 19.Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, Flavell RA. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–1316. doi: 10.1038/15271. doi:10.1038/15271. [DOI] [PubMed] [Google Scholar]
- 20.Lutgens E, Cleutjens KB, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJ. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci USA. 2000;97:7464–7469. doi: 10.1073/pnas.97.13.7464. doi:10.1073/pnas.97.13.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A, Seijkens T, Engel D, Cleutjens J, Keller AM, Naik SH, Boon L, Oufella HA, Mallat Z, Ahonen CL, Noelle RJ, de Winther MP, Daemen MJ, Biessen EA, Weber C. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391–404. doi: 10.1084/jem.20091293. doi:10.1084/jem.20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK, Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–180. doi: 10.1038/nm1343. doi:10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 23.Klingenberg R, Lebens M, Hermansson A, Fredrikson GN, Strodthoff D, Rudling M, Ketelhuth DF, Gerdes N, Holmgren J, Nilsson J, Hansson GK. Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:946–952. doi: 10.1161/ATVBAHA.109.202671. doi:10.1161/ATVBAHA.109.202671. [DOI] [PubMed] [Google Scholar]
- 24.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6:600–607. doi: 10.1038/ni1197. doi:10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 26.Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF- signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, Choi J, Perkmann T, Backhed F, Miller YI, Horkko S, Corr M, Witztum JL, Binder CJ. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119:1335–1349. doi: 10.1172/JCI36800. doi:10.1172/JCI36800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor- controls T helper type 1 cell development through regulation of natural killer cell interferon. Nat Immunol. 2005;6:600. doi: 10.1038/ni1197. doi:10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 29.Wu H, Gower RM, Wang H, Perrard XY, Ma R, Bullard DC, Burns AR, Paul A, Smith CW, Simon SI, Ballantyne CM. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–2717. doi: 10.1161/CIRCULATIONAHA.108.823740. doi:10.1161/CIRCULATIONAHA.108.823740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. doi:10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 31.Lutgens E, Gijbels M, Smook M, Heeringa P, Gotwals P, Koteliansky VE, Daemen MJ. Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler Thromb Vasc Biol. 2002;22:975–982. doi: 10.1161/01.atv.0000019729.39500.2f. doi:10.1161/01.ATV.0000019729.39500.2F. [DOI] [PubMed] [Google Scholar]
- 32.Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamate C, Merval R, Fradelizi D, Tedgui A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. 2001;89:930–934. doi: 10.1161/hh2201.099415. doi:10.1161/hh2201.099415. [DOI] [PubMed] [Google Scholar]
- 33.Frutkin AD, Otsuka G, Stempien-Otero A, Sesti C, Du L, Jaffe M, Dichek HL, Pennington CJ, Edwards DR, Nieves-Cintron M, Minter D, Preusch M, Hu JH, Marie JC, Dichek DA. TGF-[beta]1 limits plaque growth, stabilizes plaque structure, and prevents aortic dilation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2009;29:1251–1257. doi: 10.1161/ATVBAHA.109.186593. doi:10.1161/ATVBAHA.109.186593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342–1350. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ovchinnikova O, Robertson AK, Wagsater D, Folco EJ, Hyry M, Myllyharju J, Eriksson P, Libby P, Hansson GK. T-cell activation leads to reduced collagen maturation in atherosclerotic plaques of Apoe(−/−) mice. Am J Pathol. 2009;174:693–700. doi: 10.2353/ajpath.2009.080561. doi:10.2353/ajpath.2009.080561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gojova A, Brun V, Esposito B, Cottrez F, Gourdy P, Ardouin P, Tedgui A, Mallat Z, Groux H. Specific abrogation of transforming growth factor-beta signaling in T cells alters atherosclerotic lesion size and composition in mice. Blood. 2003;102:4052–4058. doi: 10.1182/blood-2003-05-1729. doi:10.1182/blood-2003-05-1729. [DOI] [PubMed] [Google Scholar]
- 37.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. doi:10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 38.Halliday GM, Le S. Transforming growth factor-beta produced by progressor tumors inhibits, while IL-10 produced by regressor tumors enhances, Langerhans cell migration from skin. Int Immunol. 2001;13:1147–1154. doi: 10.1093/intimm/13.9.1147. doi:10.1093/intimm/13.9.1147. [DOI] [PubMed] [Google Scholar]
- 39.Geissmann F, Revy P, Regnault A, Lepelletier Y, Dy M, Brousse N, Amigorena S, Hermine O, Durandy A. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol. 1999;162:4567–4575. [PubMed] [Google Scholar]
- 40.Yamaguchi Y, Tsumura H, Miwa M, Inaba K. Contrasting effects of TGF-beta 1 and TNF-alpha on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells. 1997;15:144–153. doi: 10.1002/stem.150144. doi:10.1002/stem.150144. [DOI] [PubMed] [Google Scholar]
- 41.Strobl H, Knapp W. TGF-beta1 regulation of dendritic cells. Microbes Infect. 1999;1:1283–1290. doi: 10.1016/s1286-4579(99)00256-7. doi:10.1016/S1286-4579(99)00256-7. [DOI] [PubMed] [Google Scholar]
- 42.Felker P, Sere K, Lin Q, Becker C, Hristov M, Hieronymus T, Zenke M. TGF-beta1 accelerates dendritic cell differentiation from common dendritic cell progenitors and directs subset specification toward conventional dendritic cells. J Immunol. 2010;185:5326–5335. doi: 10.4049/jimmunol.0903950. doi:10.4049/jimmunol.0903950. [DOI] [PubMed] [Google Scholar]
- 43.Daissormont IT, Christ A, Temmerman L, Sampedro Millares S, Seijkens T, Rousch M, Poggi M, Boon L, van der Loos C, Daemen M, Lutgens E, Halvorsen B, Aukrust P, Janssen E, Biessen EA. Plasmacytoid dendritic cells protect against atherosclerosis by tuning T-cell proliferation and activity. Circ Res. 2011;109:1387–1395. doi: 10.1161/CIRCRESAHA.111.256529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. doi:10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith E, Prasad KM, Butcher M, Dobrian A, Kolls JK, Ley K, Galkina E. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2010;121:1746–1755. doi: 10.1161/CIRCULATIONAHA.109.924886. doi:10.1161/CIRCULATIONAHA.109.924886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Es T, van Puijvelde GH, Ramos OH, Segers FM, Joosten LA, van den Berg WB, Michon IM, de Vos P, van Berkel TJ, Kuiper J. Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem Biophys Res Commun. 2009;388:261–265. doi: 10.1016/j.bbrc.2009.07.152. doi:10.1016/j.bbrc.2009.07.152. [DOI] [PubMed] [Google Scholar]
- 47.Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, Wang Y, Bockler D, Katus HA, Dengler TJ. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol. 2009;183:8167–8175. doi: 10.4049/jimmunol.0901126. doi:10.4049/jimmunol.0901126. [DOI] [PubMed] [Google Scholar]
- 48.Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, Esposito B, Perez N, Yasukawa H, Van Snick J, Yoshimura A, Tedgui A, Mallat Z. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med. 2009;206:2067–2077. doi: 10.1084/jem.20090545. doi:10.1084/jem.20090545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13(18 Pt 1):5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. doi:10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 50.Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 51.Terrell TG, Working PK, Chow CP, Green JD. Pathology of recombinant human transforming growth factor-beta 1 in rats and rabbits. Int Rev Exp Pathol. 1993;34(Pt B):43–67. doi: 10.1016/b978-0-12-364935-5.50009-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.