

Abstract
Purpose
To identify an allele-specific siRNA, against the common KRT12 mutation Arg135Thr in Meesmann epithelial corneal dystrophy (MECD) as a personalized approach to treatment.
Methods
siRNAs against the K12 Arg135Thr mutation were evaluated using a dual luciferase reporter gene assay and the most potent and specific siRNAs were further screened by western blot. Off-target effects on related keratins were assessed and immunological stimulation of TLR3 was evaluated by RT-PCR. A modified 5′ rapid amplification of cDNA ends (RACE) method was used to confirm siRNA-mediated mutant knockdown. Allele discrimination was confirmed by quantitative infrared immunoblotting.
Results
The lead siRNA, with an IC50 of thirty picomolar, showed no keratin off-target effects or activation of TLR3 in the concentration ranges tested. We confirmed siRNA-mediated knockdown by the presence of K12 mRNA fragments cleaved at the predicted site. A dual tag infrared immunoblot showed knockdown to be allele-specific with 70-80% silencing of the mutant protein.
Conclusions
A potent allele-specific siRNA against the K12 Arg135Thr mutation was identified. In combination with efficient eye drop formulation delivery, this would represent a personalized medicine approach, aimed at preventing the pathology associated with MECD and other ocular surface pathologies with dominant-negative or gain-of-function pathomechanisms.
INTRODUCTION
Meesmann epithelial corneal dystrophy (MECD) is an eye disorder that is inherited in an autosomal dominant manner and is caused by point mutations in either of the KRT3 or KRT12 genes which encode keratins K3 or K12 respectively 1, 2. It can cause foreign body sensation and photophobia or can be asymptomatic and only detected in the course of routine eye examination. Slit lamp observation often shows multitudinous microcysts within the anterior epithelium, evident even in symptomless individuals.
We recently reported a severe presentation of MECD with corneal erosions and scarring leading to significant loss of visual acuity caused by the Leu132Pro mutation in K12, suggesting a molecular mechanism for differing severity within MECD 3. Although the K12 mutation Arg135Thr generally results in a less severe phenotype, it is the much more common MECD mutation throughout Europe. Presently the mainstay of treatment for MECD, in the more severe cases, and for other corneal dystrophies, includes keratoplasty or corneal grafting 4-6. Unfortunately the resident mutation-containing corneal epithelial stem cell population, residing at the limbus, remains post-surgery and within a few weeks to months, corneal surface re-growth results in a reappearance of pathology associated with the mutation. To date, no therapy has been developed which addresses the underlying pathology associated with any of the corneal dystrophies.
Since its discovery, RNA interference (RNAi) has given much promise for the design of new therapy approaches based on gene silencing 7-9. The ability to hijack the endogenous post-transcriptional gene regulatory RNAi pathway, through the use of synthetic siRNA, theoretically provides an ideal route to treat dominant-negative diseases 10, 11. Gene silencing by siRNA is highly efficacious compared with traditional antisense methods, as the processive nature of the RNA-induced silencing complex (RISC) allows cleavage of multiple copies of the target mRNA by a single siRNA molecule. In comparison, the one-to-one ratio of binding by an antisense oligonucleotide to its target can require the cell to be saturated with a dangerously high concentration of an antisense drug. In addition, the irreversible binding to their target by traditional antisense molecules makes these systems poorly discriminating for disease alleles that differ by as little as one nucleotide. siRNAs have unprecedented potency and specificity in terms of silencing genes in a user-defined manner. Their use in sequence specific, post-transcriptional inhibition of gene expression has great potential to be developed as a novel therapeutic approach for a number of disorders where gene inhibition is predicted to be therapeutic. A few siRNAs have now entered clinical trials for diseases including age-related macular degeneration, melanoma and viral infections with many others at preclinical development stages 12-19.
In dominant-negative or gain-of-function genetic disorders, design of allele-specific silencing reagents is required for each individual mutation 20, 21. Most MECD patients in Europe have the common founder mutation Arg135Thr 22, 23. Here we used a K12-luciferase reporter gene assay to systematically perform a sequence walk across the Arg135Thr point mutation in order to identify siRNAs that potently and specifically knocked down the mutant allele without any gene silencing effect on the normal wild type K12 allele. Off target effects on closely related keratin genes were not detected. Unwanted immunostimulation of Toll-like receptor 3 (TLR 3) 24 on a spontaneously immortalised cell line 25 was not found. Concurrently the mechanism of action of the gene silencing observed was confirmed by 5′ rapid amplification of cDNA ends (RACE). We also developed a dual tag infrared immunoblot to confirm >70% knock down of the mutant K12 allele.
METHODS
Constructs
Full-length human wild type K12 and K3 coding sequences were PCR amplified, cloned into pCR2.1 (Invitrogen, Paisley, UK) and fully sequenced as previously described 3. Mutation Arg135Thr was introduced into the K12 cDNA clone (in pCR2.1) with the QuickChange™ Site-Directed Mutagenesis kit (Stratagene; Agilent Technologies, Cheshire, UK), using primer pairs 5′-AAT CTT AAT GAT ACA TTA GCT TCC TAC-3′ and 5′-GTA GGA AGC TAA TGT ATC ATT AAG ATT-3′.
To generate luciferase reporter constructs for siRNA screening, the full length wild type and Arg135Thr mutant pCR2.1 K12 cDNA fragments described above were subcloned into psiTEST-LUC-target vector (York Bioscience Ltd, York, UK) using Not I and BamH I. A Renilla luciferase expression construct (pRL-CMV, Promega, Southampton, UK) was used as an internal control for both cell viability and transfection efficiency.
Wild type and Mutant K12 eGFP tagged constructs were made as previously described 3. For the Dual-Flag/Strep Tag II quantitative infrared immunoblotting assay, a K12 cDNA clone in pCR3.1 was used to generate a K12 wild type FlagHA tagged clone (pKRT12-WT/FlagHA) and a K12 Arg135Thr StrepHA tagged clone (pKRT12-Arg135Thr/StrepHA). The FlagHA (DYKDDDDK-LDGG-YPYDVPDYA) and StrepHA (WSHPQFEK-LDGG-YPYDVPDYA) tandem epitope tags were generated by introducing their corresponding nucleotide sequences immediately 3′ of the K12 cDNA. The epitope tags were introduced using PCR where the +strand primer sequences were 5′-ATT TAG GGA TCC ATC TGC AGA ATT CGG CTT CCT TCC CCA GGC CAT GGA TCT CTC CAA CAA CAC CAT GTC ACT-3′ (FLAG-HA tag) and 5′GTT CAG GAA ATT GAA GAA CTA ATG GAC TAC AAG GAC GAC GAT GAC AAG CTC GAT GGA GGA TAC CCA TAC GAC GTC CCA GAC TAC GCT TGA CTC GAG CTA AAT-3′ (Strep-HA tag).
siRNA design
All siRNAs (19+2 geometry: 19-nucleotide duplex with a 3′ dTdT nucleotide overhang) were synthesized by MWG Biotech AG (Ebersberg, Germany). The K12 Arg135Thr mutation is a G>C change occuring at position 404 in the K12 cDNA sequence, numbering from the A in the initiating ATG codon (c.404G>C). All 19 possible siRNAs that could target this mutation were designed; the sense strands for each are listed in Table 1. The sequences for positive control siRNA (siLuc) targeting the firefly luciferase gene and for the non-specific control siRNA (NSC4; an inverted bacterial β-galactosidase sequence) are also listed in Table 1.
Table 1. siRNA sequence walk against K12 mutation Arg135Thr (DNA mutation c.404G>C).

|
Bold denotes mutated nucleotide. The sequences of a non-specific control siRNA (NSC4) and a positive control against firefly luciferase (siLuc) are also shown
Cell culture
All cell lines were of human origin. AD293 embryonic kidney cells (Invitrogen, Paisley, UK) and HaCaT cells, an immortalized keratinocyte line 26 were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Paisley, UK) supplemented with 10% fetal calf serum (FCS; Invitrogen, Paisley, UK). The spontaneously transformed corneal epithelial cell line, HCE-S (a gift from J.T. Daniels, Institute of Ophthalmology, University College London, UK)25 was maintained in DMEM GlutaMAX medium (Invitrogen, Paisley, UK) with 10% FCS. Cells were incubated at 37°C with 5% CO2 and passaged following standard laboratory procedures.
Luciferase Reporter Assay
The luciferase reporter assay was performed as previously reported 3, 27
Western blot analysis
Western blot analysis was performed as previously reported 3, 27.
Dual Flag/Strep Tag II Quantitative Infrared Immunoblot
An infrared quantitative western blot assay was used as previously described 28. AD293 cells were cotransfected in triplicate with 100ng of pKRT12-WT/FlagHA and 100ng of pKRT12-Arg135Thr/StrepHA as well as siRNA at concentrations of 0.25nM and 5nM using 1.25μl of Lipofectamine 2000 in a final volume of 200μl of OptiMEM. 48 h after transfection, cells were processed for immunoblotting, imaged and quantified as described previously 28. Briefly, membranes were cut at 50kDa to separate the K12 and β-actin protein bands. After blocking the K12 portion was simultaneously probed with mouse α-Flag and rabbit α-Strep-tag II antibodies. The β-actin portion was simultaneously probed with rabbit α-Strep-tag II and mouse α-β-actin antibodies. After washing membranes were probed with secondary antibodies, Alexa Fluor 680 goat α-rabbit IgG (A-21076; Invitrogen, Paisley, UK) and IRDye® 800-conjugated goat α-mouse IgG (610-132-121; Rockland Immunochemicals Inc., Gilbertsville, USA), washed and scanned in the 700 nm and 800 nm channels using the Odyssey® Infrared Imaging System (LI-COR Biotechnology UK Ltd., Cambridge, UK). Coloured images of each membrane were generated from the 700 nm and 800 nm wavelength scans using Photoshop.
Transient transfection of HaCaT cells and off-target effects
The lead inhibitor was investigated for off-target effects on other keratins. HaCaT cells, known to express a wide range of keratins, were used as described previously 3.
Cytoskeletal extractions
Cytoskeletal protein extracts were made 72h post-transfection as previously described 29.
Assessment of unwanted immunostimulatory effects
HCE-S cells were treated with K12-Arg135Thr-5 and NSC4 siRNA at 0.5 and 5.0 nM concentrations for 24h or 48h and cDNA assessed for TLR3 activation by RT-PCR using primers 5′-GGG CAA GAA CTC ACA GGC CAG G-3′ and 5′-AAG GGC CAC CCT TCG GAG CA-3′ (amplicon size 147bp). GAPDH primers 5′-GGA GCC AAA AGG GTC ATC AT-3′ and 5′-AGC GTC AAA GGT GGA GGA GT-3′ (amplicon size 549bp) were used as a normalization control. As evidence of positive stimulation of TLR3, HCE-S cells were treated with Poly (I:C) LMW TLR3 agonist (Invivogen, San Diego, USA) at concentrations of 0, 0.05, 0.5 and 5μg/ml for 24h. All PCR reactions were carried out using GoTaq Flexi DNA polymerase system (Promega, Southampton, UK) according to manufacturer’s instructions using the following cycling conditions: 94°C for 4 min, then 32 cycles of 95°C for 30s, 62°C for 30s and 72°C for 15s, followed by 5 min at 72°C and a 4°C hold.
5′RACE analysis
3.5 × 105 AD293 cells were seeded in 2ml DMEM (10% FBS) in a 6 well plate, incubated for 24h and transfected with 1μg of plasmid DNA expressing K12-Arg135Thr-eGFP fusion protein using 3.3μl Lipofectamine 2000 (Invitrogen, Paisley, UK) in Optimem serum-free medium following manufacturer’s instructions. After 24h the medium was replaced and the cells transfected with siRNA K12-Arg135Thr-5 to a final concentration of 3nM using 6μl of Lipofectamine RNAiMAX (Invitrogen, Paisley, UK) according to the manufacturer’s instructions for 6h. Total RNA was extracted using RNeasy minikit (Qiagen, West Sussex, UK). 5μg of RNA was used to perform a modified 5′ RACE using the Generacer kit (Invitrogen, Paisley, UK) following the manufacturer’s instructions, with 5′ Generacer oligo ligated directly to total RNA without pretreatment. DNA amplification was carried out using the Generacer touchdown PCR protocol with a final annealing temperature of 64°C and a 45s elongation at 72°C for 25 cycles in the final cycling step. Forward primer was the Generacer 5′ primer and the reverse was a K12 gene specific primer, Race.GSP.K12.1R-5′-CAT ACT GCG CCC GCA TAT CAT TG-3′ (MWG, Ebersberg, Germany). PCR products were separated on an agarose gel and the major band corresponding to the predicted 562bp product was excised and gel purified using QiaQuick Gel extraction kit (Qiagen, West Sussex, UK) and ligated into pCR-4-TOPO vector (Invitrogen, Paisley, UK) according to the manufacturer’s instructions. 11 out of 12 colonies were positive for the insert when assessed by PCR and 6 were sequenced (DNA Sequencing Services, University of Dundee, UK).
Cell Viability Assay
Toxicity of siRNA treatment was assessed using the PrestoBlue™ Cell Viability Reagent (Invitrogen, Paisley, UK). Six replicates of AD293 cells were seeded at 6.5×103 cells per well and transfected with mutant or wild type K12 expression constructs and treated with K12-Arg135Thr-5 or NSC4 siRNA at a concentration range of 10pM to 5nM for 24h. PrestoBlue™ was added to the wells following the manufacturer’s instructions and incubated for 30 min. The optical density (OD) was measured using a 544nm excitation filter and 590nm emission filter using the FLUOstar OPTIMA (BMG Labtech, Aylesbury, UK).
Statistics
Statistical analysis undertaken for data from the infrared immunoassay included an analysis of variance using Levenes followed with a Brown Forsythe test. Posthoc test carried out using Dunnet C. Statistical significance was set at 0.05.
RESULTS
Design and development of optimal siRNA
A sequence walk was performed whereby all 19 possible siRNA inhibitors were designed to span and include the K12 Arg135Thr point mutation 3, 27 as shown in Table 1. For each siRNA inhibitor investigated, the effect on K12 firefly luciferase reporter gene expression was determined in four replicate experiments over a range of siRNA molar concentrations (0, 0.01, 0.05, 0.25, 1.25 and 6.25 nM) against the wild type and mutant K12 constructs in AD293 cells. In each case, the data was normalized against co-transfected Renilla luciferase as a control for cell viability and transfection efficiency. Representative results are presented graphically in Fig. 1. A non-specific negative control siRNA (NSC4− an inverted bacterial β-galactosidase sequence) had no inhibitory effect on either the wild type K12 reporter or the mutant reporters, while a known potent siRNA against firefly luciferase (siLuc), knocked down the expression of both wild type and mutant mRNA almost equally and over a wide range of siRNA concentrations.
Figure 1. A comprehensive siRNA sequence walk for the K12 mutation Arg135Thr.
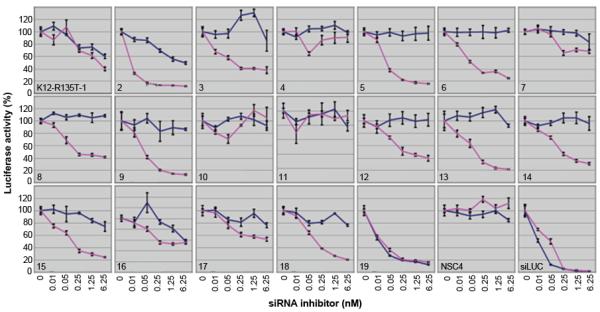
This sequence walk shows the best siRNA was at position 5 (K12-Arg132Thr-5), which was very potent against the mutant reporter (pink line on plots) but did not significantly inhibit wild type (blue line on plots) at concentrations as high as 6.25nM. siRNA inhibitor K12-Arg135Thr-4 and K12-Arg135Thr-11 did not affect wild type or mutant reporter while K12-Arg135Thr-16 and K12-Arg135Thr-19 knocked down both alleles to varying degrees. NSC4 is a non-specific control siRNA, (negative control). siLUC is an siRNA specific for luciferase (positive control).
Allele specific knockdown of K12
Of the mutant-specific siRNAs under test, some were not discriminatory, inhibiting both wild type and mutant K12 equally and over a range of concentrations, such as K12-Arg135Thr-16, or K12-Arg135Thr-19 (Fig. 1). Some test inhibitors had little or no effect on expression of either the wild type or mutant reporter gene expression, even at high concentrations, such as K12-Arg135Thr-10 or K12-Arg135Thr-11. The optimal positions of lead inhibitor siRNAs was compared to our previously published K12, K5, K6a and K9 gene walk data 3, 27, 28 and again showed no pattern to help predict optimal mutation specific siRNAs.
From the 19 inhibitors tested for K12 Arg135Thr mutant, a number were able to specifically and potently inhibit the mutant allele with little or no effect on the wild type allele, such as K12-Arg135Thr-2, 5, 6, 8, 9, 13 and 14. Although all of these had good potential for therapeutic use, arguably the best inhibitor for further development was K12-Arg135Thr-5, where expression of the wild type allele is not significantly knocked down, even at the highest concentrations, whereas the mutant allele is potently knocked down even at the lower siRNA concentration of 0.01 nM (Fig. 1). K12-Arg135Thr-5 had a half maximal inhibitory concentration (IC50) of approximately 30pM: meaning 50% knock down of the mutant Arg135Thr allele was achieved with 30pM of the K12-Arg135Thr-5 siRNA.
Cell viability was confirmed using PrestoBlue™ on AD293 cells transfected with mutant or wild type K12 expression constructs and treated with K12-Arg135Thr-5 siRNA or NSC4 siRNA at concentrations ranging from 10pM to 5nM for 24 h. No toxicity was noted at these concentrations at this time point (data not shown).
Allele specific knockdown of K12 at protein level
To confirm and quantify the highly differential inhibitory effect of lead siRNA at the level of protein expression, western blot analysis was performed. AD293 cells were transfected with wild type or mutant K12 expression constructs and treated with a number of representative discriminatory siRNAs at a final concentration of 5nM. Non-specific siRNA NSC4 was used as a negative control and Opti-MEM alone was used as an “untreated” control. Western blot data confirm that a number of the siRNAs are highly potent and highly specific for this K12 mutation at the level of protein expression with almost complete knock down of the mutant K12 protein expression and negligible knock down of the wild type K12 protein (Fig. 2). Lead inhibitor (K12-Arg135Thr-5) was chosen for investigations into off-target effects on other keratins in HaCaT cells, which endogenously express a range of keratins, including K14, which is closely related to K12 in terms of sequence conservation. SimplyBlue stained gels showed no reduction on any keratins expressed in this keratinocyte cell line (Fig. 3). In contrast, a positive control siRNA against K6a 17 knocked down K6a protein expression.
Figure 2. Differential inhibition of mutant K12 protein versus wild type by allele-specific siRNAs in cultured cells.

AD293 cells were transfected with wild type or mutant K12-eGFP construct and each of the siRNAs: K12-Arg135Thr-5, -6, -8,-9,-13,-14 at a final concentration of 5 nM. To confirm the highly differential inhibitory effect of K12-Arg135Thr siRNA on mutant K12, western blot analysis was performed. When cells were treated with K12-Arg135Thr-5, the mutant K12-eGFP was almost completely knocked down, while expression of wild type K12 construct treated with the same siRNA showed a negligible reduction compared with NSC4 treatment. Wt = wild type and mut = mutant.
Figure 3. Lack of off-target silencing of other keratins by lead inhibitor K12-Arg135Thr-5.
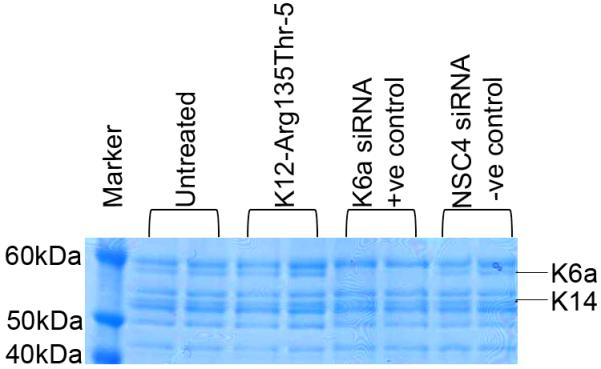
SimplyBlue staining of cytoskeletal protein extracts from HaCaT cells revealed allele specific K12-Arg135Thr-5 siRNA had negligible effect on endogenous keratins expressed in the HaCaT cell line, most notably K14 which is closely related to K12 in terms of sequence conservation. In contrast, a positive control siRNA directed against wild type K6a demonstrated a dramatic decrease in K6a expression. Non-specific control siRNA NSC4 had no effect on keratin protein expression profile.
siRNA inhibition of mutant K12 protein in the presence of wild type K12
One limitation of the dual-luciferase reporter assay and the western blot described above is that they only test the ability of K12-Arg135Thr-5 siRNA to inhibit mutant allele expression in isolation and not when wild type K12 is co-expressed in the same cell. This is not representative of the endogenous cellular situation in MECD patients whereby both wild type and mutant alleles of KRT12 are co-expressed. Since patient primary corneal epithelial cells expressing the Arg135Thr mutation were not available to test K12-Arg135Thr-5 siRNA in this scenario, a CMV promoter driven K12 Flag-HA epitope-tagged allele (K12-WT) and a CMV promoter-driven dominant-negative mutant KRT12 Strep-HA epitope-tagged allele (K12-MUT) were generated. AD293 cells were co-transfected with equal amounts of each along with 0, 0.5, or 5nM of NSC4 or K12-Arg135Thr-5 siRNA. A dual-tag quantitative immunoblotting assay was developed, using anti-Flag monoclonal and anti-Strep-tag II polyclonal antibodies, to confirm inhibition of K12 protein translation. Quantification of protein expression was achieved by digital integration of infrared fluorochromes linked to secondary antibodies (Li-Cor Odyssey system). NSC4 siRNA had no effect on wild type or mutant K12 protein production at either of the concentrations tested, while the K12-Arg135Thr-5 siRNA showed approximately 70-80% knockdown of the mutant K12 Strep fusion protein (green bands in Fig. 4; p<0.05) with no effect on the wild type K12 Flag fusion protein (red bands in middle panel in Fig. 4). β actin protein expression shown in the bottom panel of Fig. 4 confirmed equal loading.
Figure 4. Dual Flag Strep Tag II quantitative infrared immunoblotting confirming allele specific silencing.

AD293 cells were transiently co-transfected with pKRT12-WT/FlagHA (K12-WT) and pKRT12-Arg135Thr/StrepHA (K12-Arg135Thr) and 0, 0.5 or 5 nM K12-Arg135Thr-5 or NSC4 siRNA, immunoblotting of whole cell lysates with α-Flag and α-StrepTagII antibodies and differential fluorescence visualization using a LI-COR Odyssey scanner revealed preferential inhibition of mutant K12 StrepTagII fusion protein synthesis by the K12-Arg135Thr-5 siRNA. Semiquantitative analyses of α-StrepTagII (mutant top panel, green bands) and α-Flag (wild-type, middle panel, red bands) immunoblot signal intensities, relative to β-actin (bottom panel), were used to quantify siRNA inhibition specificities and potencies. Mean relative abundances (n=9) are shown and error bars indicate signal intensity standard deviations. Approximately 70-80% knock down of the mutant allele was noted at both concentrations of siRNA assessed and no knock down was noted with the NSC4 negative control siRNA (p<0.05).
Immunostimulation by siRNAs
To investigate the immunostimulatory effect of adding exogenous double-stranded RNA to cells in the form of short synthetic therapeutic siRNAs, the main mediator involved in the immune response to viral RNA was assessed. RT-PCR primers were designed to assess Toll-like receptor 3 (TLR3). HCE-S cells were treated with lead K12-Arg135Thr-5 siRNA at 0.5 and 5nM for 24h or 48h (Fig. 5). A synthetic analog of dsRNA (poly I:C-LMW), was used as a positive control for immunostimulation of TLR3 in HCE-S cells. RT-PCR showed increased levels of TLR3 in the presence of the agonist while no obvious increase was noted with siRNA treatment at the times and concentrations assessed. An earlier time point of 6h was also assessed and no obvious increase in TLR3 was seen (data not shown).
Figure 5. Immunostimulation of TLR3 by siRNAs.
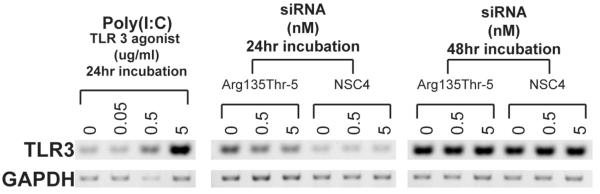
A synthetic analog of dsRNA (poly I:C-LMW), was used as a positive control for immunostimulation of TLR3 in the corneal epithelial cells. RT-PCR showed increased levels of TLR3 in the presence of this agonist while cells treated with K12-Arg135Thr-5 siRNA at 0.5 and 5nM for 24h or 48h show no obvious increase at the times and concentrations assessed. An earlier time point of 6h was also assessed and no obvious increase in TLR3 was noted (data not shown).
5 RACE determination of mRNA cleavage site
To confirm the observed mutant K12 knockdown was due to siRNA-mediated cleavage, AD293 cells transiently expressing K12 Arg135Thr mutant protein were treated with 3nM siRNA K12-Arg135Thr-5 for 6h. Total RNA was extracted and subjected to a modified 5′RACE technique 30-32. Cleaved mRNA was separated from total RNA by ligating a generacer adapter oligo to RNA with free, cleaved 5′ ends. Further selection for K12 specific cleaved mRNA was achieved by PCR using a K12 specific reverse PCR primer. K12 specific PCR products were ligated into the pCR4.0 topo plasmid vector for sequencing. Sequencing showed that the generacer adapter oligo had ligated to the 5′ end of the cleaved K12 mRNA at the predicted position indicative of siRNA-RISC mediated cleavage between nucleotides 398 and 399 of the K12 mRNA coding sequence (Fig. 6).
Figure 6. 5′RACE verifies siRNA-mediated cleavage at the predicted site on K12 mRNA.

K12 specific mRNA cleavage products of AD293 cells exogenously expressing K12-Arg135Thr and treated with 3nM siRNA K12-Arg135Thr-5 were assessed by a modified 5′ Rapid Amplification of cDNA Ends (5′RACE). The generacer oligo adapter was ligated to the free 5′ end of cleaved mRNA products. K12 specific cleaved mRNAs were selected by PCR amplification with a K12 Gene Specific Primer (GSP). The major K12 specific PCR product was purified, ligated into pCR4.0 topo plasmid and sequenced. Sequencing shows that the major product had the generacer adapter ligated to the 399th nucleotide of the K12 CDS, this corresponding to the predicted cleavage site between nucleotides 10 and 11 of the hybridized guide strand of siRNA K12-Arg135Thr-5.
DISCUSSION
RNA interference (RNAi) is an ideal approach to treatment of diseases like MECD where heterozygous single nucleotide mutations present in a dominant negative manner. In the field of keratin disorders affecting the skin and cornea, we have previously shown that it is possible to design short interfering RNAs (siRNA) which specifically and potently knockdown a mutated allele while having neglible impact on the wild-type allele3, 8, 17, 27, 28. We hypothesize this approach is clinically valid with complete or partial (ca. 50%) knockdown of the mutant allele allowing restoration of a normal phenotype. Cao and colleagues observed no epidermal fragilty when the mutant to wild-type expression ratio of K14 in a transgenic mouse model was 1:2 33, 34. The first-in-human trial for siRNA, aimed at dominant K6a mutation causing the skin disorder pachyonychia congenita, confirmed that epidermal lesions reduce in size over time with mutation-specific siRNA treatment 35. Here we have extended this approach to the design and validation of allele-specific siRNAs targeting the common founder mutation in MECD, specifically Arg135Thr in the KRT12 gene 1, 23. Using a dual luciferase reporter gene assay and siRNA sequence walk approach 3 we tested all 19 possible siRNAs targeting the K12 Arg135Thr mutation and identified 6 mutant specific siRNAs, namely K12-Arg135T-5, 6, 8, 9, 13 and 14 (Fig. 1); of these K12-Arg135Thr-5 exhibited the highest potency with a half-maximal inhibitory concentration (IC50) of approximately 30 pM, on a par with the best small molecule drugs. All mutant-specific siRNAs identified were then screened by western blot for their ability to specifically knockdown exogenously expressed K12 Arg135Thr protein (Fig. 2) K12-Arg135Thr-5 was chosen as lead inhibitor to be taken forward for further validation. With no primary corneal epithelial cells expressing the Arg135Thr mutation available we needed to create a model system to replicate endogenous conditions where the siRNA inhibitor would be required to knockdown mutant mRNA in the presence of its wild type counterpart, a dual tag Flag-Strep TagII quantitative infrared immunoblot assay was used to assess siRNA specificity. Here wild type K12-Flag and K12 Arg135Thr-Strep tagged proteins were exogenously co-expressed in AD293 cells and treated with lead inhibitor K12-Arg135Thr-5; PAGE-resolved protein extracts were assessed by infrared immunoblot assay and the results concurred with our previous assays; the K12 Arg135Thr mutant protein is specifically knocked down by approximately 70-80% in the presence of wild type K12, which was unaffected (Fig. 4). Taken together these data confirm that in K12-Arg135Thr-5 we have identified a potent, specific siRNA targeting the K12 Arg135Thr mutant.
We predict that using similar methodology mutant-allele-specific siRNAs can be identified for the vast majority of MECD causative mutations in the keratins K3 and K12 8, 36. This methodology could similarly be taken forward to identify mutation specific siRNAs aimed at the more clinically significant TGFBI gene (formerly known as BIGH3), responsible for many severe forms of corneal dystrophy 37-39. Like the keratins, the majority of TGFBI mutants exhibit with a dominant-negative pathomechanism making this allele specific approach similarly relevant.
One important caveat of siRNAs is off-target effects 11. Firstly unrelated genes with similar target homology could potentlially be targeted for knockdown by the siRNA. We have demonstrated that the K12-Arg135Thr-5 siRNA does not target the wild-type allele, this differing only by a single nucleotide from the mutated allele, thus effects against unrelated targets are unlikely, however we used HaCaT epithelial cells to initially confirm that keratins with close sequence homology to K12, notably K14, were not affected by K12-Arg135Thr-5 siRNA treatment (Fig. 3). Further in-depth study into the global effects of the siRNA treatment is planned in future in-vitro and in-vivo studies. A second consideration is that with siRNAs being short double-stranded RNA (dsRNA), they could potentially trigger an innate immune response via activation of toll-like receptor 3 (TLR3) in an attempt to protect against suspected pathogen-derived RNA. Human corneal and conjunctival epithelial cells are known to express TLR3 and recent studies in the retina suggest that siRNA treatment results in sequence independent activation of TLR3 and activation of an interferon response, with this TLR3 response misinterpreted as an siRNA specific effect 24, 40, 41. Here, though MECD has no immunological related etiology and a TLR3 response cannot be mistaken for an effective siRNA therapy, it nevertheless required further investigation, therefore we assessed the effect of our lead siRNA on HCE-S cells, a spontaneously immortalised human corneal cell line 25. Semi-quantitative RT-PCR showed no obvious TLR3 activation for treated cells, with a specific TLR3 agonist confirming presence and ability to activate this toll like receptor (Fig. 5). Future work will assess these effects more fully on primary cell lines. Thirdly, we confirmed a siRNA-RISC-mediate mechanism of mutant allele knockdown using a modified 5′ RACE assay to identify the predicted cleavage products of the K12-Arg135Thr-5 guide strand. Finally, cell viability was assessed throughout all experimentation using a resazurin-based assay and no evidence of cell toxicity was observed at any of the concentrations tested. In combination, these data provide evidence that unwanted off-target effects have been minimised, though further in-vitro patient corneal epithelial cell line and in-vivo experimentation on an intact ocular surface will be required to fully assess all possible off-target effects, immunological or otherwise. Beneficial to this cause is the superb potency of the K12-Arg135Thr-5 siRNA, allowing lower concentrations to be employed, minimising potential RISC saturation and other off target effects associated with having to use high concentrations of siRNA. Further preclinical assessment featuring good manufacturing practice (GMP) production and toxicology assessment would fully pave the way for a clinical application.
The identification of siRNAs that specifically target a gene or allele is only one hurdle that must be overcome for these molecules to be of benefit therapeutically. Although it is already a great advantage that siRNAs are closer in size to a small molecule drug than gene therapy vectors, many challenges must yet be overcome to allow their delivery to their target cellular compartments. The natural respective negative charges of the siRNA backbone and the phospholipid bi-layer of each cell membrane, through which they must pass, make this a challenge that has resulted in great international efforts from both academic and industrial research groups alike, with many approaches being investigated 42, 43. Given their transient mode of function and the long-term necessity for repeated application, an efficient delivery system is key to the therapeutic success of siRNA. Ideally we envisage that for symptomatic forms of MECD, siRNA would be applied topically on a long-term basis in the form of eye drops; the more severe corneal dystrophies associated with TGFBI, however, may require surgical graft intervention to clear aggregate build-up followed by mutation-specific siRNA to assuage their reoccurrence.
In more general terms the cornea provides a highly attractive target tissue for siRNA therapy in terms of optimization of delivery strategies. With its anterior epithelium a thin, stratified but non-cornified four cell layer, delivery should be easier than in the epidermis where the stratum corneum provides additional challenges. The cornea is therefore ideal as a proof-of-concept tool for translation of siRNA therapy into clinical practice, being so small, easily accessible and with disease status easily monitored by external examination. With TGFBI disorders generally taking longer to exhibit, making cell culture and animal modeling more of a challenge, the rapid turnover of keratin aggregates and the anterior corneal epithelium 44, make MECD a more relevant model for rapid assessment of the efficacy of siRNA treatment in vivo.
We have now identified highly potent and specific siRNA therapy reagents that target MECD-causing K12 mutations Arg135Thr and Leu132Pro; the next step will be assessment of lead inhibitors in clinically relevant mouse models, monitoring the efficacy of siRNA delivery and function in combination with more detailed assessment of off-target effects. siRNAs coupled with a suitable delivery system, have great potential for treating corneal dystrophies.
Acknowledgments
This work was funded by the Medical Research Council (grant numbers G0801742 and G0802780; to FJDS and WHIM) and Fight for Sight project grant (to CBTM and WHIM).
REFERENCES
- 1.Meesmann A. Über eine bisher nicht beschriebene dominant vererbte Dystrophia epithelialis corneae. Ber Zusammenkunft Dtsch Ophthalmol Ges. 1938;52:154–158. [Google Scholar]
- 2.Irvine AD, McLean WHI. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation. Br J Dermatol. 1999;140:815–828. doi: 10.1046/j.1365-2133.1999.02810.x. [DOI] [PubMed] [Google Scholar]
- 3.Liao H, Irvine AD, Macewen CJ, et al. Development of allele-specific therapeutic siRNA in Meesmann epithelial corneal dystrophy. PloS one. 2011;6:e28582. doi: 10.1371/journal.pone.0028582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiou AG, Florakis GJ, Copeland RL, Williams VA, McCormick SA, Chiesa R. Recurrent Meesmann’s corneal epithelial dystrophy after penetrating keratoplasty. Cornea. 1998;17:566–570. doi: 10.1097/00003226-199809000-00017. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg A, Schlotzer-Schrehardt U, Seiler T. Unilateral Meesmann’s dystrophy. Int Ophthalmol. 1997;21:117–120. doi: 10.1023/a:1026456232212. [DOI] [PubMed] [Google Scholar]
- 6.Irvine AD, Coleman CM, Moore JE, et al. A novel mutation in KRT12 associated with Meesmann’s epithelial corneal dystrophy. Br J Ophthalmol. 2002;86:729–732. doi: 10.1136/bjo.86.7.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson BL, McCray PB., Jr. Current prospects for RNA interference-based therapies. Nature reviews Genetics. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLean WH, Moore CB. Keratin disorders: from gene to therapy. Human molecular genetics. 2011;20:R189–197. doi: 10.1093/hmg/ddr379. [DOI] [PubMed] [Google Scholar]
- 9.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 10.Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711–719. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hickerson RP, Smith FJD, Reeves RE, et al. Single-nucleotide-specific siRNA targeting in a dominant-negative skin model. J Invest Dermatol. 2008;128:594–605. doi: 10.1038/sj.jid.5701060. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen T, Menocal EM, Harborth J, Fruehauf JH. RNAi therapeutics: an update on delivery. Current opinion in molecular therapeutics. 2008;10:158–167. [PubMed] [Google Scholar]
- 13.Dykxhoorn DM, Lieberman J. Knocking down disease with siRNAs. Cell. 2006;126:231–235. doi: 10.1016/j.cell.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haussecker D. The business of RNAi therapeutics. Human gene therapy. 2008;19:451–462. doi: 10.1089/hum.2008.007. [DOI] [PubMed] [Google Scholar]
- 15.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nature reviews Drug discovery. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeVincenzo J, Lambkin-Williams R, Wilkinson T, et al. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8800–8805. doi: 10.1073/pnas.0912186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith FJ, Hickerson RP, Sayers JM, et al. Development of therapeutic siRNAs for pachyonychia congenita. The Journal of investigative dermatology. 2008;128:50–58. doi: 10.1038/sj.jid.5701040. [DOI] [PubMed] [Google Scholar]
- 18.Davis ME, Zuckerman JE, Choi CH, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nature reviews Drug discovery. 2007;6:443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaspar RL. Challenges in developing therapies for rare diseases including pachyonychia congenita. J Investig Dermatol Symp Proc. 2005;10:62–66. doi: 10.1111/j.1087-0024.2005.10208.x. [DOI] [PubMed] [Google Scholar]
- 21.Lewin AS, Glazer PM, Milstone LM. Gene therapy for autosomal dominant disorders of keratin. J Investig Dermatol Symp Proc. 2005;10:47–61. doi: 10.1111/j.1087-0024.2005.10207.x. [DOI] [PubMed] [Google Scholar]
- 22.Corden LD, Swensson O, Swensson B, et al. Molecular genetics of Meesmann’s corneal dystrophy: ancestral and novel mutations in keratin 12 (K12) and complete sequence of the human KRT12 gene. Experimental eye research. 2000;70:41–49. doi: 10.1006/exer.1999.0769. [DOI] [PubMed] [Google Scholar]
- 23.Irvine AD, Corden LD, Swensson O, et al. Mutations in cornea-specific keratins K3 or K12 cause Meesmann’s corneal dystrophy. Nat Genet. 1997;16:184–187. doi: 10.1038/ng0697-184. [DOI] [PubMed] [Google Scholar]
- 24.Kleinman ME, Kaneko H, Cho WG, et al. Short-interfering RNAs induce retinal degeneration via TLR3 and IRF3. Mol Ther. 2012;20:101–108. doi: 10.1038/mt.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Notara M, Daniels JT. Characterisation and functional features of a spontaneously immortalised human corneal epithelial cell line with progenitor-like characteristics. Brain research bulletin. 2010;81:279–286. doi: 10.1016/j.brainresbull.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atkinson SD, McGilligan VE, Liao H, et al. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. The Journal of investigative dermatology. 2011;131:2079–2086. doi: 10.1038/jid.2011.169. [DOI] [PubMed] [Google Scholar]
- 28.Leslie Pedrioli DM, Fu DJ, Gonzalez-Gonzalez E, et al. Generic and personalized RNAi-based therapeutics for a dominant-negative epidermal fragility disorder. J Invest Dermatol. 2012;132:1627–1635. doi: 10.1038/jid.2012.28. [DOI] [PubMed] [Google Scholar]
- 29.Rugg EL. Detection and characterization of keratins by immunocytochemistry and immunoblotting. In: Leigh IM, Lane EB, Watt FM, editors. Keratinocyte Methods. Cambridge University Press; Cambridge: 1994. pp. 127–148. [Google Scholar]
- 30.Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 31.Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. The EMBO journal. 2001;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hickerson RP, Leachman SA, Pho LN, et al. Development of quantitative molecular clinical end points for siRNA clinical trials. The Journal of investigative dermatology. 2011;131:1029–1036. doi: 10.1038/jid.2010.372. [DOI] [PubMed] [Google Scholar]
- 33.Rugg EL, McLean WH, Lane EB, et al. A functional “knockout” of human keratin 14. Genes Dev. 1994;8:2563–2573. doi: 10.1101/gad.8.21.2563. [DOI] [PubMed] [Google Scholar]
- 34.Cao T, Longley MA, Wang XJ, Roop DR. An inducible mouse model for epidermolysis bullosa simplex: implications for gene therapy. The Journal of cell biology. 2001;152:651–656. doi: 10.1083/jcb.152.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leachman SA, Hickerson RP, Schwartz ME, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther. 2010;18:442–446. doi: 10.1038/mt.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szeverenyi I, Cassidy AJ, Chung CW, et al. The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat. 2008;29:351–360. doi: 10.1002/humu.20652. [DOI] [PubMed] [Google Scholar]
- 37.Boutboul S, Black GC, Moore JE, et al. A subset of patients with epithelial basement membrane corneal dystrophy have mutations in TGFBI/BIGH3. Hum Mutat. 2006;27:553–557. doi: 10.1002/humu.20331. [DOI] [PubMed] [Google Scholar]
- 38.Munier FL, Frueh BE, Othenin-Girard P, et al. BIGH3 mutation spectrum in corneal dystrophies. Invest Ophthalmol Vis Sci. 2002;43:949–954. [PubMed] [Google Scholar]
- 39.Korvatska E, Henry H, Mashima Y, et al. Amyloid and non-amyloid forms of 5q31-linked corneal dystrophy resulting from kerato-epithelin mutations at Arg-124 are associated with abnormal turnover of the protein. J Biol Chem. 2000;275:11465–11469. doi: 10.1074/jbc.275.15.11465. [DOI] [PubMed] [Google Scholar]
- 40.Ueta M, Hamuro J, Kiyono H, Kinoshita S. Triggering of TLR3 by polyI:C in human corneal epithelial cells to induce inflammatory cytokines. Biochemical and biophysical research communications. 2005;331:285–294. doi: 10.1016/j.bbrc.2005.02.196. [DOI] [PubMed] [Google Scholar]
- 41.Li J, Shen J, Beuerman RW. Expression of toll-like receptors in human limbal and conjunctival epithelial cells. Molecular vision. 2007;13:813–822. [PMC free article] [PubMed] [Google Scholar]
- 42.Hao J, Li SK, Kao WW, Liu CY. Gene delivery to cornea. Brain Res Bull. 2010;81:256–261. doi: 10.1016/j.brainresbull.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leachman SA, Hickerson RP, Hull PR, et al. Therapeutic siRNAs for dominant genetic skin disorders including pachyonychia congenita. J Dermatol Sci. 2008;51:151–157. doi: 10.1016/j.jdermsci.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanna C, O’Brien JE. Cell production and migration in the epithelial layer of the cornea. Archives of ophthalmology. 1960;64:536–539. doi: 10.1001/archopht.1960.01840010538009. [DOI] [PubMed] [Google Scholar]