

Abstract
Cyanobacterial aldehyde-deformylating oxygenases (ADOs) belong to the ferritin-like diiron-carboxylate superfamily of dioxygen-activating proteins. They catalyze conversion of saturated or mono-unsaturated Cn fatty aldehydes to formate and the corresponding Cn-1 alkanes or alkenes, respectively. This unusual, apparently redox-neutral transformation actually requires four electrons per turnover to reduce the O2 co-substrate to the oxidation state of water and incorporates one O-atom from O2 into the formate co-product. We show here that the complex of the diiron(II/II) form of ADO from Nostoc punctiforme (Np) with an aldehyde substrate reacts with O2 to form a colored intermediate with spectroscopic properties suggestive of a Fe2III/III complex with a bound peroxide. Its Mössbauer spectra reveal that the intermediate possesses an antiferromagnetically (AF) coupled Fe2III/III center with resolved sub-sites. The intermediate is long-lived in the absence of a reducing system, decaying slowly (t1/2 ~ 400 s at 5 °C) to produce a very modest yield of formate (< 0.15 enzyme equivalents), but reacts rapidly with the fully reduced form of 1-methoxy-5-methylphenazine (MeOPMS) to yield product, albeit at only ~ 50% of the maximum theoretical yield (owing to competition from one or more unproductive pathway). The results represent the most definitive evidence to date that ADO can use a diiron cofactor (rather than a homo- or hetero-dinuclear cluster involving another transition metal) and provide support for a mechanism involving attack on the carbonyl of the bound substrate by the reduced O2 moiety to form a Fe2III/III-peroxyhemiacetal complex, which undergoes reductive O-O-bond cleavage, leading to C1–C2 radical fragmentation and formation of the alk(a/e)ne and formate products.
INTRODUCTION
Cyanobacterial aldehyde-deformylating oxygenases (ADOs), originally designated by Schirmer, et al. as “aldehyde decarbonylases,”1 catalyze conversion of Cn fatty aldehydes to the corresponding Cn-1 alk(a/e)nes and formate in a reaction requiring one molecule of O2 and four electrons per turnover.2-4 The ADO substrate is provided in vivo by the preceding enzyme in the pathway, acyl-acyl carrier protein (ACP) reductase, which reductively cleaves its fatty acid thioester substrate using a reduced nicotinamide.1 Because ADOs catalyze the final step in a pathway that converts physiologically abundant fatty acids into combustible hydrocarbons, they have received much recent attention for their potential biotechnological applications.1,5-10 Insight into their structure and mechanism might facilitate their deployment in, for example, bioprocesses to produce renewable hydrocarbon fuels in cyanobacteria or other heterologous bacterial hosts.5,11,12 Particularly for the latter approach, an understanding of the details of the reaction, including the identities and modes of participation of accessory factors, might be necessary to make the bioprocess efficient enough to be economically viable.
ADOs belong to the ferritin-like protein structural superfamily (see pdb accession 2OC5 for structural details), which comprises a number of well-characterized oxidases and oxygenases known to employ their carboxylate bridged non-heme-diiron cofactors to activate O2 for oxidation (in many cases, oxygenation) of their substrates.13-19 The ADO reaction is, however, unusual for a member of this enzyme superfamily, in that it results in no net oxidation of its primary substrate. Indeed, its similarity to the reactions of the other members is revealed only by the obligatory requirement for a reducing system1-4 and incorporation of a single atom of oxygen from O2 into the formate co-product.3,4 Despite these idiosyncrasies, it has been presumed that ADO also employs a diiron cofactor in catalysis. The fact that no ADO has, to our knowledge, been isolated from its native source, and the growing recognition that members of the structural superfamily can employ metal ions other than iron,20-23 have left the nature of the cofactor still somewhat in doubt. Obviously, knowledge of the identity of the cofactor could impact efforts to make the enzyme function at high efficiency in an engineered bioprocess.
In this study, the Nostoc punctiforme (Np) ADO has been over-expressed in Escherichia coli under culture conditions designed to favor incorporation of exclusively iron (minimal medium supplemented with Fe). A combination of metal analyses and Mössbauer spectroscopy has been used to show that these preparations do indeed harbor predominately (>95%) diiron clusters. Combined with the demonstration that the ADO prepared in this manner reacts, under the proper conditions, almost homogeneously with O2 to form an intermediate that is capable of yielding products, these results provide the best evidence to date for the previously presumed catalytic competence of the diiron form of ADO.
Iron-dependent oxidases and oxygenases often exhibit the apparently adaptive characteristic of becoming activated toward reaction with O2 upon binding of either the substrate [as in the FeII/α-ketoglutarate-dependent hydroxylases and halogenases24-26 and stearoyl-ACP Δ9 desaturase (Δ9D)27] or an effector protein [as in soluble methane monooxygenase (sMMO) and toluene/o-xylene monooxygenase (ToMOH)28-32]. We have adopted the term “substrate triggering” to refer to the cases in which the substrate acts as the relevant activator.26 This effect may have evolved to limit self-inflicted damage to the enzyme resulting from formation of potently oxidizing intermediates in the absence of the oxidation target. The activating binding event is typically associated with a change in cofactor coordination. In many cases, dissociation of a water ligand creates an open coordination site to which O2 can add to initiate the reaction.33-35
In this study, we show that the Np ADO is triggered by short-chain (C6-C10) n-aldehyde substrates4 for addition of O2 to its Fe2II/II cofactor. The reaction yields an intermediate with an absorption feature at ~ 450 nm, a diamagnetic ground state (S = 0), and Mössbauer parameters indicative of two distinct, AF coupled, high-spin FeIII sub-sites. Its spectroscopic properties and reactivity suggest that the intermediate is a Fe2III/III complex with a bound peroxide. This intermediate is long-lived (t1/2 ~ 400 s at 5 °C) in the absence of a reductant, but effects rapid (t1/2 ~ 0.14 s) oxidation of the reduced form of 1-methoxy-5-methylphenazinium methylsulfate (MeOPMS). Its decay in the absence of reductant produces little formate (< 0.15 equivalents), but its reductant-promoted decay generates formate with a stoichiometry of ~ 0.5/MeOPMS (approximately half the theoretical maximum yield). These results provide strong support for two key features of our previously proposed mechanism for ADO (Scheme 1):3,4 (1) formation of a Fe2III/III intermediate with a bound peroxide moiety by addition of O2 to the reduced Fe2II/II cofactor of the ADO•aldehyde complex; and (2) reductive cleavage of the peroxide to initiate a free-radical C1-C2 β scission reaction. The results emphasize the importance of controlled delivery of electrons to the ADO cofactor during turnover and thereby identify another aspect of its function that might be tunable for increased efficiency in associated bioprocesses.
Scheme 1.

Proposed catalytic mechanism for the conversion of aldehyde to alk(a/e)ne and formate by ADO.3,4 The new hydrogen atom incorporated into the alk(a/e)ne product ultimately comes from solvent, but the proximal donor could be either a solvent ligand (green arrows) or an amino acid residue (depicted as H-X; purple arrows).
EXPERIMENTAL SECTION
Materials
Tris-(2-carboxyethyl)phosphine (TCEP), technical grade (> 85% purity) sodium hydrosulfite (dithionite), β-nicotinamide adenine dinucleotide, reduced disodium salt hydrate (≥ 97%) (NADH), 2-nitrophenylhydrazine (97%), and short-chain aldehyde substrates (n-hexanal, n-octanal, and n-decanal) were purchased from Sigma-Aldrich (St. Louis, MO). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide (98%) was purchased from Alfa Aesar (Ward Hill, USA). 1-Methoxy-5-methylphenazinium methylsulfate (MeOPMS) was obtained from Acros Organics (New Jersey, USA). 1-[13C]-octanal was synthesized according to a previously described procedure.3 All other chemicals used for cell growth and protein purification were purchased from Sigma-Aldrich (St. Louis, USA), unless stated otherwise.
Preparation of Np ADO
The plasmid used to direct over-expression of the ADO protein from Nostoc punctiforme PCC 73102 (Npun_R1711; accession number YP_001865325) has previously been described.3 The protein was produced in Escherichia coli BL21(DE3) (Invitrogen; Carlsbad, CA) by a procedure modified from the original, published procedure, as noted below. The cells were grown at 37 °C in M9 minimal medium containing 50 mg/L kanamycin, 2 mM MgSO4, 0.2% (v/v) glucose, 100 μM CaCl2 and 250 μM (NH4)2Fe(SO4)2·6H2O until they reached an OD600 of 0.6-0.8. Expression of the ADO was induced by addition of 250 μM isopropyl-β-D-1-thiogalactopyranoside (IPTG), and the cultures were incubated at 18 °C for 20-24 h. All buffers used in the anaerobic purification of Np ADO contained 1 mM tris-(2-carboxyethyl)phosphine (TCEP). The anaerobically isolated ADO (shown below to contain primarily the Fe2II/II form of the cofactor) was obtained by carrying out cell lysis and immobilized metal-ion affinity chromatography in a Coy anaerobic chamber (Michigan, USA). Cell lysis by sonication of ~ 40 g of cells suspended in ~ 200 mL lysis buffer (50 mM sodium phosphate buffer pH 8.0, 10 mM imidazole, 300 mM NaCl) was followed by ultracentrifugation in sealed, gas-tight tubes at 50,000 × g for 40 min at 4 °C. The supernatant was applied to a NiII-nitrilo-tris-acetate (NTA) column (30 mL resin per 200-220 mL crude lysate). The application was followed by washing with 5 column volumes of 50 mM sodium phosphate buffer (pH 8.0) containing 40 mM imidazole and 300 mM NaCl. The protein was eluted with 50 mM sodium phosphate buffer (pH 8.0) containing 250 mM imidazole and 100 mM NaCl. The protein was concentrated, buffer-exchanged by using a pre-packed PD-10 desalting column (G-25 Sephadex medium, GE Healthcare) in 50 mM sodium HEPES buffer (pH 7.5, 10% glycerol), transferred into gas-tight Eppendorf tubes, and flash frozen in liquid N2, where it was stored until it was used in experiments. Aerobically isolated enzyme (shown below to contain primarily the Fe2III/III form of the cofactor) was obtained by carrying out the same procedure in air. Iron and other trace metals in the samples were quantified by inductively-coupled plasma atomic emission spectroscopy (ICP-AES) by Mr. Henry Gong at the Penn State Materials Research Institute. Purity of the ADO was estimated on the basis of SDS-PAGE and Coomassie staining to be > 90%, and the concentration was calculated by measuring the absorbance at 280 nm and assuming a molar absorption coefficient of 22,920 mM−1cm−1, which was calculated from the amino acid sequence using the ProtParam tool (http://web.expasy.org/protparam/).
Stopped-Flow Absorption (SF-Abs) Experiments and Data Analysis
SF-Abs measurements were carried out with a SX20 stopped-flow spectrophotometer from Applied Photophysics Ltd (Leatherhead, UK) that was housed in an anoxic chamber (Labmaster, MBraun, Stratham, USA). All experiments were carried out at 5 °C. In single-mix (two-syringe) experiments, the ADO reactant solution was diluted by two-fold, and in the sequential-mix (four-syringe) experiments, the protein was diluted by 4-fold. In the single-mix experiments, an O2-free solution of Fe2II/II-ADO (0.20 - 2.5 mM), with or without aldehyde substrate, was mixed with O2-saturated (on ice) 50 mM sodium HEPES buffer (pH 7.5) containing 10% glycerol, giving an estimated O2 concentration after mixing of 0.9 mM.36 The optical path length used was 10 mm or 2 mm, depending on the protein concentration. For initial characterization of the reaction, the photodiode array detector (PDA) was used to acquire time resolved absorption spectra. However, the Fe2III/III-peroxide intermediate was found to be photolytically labile under the intense white light required for use of the multi-wavelength detector. Therefore, the true kinetics of the intermediate were determined by using monochromatic 450-nm light and the photomultiplier tube (PMT) detector.
For a system of two consecutive, irreversible reactions converting reactant, R, to product, P, via intermediate, I, Beer's law gives the absorbance of the solution at 450 nm as a function of time as:
| (1) |
The time dependencies of the reaction components obtained by integrating the relevant differential rate equations are obtained as:37,38
| (2) |
| (3) |
| (4) |
from which the expression for ΔA450nm as a function of time (Eq. 5) can be obtained as:
| (5) |
Because the kinetic traces suggest that the reactant exists in two forms, R and R′, which can both convert to intermediate I, Equation 5 can be expanded to include an additional term for the step R′ → I with its unique rate constant, k′. Alternatively, neglecting the minor absorption at 450 nm from the product complex (εP ~ 0) yields the simpler expression:
| (6) |
which, when used in the regression analyses, was found to return essentially the same values of the rates constants as the more rigorous, complex expression that takes into account the minor absorbance of the product complex.
ΔA388-versus-time traces reporting oxidation of the MeOPMS reductant in sequential mixing stopped-flow experiments were analyzed according to equation (7), which corresponds to two parallel first-order reactions with different amplitudes (ΔA1 and ΔA2) and rate constants (k1 and k2).
| (7) |
Preparation of Freeze-Quench (FQ) Samples
Freeze-quench experiments were carried out at 5 °C as previously described.39
Assays for Formate Production
Formate produced in the ADO reaction was quantified as its 2-nitrophenylhydrazide derivative by liquid chromatography and mass spectrometry (LC-MS), as previously described.3,40 The reactions were carried out in the anoxic chamber by a procedure designed to mimic the sequential-mixing stopped-flow experiments. A 50-μL aliquot containing 1.49 mM anaerobically isolated Fe2II/II-ADO and 16.6 mM 1-[13C]-octanal was mixed in a sealed Eppendorf tube via a gas-tight syringe with an equal volume of O2-saturated buffer. After a 32 second delay for accumulation of the intermediate, a 100 μL aliquot of MeOPMS that had previously been reduced by equimolar sodium dithionite was rapidly injected into this solution. Immediately (one to two seconds later), 10 μL of this solution was quenched into 90 μL of a solution containing 0.032 mM 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide, 0.013 mM 2-nitrophenylhydrazine (2NPH) and 28 mM HCl. A 10-μL aliquot of 1 mM propionic acid was then added as an internal standard, giving a final concentration of 0.1 mM. The reaction was incubated for 15 min at 70 °C. The precipitated protein was pelleted by centrifugation at 17,000 × g, and the supernatant was then filtered through a 0.2 μM PVDF filter (National Scientific, USA) by centrifugation. The solution was analyzed on a 6410 LC/MS Agilent QQQ spectrometer by injecting 2 μL onto a Hamilton PRP-1 analytical column. An isocratic mobile phase (2:1 methanol:H2O containing 0.05% acetic acid) was used. Single-ion monitoring (SIM) was carried out at m/z values of 180 (12CHO-2NPH), 181 (13CHO-2NPH), and 208 (propionyl-2NPH).3
Mössbauer spectroscopy
Mössbauer spectra were recorded on spectrometers from WEB Research (Edina, MN) that have been described previously.41 The spectrometer used to acquire the weak field spectra is equipped with a Janis SVT-400 variable-temperature cryostat, whereas the spectrometer used to acquire the strong field spectra is equipped with a Janis 8TMOSS-OM-12SVT variable-temperature cryostat. The external magnetic field was applied parallel to the γ beam. All isomer shifts quoted are relative to the centroid of the spectrum of α-iron metal at room temperature. Simulation of the Mössbauer spectra was carried out by using the WMOSS spectral analysis software (www.wmoss.org, WEB Research, Edina, MN).
RESULTS
Expression and Purification of Np ADO to Favor Iron Incorporation
Since the initial report on the cyanobacterial alk(a/e)ne-production pathway ending with the ADO (formerly aldehyde decarbonylase) reaction, the identities of the functional metal ions in the enzyme's dinuclear cofactor have remained somewhat in doubt. Initially, a diiron cofactor was presumed, because it is the most common among the ferritin-like proteins,1 but the growing recognition that manganese is functional in some members of the structural superfamily20-23 left the question open. However, our recent characterization of the reaction as a novel oxygenation process made a diiron cofactor the most likely possibility3,4 and led us to express and purify the enzyme under conditions designed both to favor incorporation of only iron and to yield a maximal fraction of the O2 reactive state.
Following its over-expression in minimal medium with 57Fe supplementation, the as-isolated Np ADO (with N-terminal His6 affinity tag comprising 20 additional amino acids) contains 1.15 ± 0.05 Fe ions per monomer, as determined both by inductively coupled plasma atomic emission spectroscopy (ICP-AES) and a spectrophotometric assay based on the FeII chelator, ferrozine.42 Incorporation of the other biologically common transition metals was minimal (0.012 equiv. Mn, 0.030 equiv. Ni, and 0.032 equiv. Zn).
Characterization of (An)aerobically Purified Np ADO by Mössbauer Spectroscopy
Confirmation of the dinuclear nature of the iron cofactor was sought by Mössbauer spectroscopy on the protein purified in the presence and absence of O2. The 4.2-K/53 mT spectrum of the aerobically purified Np ADO (Figure S1A) can be adequately analyzed as a single quadrupole doublet with an isomer shift (δ) of 0.52 mm/s and quadrupole splitting (ΔEQ) of 1.25 mm/s. These parameters are characteristic of high-spin (S = 5/2) FeIII ions.43 The spectrum of the aerobically isolated ADO containing 10 mM n-decanal (Figure 1A) is essentially identical. This observation implies either that the substrate does not bind tightly to the Fe2III/III form of the enzyme or that the aldehyde binds in a manner that does not detectably alter its Mössbauer properties. The fact that the spectrum is a quadrupole doublet is consistent with the presence of two AF coupled high-spin FeIII ions in a dinuclear cluster. Indeed, the spectrum collected with a strong (6-T) applied magnetic field (Figure 1B, vertical bars) can be simulated with δ = 0.52 mm/s, ΔEQ = −1.25 mm/s, asymmetry parameter η = 0.4, and a diamagnetic (Stotal = 0) ground state (Figure 1B, purple line).
Figure 1.

Mössbauer spectra of Np ADO isolated from Ec grown in minimal medium with 57Fe supplementation. (A) Spectrum of the aerobically isolated ADO in the presence of n-decanal recorded at 4.2 K and magnetic fields of 53 mT (vertical bars) and simulation of the experimental spectrum with parameters provided in the text (purple line). (B) Spectrum of the sample in A recorded with a magnetic field of 6 T (vertical bars) applied parallel to the γ beam and simulation thereof with parameters described in the text (purple line). (C) 4.2-K/53 mT spectrum of anaerobically isolated ADO in the presence of n-decanal (vertical bars); the 6%-contribution from the Fe2III/III complex in this sample is shown in purple.
The 4.2-K/53 mT Mössbauer spectrum of the anaerobically isolated Np ADO is dominated by a broad quadrupole doublet with (average) isomer shift (δ ~1.3 mm/s) and quadrupole splitting parameters (ΔEQ ~3.0 mm/s) characteristic of high-spin FeII ions in an octahedral O/N coordination environment (Figure S1B).43 The spectrum is characterized by broad resonances, perhaps indicating heterogeneity of the Fe2II/II cofactor, which may result in a distribution of isomer shifts and quadrupole splitting parameters. In addition, there is a minor (6%) contribution from the oxidized Fe2III/III cluster (purple sub-spectrum). Treatment of the aerobically-isolated Fe2III/III-ADO with dithionite affords a reduced state with a spectrum that is very similar to that of the anaerobically purified protein. Addition of 16.6 mM 1-[13C]-octanal to the anaerobically isolated enzyme, yielding the Fe2II/II-ADO•1-[13C]-octanal complex that is shown below to react rapidly with O2 to accumulate an intermediate, does not detectably alter its Mössbauer spectrum (Figure 1C).
SF-Abs Evidence for “Substrate Triggering” of O2 Addition and Accumulation of an Intermediate
Upon mixing of the Fe2II/II form of Np ADO (prepared either by anaerobic purification or by dithionite reduction of the aerobically purified enzyme) with oxygen-saturated buffer at 5 °C, a broad absorption band centered at ~ 350 nm develops slowly (Figure 2A), signifying oxidation of the cofactor. The absorption feature is similar to those associated with μ-oxo-Fe2III/III clusters in other ferritin-like diiron carboxylate proteins, in which it has been assigned to oxo-to-FeIII charge-transfer (CT) transitions.44-46 By contrast to the slow, monotonic development of the ~ 350 nm band in the reaction of substrate-free Fe2II/II-ADO with excess O2 (Figure S2), inclusion of an aldehyde substrate (e.g., n-decanal) with the Fe2II/II-ADO in the mix with O2 results in a much faster reaction, and a new transient absorption feature at ~ 450 nm is observed (Figure 2B). The spectrum after completion has the same ~ 350 nm absorption band seen in the reaction in the absence of substrate, suggesting formation of a μ-oxo-Fe2III/III product cluster upon decay of the transient species. The short-chain C6-10 n-aldehydes, which were previously shown to be ADO substrates,4,47 all “trigger” reaction of the Fe2II/II cofactor with O2, leading to development of the transient absorption feature (Figure S3). Corresponding n-alkyl compounds lacking a functional group or having acid or alcohol functional groups do not support accumulation of the absorbing species (Figure 3A, red, blue and purple traces). Thus, the aldehyde functional group is essential for “substrate triggering.” At a concentration of 4 mM, the shorter aldehydes (e.g., n-hexanal) are less effective at substrate triggering (Figure S3), as expected from the fact that the natural substrates are C14, C16, or C18 saturated or mono-unsaturated fatty aldehydes provided by the acyl-ACP reductase.1
Figure 2.
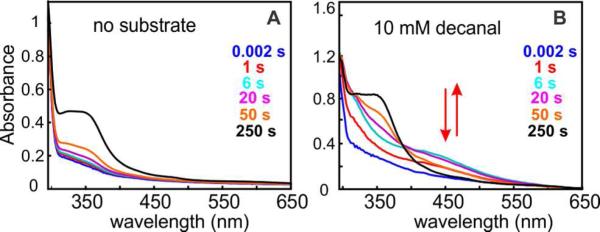
SF-Abs experiment monitoring the reaction at 5 °C of Fe2II/II-ADO (0.35 mM) with O2 (0.9 mM) (A) in the absence and (B) in the presence of 10 mM decanal. The arrows in B indicate the (dis)appearance of an intermediate absorbing at ~ 450 nm only in the presence of the aldehyde substrate.
Figure 3.

Characteristics of the formation and decay of the 450-nm-absorbing complex. (A) Requirement for the aldehyde functional group for substrate triggering of intermediate formation in reaction of Fe2II/II-ADO with O2. A solution of 200 μM Fe2II/II-ADO, 2 mM substrate or analog [n-nonanal (green), n-octanoic acid (red), n-nonan-1-ol (blue), n-nonane (purple), or no substrate (black)] was mixed in the SF-Abs instrument at 5 °C with O2-saturated (1.8 mM) buffer (50 mM sodium Hepes, 10% glycerol, pH 7.5). The reaction was monitored at 450 nm with PDA detection. (B) Assessment of the intermediate's photolytic lability by SF-Abs spectroscopy monitored at 450 nm. A solution of 700 μM Fe2II/II-ADO and 10 mM n-decanal was mixed with an equal volume of an O2-saturated buffer solution at 5 °C. ΔA450nm-vs-time traces were recorded with PDA (green trace) or PMT (blue trace) detection. Regression analysis of the latter trace was carried out according to Equation 6, giving rate constants mentioned in the text. Control experiments carried out in the absence of n-decanal with PDA and PMT detection are shown as black and grey traces, respectively.
Photolytic Lability of the Intermediate
In the SF-Abs experiments of Figure 2, multi-wavelength detection was achieved by use of a photodiode array, which requires intense, polychromatic incident light. Under these conditions, the intermediate was observed to have a half life (t1/2) of ~ 0.7 min at 5 °C (Figure 3A, 3B green traces). However, upon use of monochromatic incident light and the photo-multiplier detector (PMT), the intermediate species was found to decay ~ 10-fold less rapidly (t1/2 ~ 7 min, blue trace with superimposed red fit, Figure 3B). This observation indicates that the intermediate is photolytically labile, as has been found for O2-derived complexes in other non-heme-diiron oxygenases,21,48,49 including the peroxo-Fe2III/III complex in Δ9D,27 and related model compounds.50 Therefore, accurate kinetics for the intermediate were obtained by use of monochromatic incident light and photomultiplier detector.
Complex Kinetics of Formation of the Intermediate and Their Origin
Attempts to analyze the ΔA450nm-versus-time traces from the reaction according to a sequence of two irreversible steps gave poor agreement between the fit and experimental data, suggesting the presence of two distinct formation phases with different rate constants, each associated with a decay phase of the same rate constant. The traces were thus fit by Equation 6, which gives change in absorbance (ΔA450nm) as a function of time (t) from two converging reaction channels that form a common intermediate (I) and product (P) from two different forms of the reactant (R and R′). I forms from R and R′ with apparent first-order rate constants k1 and k1′, respectively, and decays to P with rate constant k2. R (R′), I, and P have molar absorption coefficients at 450 nm of εR (assumed to be equal for R and R′ because the Fe2II/II complexes should be essentially transparent at this wavelength), εI, and εP, respectively. The product complex was also assumed to have practically zero absorption (εP ~ 0) at 450 nm, which is justified by its spectrum (Figure 2B). This regression analysis applied to the trace in Figure 3B gave apparent first-order rate constants for formation of k1 = 0.75 s−1 and k1′ = 0.20 s−1 and a rate constant for decay of k2 = 0.0017 s−1. According to this analysis, reactant form R predominates (64%) over R′ (36%). Fitting the data so as to account for the non-zero absorption of P at 450 nm gave essentially the same results. The origin of the two formation phases and the distinction between the two reactant states (R and R′) giving rise to them were probed in two additional SF-Abs experiments. In the first, variation of the concentration of the aldehyde substrate (decanal) revealed a titration behavior, in which the relative contribution of the slow formation phase to the total amplitude of the transient is diminished with increasing substrate (Figure S4). This result suggests that incomplete formation of the Fe2II/II-ADO•substrate complex in the reactant solution could be one source of the kinetic complexity. To confirm this deduction, a second experiment, in which substrate-free Fe2II/II-ADO was mixed with an O2-saturated solution containing the substrate, was carried out (Figure S5). The markedly slower formation of the intermediate in the reaction initiated in this manner proves that substrate acquisition by Fe2II/II-ADO is slower than O2 addition to the Fe2II/II-ADO•decanal complex and provides support for the notion that sub-saturation by the substrate could be one source of the kinetic complexity in intermediate formation in the experiment of Figure 3B. Interestingly, the reaction initiated by mixing with decanal and O2 also exhibits kinetic complexity. This observation suggests that, in addition to the heterogeneity related to the presence of both substrate-free and substrate-bound forms, the free enzyme is itself heterogeneous, having two or more forms differing in their kinetics of substrate binding.
Characterization of the Intermediate by Mössbauer Spectroscopy
To obtain insight into the electronic structure of the intermediate, freeze-quench (FQ) samples were prepared for analysis by Mössbauer spectroscopy. The Fe2II/II-ADO•octanal complex was mixed with a solution of O2-saturated buffer, and the reaction was quenched after 32 s, the time of maximum A450 in a SF-Abs experiment under identical reaction conditions (Figure S6). The 4.2-K/53-mT Mössbauer spectrum of this sample (Figure 4A, vertical bars) has minor contributions from the Fe2III/III complex (6%) present in the reactant solution (purple trace) and unreacted Fe2II/II species (14%, light blue trace). After removal of these two components, the remaining spectral features (Figure 4B) can be analyzed as two partially resolved quadrupole doublets of equal intensity (40% each of the integrated intensity of the original, experimental spectrum) with parameters δ1 = 0.48 mm/s, ΔEQ1 = 0.49 mm/s (blue sub-spectrum) and δ2 = 0.55 mm/s, ΔEQ2 = 1.23 mm/s (green sub-spectrum) characteristic of high-spin FeIII sub-sites (Table 1).43 The 4.2-K/6.0-T spectrum (Figure 4C, vertical bars) can be simulated with values of δ and EQ determined from the 53-mT spectrum and η1 = 0, η2 = 0.9, and Stotal = 0. The 53-mT spectrum can be simulated equally well with parameters δ1 = 0.32 mm/s, ΔEQ1 = 0.80 mm/s and δ2 = 0.70 mm/s, ΔEQ2 = 0.93 mm/s, but this analysis is inconsistent with the 6.0-T spectrum. From the isomer shifts of the two sub-sites (0.48 and 0.55 mm/s), it can be concluded that the intermediate has a Fe2III/III core, which is more oxidized by two electrons than the cofactor in the Fe2II/II-ADO•octanal reactant complex. Because the transient complex forms in a reaction without an obvious source of additional electrons, the co-substrate O2 must be reduced by two electrons to the peroxide level, yielding, for example, peroxo-Fe2III/III state B or Fe2III/III-peroxyhemiacetal state C in Scheme 1. The transient species is, therefore, referred to hereafter as a “Fe2III/III-peroxide” complex, a designation intended to encompass both possible structural formulations. Correlation of the concentration of the intermediate determined from analysis of the Mössbauer data (80% of 0.75 mM giving [Fe2] = 0.60 mM) and the maximum value of the absorbance change (ΔA450 = 0.72) in the SF-Abs experiment carried out under identical conditions (Figure S6) provides an estimate of ε450 for the intermediate of 1,200 M−1cm−1. This value is similar to those observed for known peroxo-Fe2III/III complexes.31,51-56
Figure 4.

Characterization of the 450-nm-absorbing complex by FQ Mössbauer spectroscopy. (A) 4.2-K/53-mT Mössbauer spectrum of a FQ sample prepared by reacting Fe2II/II-ADO•1-[13C]-octanal with O2-saturated buffer for 32 s (vertical bars). The contribution of unreacted Fe2II/II-ADO•1-[13C]-octanal (14%) and the Fe2III/III-ADO (6%) are shown as light blue and purple lines, respectively. (B) Removal of the contributions from the Fe2II/II-ADO•1-[13C]-octanal and Fe2III/III-ADO from A yields the spectrum of the intermediate (vertical bars), which can be simulated as two quadrupole doublets of equal intensity (blue and green shaded areas and red line) with parameters quoted in the text. (C) 4.2-K/6-T spectrum of the 32-s sample described above (vertical bars) and simulation of the sub spectra of the Fe2III/III-peroxide intermediate (blue and green shaded areas) as described in the text. The spectrum of the minor Fe2III/III contribution is shown in purple, and the summation of the contributions from the intermediate and Fe2III/III contaminant is shown as the red line plotted on top of the data.
Table 1.
Mössbauer and optical properties of peroxo-Fe2III/III intermediates in Np ADO and other homologous carboxylate-bridged diiron proteins.
| Peroxo Species | δ (mm/s) | ΔEQ (mm/s) | λmax (nm) | Ref. |
|---|---|---|---|---|
| ADO | 0.48 | 0.49 | 450 | this work |
| 0.55 | 1.23 | |||
| MMOH | 0.66 | 1.51 | 725 | 31,66 |
| AurF | 0.55 | 0.74 | 500 | 70 |
| 0.61 | 0.35 | |||
| Δ9-ACP desaturase | 0.64 | 1.06 | 700 | 51 |
| 0.68 | 1.90 | |||
| ToMOH | 0.55 | 0.67 | n.d. | 69 |
| oxy-hemerythrin | 0.54 | 1.92 | ~500 | 86 |
| 0.51 | 1.09 | |||
| deoxyhypusine hydroxylase | 0.55 (0.49) | 1.16 (1.05) | 630 | 92 |
| 0.58 (0.63) | 0.88 (0.99) | |||
| Frog M Ferritin | 0.62 | 1.08 | 650 | 56 |
| D84E R2 | 0.63 | 1.58 | 700 | 53,82 |
The SF-Abs experiments of Figures 2 and 3 suggest that the Fe2III/III-peroxide complex accumulates only in the presence of aldehyde. This conclusion was verified by FQ Mössbauer spectroscopy (Figure S7). Upon mixing of the anaerobically isolated Np ADO with O2-saturated buffer in the absence of an aldehyde substrate, the Fe2III/III-peroxide complex was found to accumulate to a minor extent (~ 4%), if at all, after a reaction time of 15 s (Figure S7B). The spectrum reflects the presence of ~11% of the oxidized (Fe2III/III) state, which is also present in the reactant solution for this experiment (Figure S7A), but is dominated by the contribution from unreacted Fe2II/II complex. Thus, the Fe2II/II-ADO is relatively unreactive toward O2, as deduced from the SF-Abs data. By contrast, in the presence n-decanal, the Fe2III/III-peroxide complex accumulated (Figure S7C), albeit to a lesser extent (~ 44%) than in the experiment of Figure 4. The SF-Abs and FQ Mössbauer data thus confirm substrate triggering of O2 addition to form the Fe2III/III-peroxide complex.
Chemical Competence of the Intermediate for Reduction by MeOPMS
According to our working hypothesis for the mechanism of the ADO reaction (Scheme 1), the Fe2III/III-peroxide complex is expected to be analogous to state B or state C (which could also be in equilibrium). Accordingly, production of formate and the Cn-1 alkane from this complex should require a total of two electrons from the reducing system. The ability of the complex to oxidize MeOPMS, the reductant shown by Marsh and co-workers47 to support greater ADO activity than the originally employed heterologous (spinach) ferredoxin,1,3,4 was assessed in a sequential mix SF-Abs experiment. The Fe2III/III-peroxide complex was allowed to accumulate by a 32-s incubation (tdelay) at 5 °C following a first mix of the Fe2II/II-ADO•1-[13C]-octanal complex with an equal volume of O2-saturated buffer, and the solution was then mixed with an equal volume of a solution containing one equivalent of the dithionite-reduced form of MeOPMS. The reaction was monitored by the development of the spectrum of the oxidized MeOPMS57 with λmax = 388 nm (ε388 ~ 26,000 M−1 cm−1) (Figure 5A). The ΔA388-vs-time trace reveals rapid oxidation, essentially complete within 1 s (Figure 5B, green trace). Corresponding traces from control experiments omitting ADO (black), having an initial delay time insufficient to permit accumulation of the absorbing complex (tdelay = 0.01 s, blue trace), or substituting the Fe2II/II-ADO with the aerobically isolated Fe2III/III form (Figure S8) reveal much slower MeOPMS oxidation. Similarly, with tdelay = 500 s, a time sufficient to permit decay of the transient complex to ~ 50% of its maximum concentration (Figure 3B, blue trace), the fast phase of MeOPMS oxidation is diminished accordingly (Figure 5B, red trace). These observations show that the transient complex rapidly oxidizes MeOPMS, whereas neither the Fe2II/II-ADO•1-[13C]-octanal reactant nor the aerobically isolated Fe2III/III-ADO does so. The results thus establish the oxidizing nature of the complex, consistent with the hypotheses that it possesses a bound peroxide moiety and is an intermediate on the pathway to formate and alk(a/e)ne.
Figure 5.

Sequential-mixing SF-Abs experiments establishing the ability of the Fe2III/III-peroxide complex to oxidize MeOPMS. The Fe2II/II-ADO•1 [13C]-octanal reactant solution was mixed with O2-saturated buffer, the solution was allowed to react for the delay time (tdelay) indicated in the figure, the aged reaction solution was then mixed with a solution of dithionite-reduced MeOPMS, and spectra were acquired following the second mix. (A) Selected spectra from the experiment with tdelay = 32 s, reflecting the rapid oxidation of reduced MeOPMS. (B) ΔA388-vs-time traces for experiments with different delay times between the two mixes [tdelay = 0.01 s (blue), 32 s (green), 500 s (red)]. A control experiment, in which ADO was omitted is shown in black. Dashed lines are fits of Eq. 7 to the data.
Chemical Competence of the Intermediate for Formate Production and Reaction Stoichiometry
The expectation that MeOPMS-promoted decay of the Fe2III/III-peroxide intermediate should produce formate (Scheme 1) was evaluated in a chemical-quench experiment (see Materials and Methods) designed to mimic the sequential-mixing SF-Abs experiment of Figure 5. The quantity of 13C-labeled formate from 1-[13C]-octanal, determined after conversion to the formyl-2-nitrophenylhydrazide [(13CHO)-2NPH] derivative,3 was found in two independent trials to vary as an approximately linear function of the quantity of reduced MeOPMS added (Figure 6).
Figure 6.

Determination of the reductant:formate stoichiometry upon reduction of the Fe2III/III-peroxide complex with MeOPMS. The dotted fit line gives a formate:reductant ratio of ~ 0.5, which is half of the theoretical yield.
Decay of the intermediate in the absence of reductant in an eppendorf tube under ambient light produces only a low yield of formate (< 15%). The formate that is produced under these conditions could reflect electron flow from the unreacted Fe2II/II-ADO complex (~14% observed in the FQ Mössbauer experiment of Figure 1) to the Fe2III/III-peroxide complex. With increasing MeOPMS added (again, under ambient light), the yield of formate increases. Because the phenazine is a two-electron reductant, the maximum theoretical yield of formate:MeOPMS is unity. The observed deviation most likely reflects the “uncoupling” of phenazine oxidation from formate production in approximately half of the reaction events. This result is not surprising in view of the non-physiological nature of the reducing system and the possibilities for (1) unproductive two-electron reduction of the Fe2III/III-peroxide intermediate and (2) competition for reaction with the reductant from either the Fe2III/III-ADO product emerging from initial turnover of the intermediate, residual O2, or both. We conclude that the complex is chemically competent for formate production upon its reduction by MeOPMS and is an authentic intermediate in the ADO reaction.
Mössbauer Spectroscopic Characterization of the Product of Reduction of the Fe2III/III-Peroxide Intermediate by MeOPMS
The state of the ADO cofactor after the MeOPMS induced productive decay of the Fe2III/III-peroxide complex was probed by Mössbauer spectroscopy. A sample was freeze-quenched 0.56 s after the pre-formed complex was mixed with the reduced form of MeOPMS. This incubation time is sufficient to allow for the reaction to near completion (Figure 5B, green trace). The 4.2-K/53-mT Mössbauer spectrum of this sample (Figure 7A), has residual contributions from Fe2II/II-ADO (~ 13%) but is dominated by broad quadrupole doublets. Subtracting the spectrum of the sample quenched before reaction with MeOPMS (Figure 4A) from the spectrum of post-reduction sample (Figure 7A) cancels out the contributions of unreacted Fe2II/II-ADO•octanal complex and Fe2III/III impurity and illustrates that decay of the features of the transient complex (~ 46%, pink trace in Figure 7B) is associated with the appearance of a broad doublet (positive features). By adding back the 46% contribution of the FeIII/III-peroxide spectrum, the spectrum of the diamagnetic reduction product is obtained (Figure 7C). The spectrum can be adequately analyzed as two quadrupole doublets with parameters δ1 = 0.55 mm/s, ΔEQ1 = 1.80 mm/s, δ2 = 0.51 mm/s, and ΔEQ2 = 1.08 mm/s. These parameters are typical of high-spin FeIII ions 43,44 and indicative of an AF coupled high-spin Fe2III/III cluster. The spectroscopic signature of this cluster is distinct from that of the Fe2III/III complex that forms upon the slow (30 min at 5 °C) decay of the Fe2III/III-peroxide complex in the absence of MeOPMS (Figure 7D, vertical bars). This latter complex has a spectrum that is essentially identical to that of aerobically isolated ADO. The observation that reduction of the transient Fe2III/III complex by MeOPMS generates a distinct state with the cofactor still in the Fe2III/III oxidation state confirms that the oxidizing equivalents of the transient complex are in the form of a bound peroxide. The fact that the spectrum of the ADO after the productive, reductive decay of the Fe2III/III-peroxide state is distinct from those of the aerobically isolated enzyme and the spontaneous (unproductive) decay product could be explained by the presence of bound formate, alkane or both in at least a fraction of the reduction product, but other explanations are also possible.
Figure 7.

Mössbauer-spectroscopic characterization of the ADO cofactor following the productive decay of the Fe2III/III-peroxide intermediate that is initiated by reduced MeOPMS. (A) Experimental 4.2-K/53-mT spectrum after reaction of the pre-formed complex with dithionite-reduced MeOPMS for 0.56 s at 5 °C. (B) Difference spectrum (vertical bars) prepared by subtracting the spectrum of the sample quenched before reduction (Figure 4A) from spectrum A. In this presentation, the features associated with the Fe2III/III-peroxide complex point upwards (the pink line is the experimental “reference spectrum” shown in Figure 4B scaled to 46% intensity) and the features associated with the product of the productive decay point downwards. (C) “Reference spectrum” of the product of the reaction of the Fe2III/III-peroxide intermediate with reduced MeOPMS (vertical bars) and quadrupole doublet simulations with parameters quoted in the text (solid lines). (D) Spectrum of a sample prepared by allowing the Fe2III/III-peroxide intermediate to decay at 5 °C for 30 min in the absence of reduced MeOPMS.
DISCUSSION
Definitive Evidence that ADO Employs a Diiron Cofactor
A better understanding of the structure and mechanism of ADO might enable rational enhancement of its in-vivo efficiency in renewable energy applications. A key aspect of its structure is the identities of the metal ions in its functional cofactor. Although a diiron cofactor has generally been presumed, the multi-turnover enzyme assays reported to date have left open the possibility that a minor fraction of the ADO preparations, and not the major diiron component, was responsible for the alk(a/e)ne and formate produced. We sought to unequivocally test the catalytic competence of the diiron cofactor by preparing ADO as highly enriched in diiron cluster as possible and examining the reaction of these preparations in single turnover experiments with steps in the sequence resolved by controlling the delivery of the requisite electrons.
The ICP-AES metal analysis and Mössbauer spectra show that our preparations of Np ADO produced in Ec grown on minimal medium supplemented with iron contain a diiron cluster. When the enzyme is isolated aerobically, it contains an AF coupled Fe2III/III cofactor, which can be chemically reduced to the Fe2II/II form. Following isolation under anaerobic conditions, it contains a Fe2II/II cluster with spectroscopic properties identical to those of the chemically reduced form. Although the detection of a particular metal ion in a recombinant protein is, at best, very poor evidence that that metal is functional, the reactivity of the diiron-ADO characterized here indicates that it is indeed the active state. The reaction of Fe2II/II-ADO with O2 is very slow and produces the μ-oxo-Fe2III/III state without obvious accumulation of an intermediate species. The reaction of the Fe2II/II-ADO•aldehyde complex with O2 is much faster and yields a transient complex with distinct absorption and Mössbauer spectra. Accumulation of this intermediate is strictly dependent on the presence of an n-aldehyde substrate: all short-chain n-aldehydes tested (C6-C10) support its accumulation, whereas the corresponding n-alkyl compounds terminated by other functional groups (n-alkanoic acids, primary n-alcohols, and n alkanes) do not. Aldehydes with longer side chains trigger intermediate formation more efficiently. This trend is expected, because the physiological substrates are C14, C16, or C18 saturated or mono-unsaturated fatty aldehydes provided by the acyl-ACP reductase and because the longer-chain substrates should bind more tightly within the hydrophobic active-site channel.1 Triggering of O2 reactivity by the substrate or an effector protein is a common characteristic of the ferritin-like diiron-carboxylate oxidases and oxygenases, and its occurrence in our ADO preparations is consistent with the diiron form being functional. Importantly, the fact that ~ 80% of the Fe2II/II cluster can, with proper substrate triggering, convert to an intermediate that reacts rapidly with the reducing system to afford ~ 0.6 formate/ADO rules out the possibility that a minor fraction of the recombinant protein, and not the predominant enzyme form, could be responsible for the activity. Thus, the spectroscopic and reactivity data together provide the most definitive evidence to date that cyanobacterial ADOs can use a diiron cofactor in catalysis.
Nature of the Diiron-O2 Intermediate
The chemically competent intermediate state exhibits an absorption band at 450 nm (ε450 = 1,200 M−1cm−1) and two resolved Mössbauer quadrupole doublets characteristic of an AF coupled Fe2III/III cluster. Its spectroscopic properties, its formation by reaction of the Fe2II/II-ADO•aldehyde complex with O2 in the absence of reductant, and its capacity to oxidize the reduced form of MeOPMS rapidly while converting to a distinct Fe2III/III state, imply that it possesses a bound peroxide. Indeed, this two-electron reduction of O2 to peroxide is the common first step in the reactions of the carboxylate-bridged diiron oxygenases and oxidases.18 Moreover, with the exception of the complex formed in ToMOH, all peroxo-Fe2III/III complexes trapped in metalloproteins and inorganic models thereof share with the ADO intermediate the characteristics of absorption in the visible region and Mössbauer spectra reflecting AF-coupling of two FeIII ions (see Table 1).
Possible Structures of the Intermediate and Positions in the Chemical Mechanism
Scheme 1 shows the mechanistic hypothesis initially proposed by us3 and subsequently adopted by Marsh and co-workers58 upon their corrections59,60 to their claim of O2-independent ADO activity.47,61 The mechanism accommodates all published data on the reaction, including: (i) our discovery that the C1-derived co-product of Np ADO is formate2 (which was subsequently confirmed for a second ADO ortholog61) rather than the originally proposed carbon monoxide11 or carbon dioxide;5 (ii) our demonstration that the O-atom incorporated into the formate co-product is derived from O23,4 rather than from solvent;47 and (iii) recent evidence from the Marsh group for a C2 methylene radical intermediate accompanying formate production.58 The mechanism can therefore guide consideration of the structure of the diiron intermediate and its position in the reaction sequence.
The reaction begins by the binding of the aldehyde substrate with coordination of its carbonyl group to one of the two FeII ions of the reduced cofactor, yielding state A. Coordination of the substrate in this manner could displace one or more solvent ligand (not depicted), a common mechanism for substrate triggering of O2 addition.33-35,62 There is extensive precedent for triggering in iron oxygenases upon binding of the substrate merely in the vicinity of, and not directly to, the cofactor.26,33-35,62-65 Aldehyde coordination is, therefore, only a reasonable conjecture that is consistent with both the substrate triggering demonstrated here and the presence of a μ-1,3 fatty acid ligand suggested by the X-ray crystallographic data on the Pm ADO (pdb id: 2OC5).1
Analogously to the reactions of other members of the superfamily, addition of O2 to A would produce a peroxo-Fe2III/III intermediate, B, which could have the symmetrical μ-η2:η2-peroxo-FeIII/III core structure depicted in Scheme 1 or a less symmetrical peroxide binding mode (e.g., end-on to a single iron). The peroxide in B is proposed to nucleophilically attack the aldehyde carbonyl, yielding the peroxyhemiacetal state C. This step would distinguish the ADO mechanism from those of related diiron oxygenases studied to date, in which peroxo-Fe2III/III complexes undergo redox-neutral or reductive O-O bond cleavage to yield high-valent states (in sMMO66 and RNR-R2,67,68 respectively) or are attacked by a nucleophilic substrate to yield oxidized (most often hydroxylated) products (in ToMOH69 and perhaps AurF54,70). The proposed nucleophilicity of the ADO peroxide complex is thus reminiscent of the ambivalent electrophilicity/nucleophilicity of C4a-(hydro)peroxyflavin intermediates in flavin dependent monooxygenases.71 The demonstrated protonolyses of an inorganic peroxo-FeIII/III complex72 and the corresponding intermediate in ferritin73 to produce H2O2 suggest that such complexes can indeed also be basic/nucleophilic. Moreover, the nucleophilic attack by a peroxo-Fe intermediate on a carbonyl carbon has been proposed as a key step in the C-C-bond cleaving reactions of the mixed-valent non-heme diiron enzyme, myo-inositol oxygenase (MIOX),74,75 the mononuclear non-heme-iron enzymes, 2 hydroxyethyl-1-phosphonate dioxygenase (HEPD)76,77 and methylphosphonate synthase (MPnS),78 as well as the heme-dependent cytochrome P450 sterol 14a-demethylase (CYP51).7,79
The next step in the proposed mechanism is the one-electron reductive cleavage of the O-O bond of the Fe2III/III-peroxyhemiacetal complex in C by the reducing system, which yields a μ-oxo-Fe2III/III cluster and gem-diolyl radical (D). The radical undergoes a β-scission of the C1-C2 bond, generating formate and a C2 methylene radical (E). Support for the formation of this radical was recently provided by the Marsh group, who found evidence for the expected ring opening accompanying formate production from a C3-C4 cyclopropanated substrate analog.58 The C2 methylene radical then abstracts a hydrogen atom from a nearby amino acid (XH in Scheme 1, pink arrows) or a solvent ligand to the cofactor (green arrows), and quenching of the resultant oxidized species (amino acid radical or Fe2III/IV cluster) by another electron from the reducing system and a proton gives the product state, F. The fact that the Mössbauer spectrum of the samples quenched immediately after the productive (reductive) decay of the Fe2III/III-peroxide complex differs significantly from the spectra of both the aerobically purified protein and the product of spontaneous decay of the intermediate is consistent with the formation of a downstream intermediate such as state F. Release of the products from state F and two-electron reduction of the cofactor by the reducing system completes the cycle.
The peroxo-Fe2III/III state, B, and Fe2III/III-peroxyhemiacetal state, C, in Scheme 1 both have the requisite characteristics of a III/III oxidation state and a bound peroxide equivalent and are, therefore, both viable representations of the ADO intermediate. Three characteristics of the intermediate favor a C-like structure but can, with additional rationalization, also accommodate a B-like structure. First, formation of the intermediate requires a bound aldehyde. The carbonyl group of the substrate would not be directly involved in formation of B but would be indispensable for formation of C. The observation that n-alkyl compounds lacking the carbonyl uniformly fail to support intermediate accumulation could thus be readily understood in terms of structure C. However, an alternative explanation is that the structural requirements for activating the Fe2II/II cofactor for addition of O2 to form B are just unusually stringent and fulfilled only by the aldehyde. Second, the intermediate persists for minutes in the absence of reductant but oxidizes the reduced form of MeOPMS in hundreds of milliseconds. Progression of B to C in Scheme 1 is redox neutral, but conversion of C to D requires an electron. The promotion of its decay by reduced MeOPMS (yielding formate) is thus most naturally rationalized by a C-like structure. However, B and C could interconvert in a rapid equilibrium favoring B. In this case, decay of B would be fast only under conditions favoring rapid reductive trapping of C. Alternatively, binding of reduced MeOPMS to state B could induce its conversion to C and subsequent fast reduction to D. Indeed, the Marsh group provided evidence for the tight binding of a similar phenazine to the enzyme.61 Either of these idiosyncratic kinetic scenarios could rationalize both the stability of a complex similar to B in the absence of reductant and the acceleration of its decay by reduced MeOPMS.
The third characteristic of the intermediate that is also most easily explained by a Fe2III/III-peroxyhemiacetal rather than a μ-peroxo-Fe2III/III formulation is its Mössbauer spectrum, which exhibits more pronounced site-differentiation than has often been seen among μ-peroxo-Fe2III/III complexes analogous to B (Table 1). An inorganic complex originally prepared by Kitajima and the intermediate trapped in the D84E variant of Ec ribonucleotide reductase β subunit are the most extensively studied among the known inorganic and protein complexes (respectively) of this type. Each has been shown to possess a cis-μ-1,2-peroxo-Fe2III/III core structure.53,80-82 Each exhibits an absorption band at ~ 700 nm, assigned to a peroxide→FeIII charge transfer transition,81 but only a single Mössbauer quadrupole doublet with a rather high isomer shift (> 0.60 mm/s) for both Fe sites. Peroxide complexes in the wild-type RNR-β proteins from Ec and mouse,67,68, soluble methane monooxygenase,31,66 ToMOH,69 and frog M ferritin,56 as well as several inorganic complexes,52,80,83,84 also have iron sites that are unresolved by Mössbauer and thought to contain the cis-μ-1,2-peroxo-Fe2III/III core, though calculations on the intermediate from sMMO suggest that a μ-η2:η2-peroxo binding mode is also possible.85
There are, however, several examples of protein 51,54,86 and inorganic (hydro)peroxo-Fe2III/III complexes 87,88 that do exhibit site resolution in their Mössbauer spectra (Table 1). The peroxo-Fe2III/III complexes in oxy-hemerythrin, the N-oxygenase designated AurF, and Δ9D desaturase are such examples. Differentiation of the two iron subsites in these complexes arises either from asymmetric coordination of the peroxide moiety (e.g., to a single Fe), from asymmetry imposed by the protein or solvent ligands, or from a combination of these factors. For example, in oxyhemerythrin, coordinative saturation of one iron with a μ-hydroxo, two μ-1,3 carboxylate, and three histidine ligands enforces addition of O2 terminally only to the other FeII ion,89 which has one fewer histidine and thus a single open coordination site. The structure of the (hydro)peroxo-Fe2III/III intermediate in AurF is not known, but here again the protein provides one more histidine ligand to the FeII in site 190 and may therefore enforce O2 addition either only to the site 2 FeII or in a manner that more intimately involves and profoundly affects site 2. On the other hand, for the peroxo-Fe2III/III state in Δ9D, recent quantum mechanical calculations suggest that the asymmetry reflected by its Mössbauer spectrum arises in spite of an inherently symmetrical μ-1,2 mode of peroxide coordination by virtue of a bidentate-to-monodentate carboxylate shift at one of the FeIII ions, which makes that site five-coordinate while leaving the other FeIII six-coordinate.91
The amino-acid iron ligands of ADO are, at least in terms of their identities, inherently site-symmetrical (two His and four Glu residues), as they are in Δ9D. The site resolution in the Mössbauer spectrum of the Fe2III/III-peroxide complex in ADO is thus most likely associated either with rearrangement of protein ligand(s) in the first coordination sphere (as proposed for Δ9D), asymmetrical coordination of the peroxide moiety, direct, asymmetric coordination of the substrate (or an intermediate derived from it), or a combination of these factors. As depicted in Scheme 1, the postulated Fe2III/III-peroxyhemiacetal state C could potentially possess both an asymmetrically coordinated hemiacetal anion and an asymmetrically bound peroxide. In this respect, state C provides a potential rationale for the spectroscopic properties of the Fe2III/III-peroxide intermediate in ADO. More detailed structural studies on the intermediate will be required to test our hypothesis that it is a unique type of peroxide (peroxyhemiacetal) adduct to its non-heme diiron cofactor.
Conclusions
Our results verify that cyanobacterial ADO can employ a diiron cofactor rather than a homo- or heterodinuclear cofactor involving one or more transition metal other than iron. Upon reaction of the Fe2II/II-ADO•substrate complex with O2 in the absence of the reducing system, an intermediate with a Fe2III/III cluster and bound peroxide equivalent accumulates to ~ 80% yield. It is chemically competent to generate formate upon reacting with the reducing system, and this reaction generates a stable Fe2III/III cluster that is spectroscopically distinct from the aerobically isolated ‘resting’ state. The identification of the intermediate provides support for our previously proposed mechanism involving a novel attack of the peroxide of a peroxo-Fe2III/III complex upon the substrate carbonyl and the reductant promoted decomposition of the resultant peroxyhemiacetal complex, leading to formate and alkane production via a free-radical β-scission mechanism.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Science Foundation (MCB-1122079 to C.K., S.J.B. and J.M.B.) and the National Institutes of Health (GM-055365 to J.M.B. and C.K.).
Footnotes
This work was supported by the National Institutes of Health (GM-55365 to JMB and CK) and the National Science Foundation (MCB-1122079 to CK, SJB, and JMB).
ABBREVIATIONS: ADO, aldehyde-deformylating oxygenase; Np, Nostoc punctiforme; Pm, Prochlorococcus marinus; Ec, Escherichia coli; AF, antiferromagnetic(ally); MeOPMS, 1-methoxy-5-methylphenazine; SF-Abs, stopped-flow absorption; FQ, freeze-quench(ed)
Supporting Information
Figures S1-S8 contain spectra of stopped-flow absorption and Mössbauer experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Schirmer A, Rude MA, Li XZ, Popova E, del Cardayre SB. Science. 2010;329:559. doi: 10.1126/science.1187936. [DOI] [PubMed] [Google Scholar]
- 2.Warui DM, Li N, Nørgaard H, Krebs C, Bollinger JM, Jr., Booker SJ. J. Am. Chem. Soc. 2011;133:3316. doi: 10.1021/ja111607x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li N, Nørgaard H, Warui DM, Booker SJ, Krebs C, Bollinger JM., Jr. J. Am. Chem. Soc. 2011;133:6158. doi: 10.1021/ja2013517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li N, Chang WC, Warui DM, Booker SJ, Krebs C, Bollinger JM., Jr. Biochemistry. 2012;51:7908. doi: 10.1021/bi300912n. [DOI] [PubMed] [Google Scholar]
- 5.Reppas NB, Ridley CP. Methods and compositions for the recombinant biosynthesis of n-alkanes. Joule Unlimited Inc.; 2010. U.S. Patent 7794969. [Google Scholar]
- 6.Ducat DC, Way JC, Silver PA. Trends Biotech. 2011;29:95. doi: 10.1016/j.tibtech.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Patra T, Manna S, Maiti D. Angew. Chem. Int. Ed. 2011;50:12140. doi: 10.1002/anie.201103860. [DOI] [PubMed] [Google Scholar]
- 8.Zhang FZ, Rodriguez S, Keasling JD. Curr. Opin. Biotech. 2011;22:775. doi: 10.1016/j.copbio.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 9.Robertson DE, Jacobson SA, Morgan F, Berry D, Church GM, Afeyan NB. Photosynth. Res. 2011;107:269. doi: 10.1007/s11120-011-9631-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serrano-Ruiz JC, Ramos-Fernandez EV, Sepulveda-Escribano A. Energy Environ. Sci. 2012;5:5638. [Google Scholar]
- 11.Schirmer A, Rude MA, Brubaker S. Methods and compositions for producing hydrocarbons. 2009:LS9. U.S. Patent 140696.
- 12.Akhtar MK, Turner NJ, Jones PR. Proc. Natl. Acad. Sci. USA. 2013;110:87. doi: 10.1073/pnas.1216516110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallar BJ, Lipscomb JD. Chem. Rev. 1996;96:2625. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 14.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Muller J, Lippard SJ. Angew. Chem. Int. Ed. 2001;40:2782. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 15.Stubbe J. Curr. Opin. Chem. Biol. 2003;7:183. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 16.Fox BG, Lyle KS, Rogge CE. Acc. Chem. Res. 2004;37:421. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 17.Andrews SC. BBA GEN Subjects. 2010;1800:691. doi: 10.1016/j.bbagen.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Krebs C, Bollinger JM, Jr., Booker SJ. Curr. Opin. Chem. Biol. 2011;15:291. doi: 10.1016/j.cbpa.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nordlund P, Eklund H. Curr. Opin. Struc. Biol. 1995;5:758. doi: 10.1016/0959-440x(95)80008-5. [DOI] [PubMed] [Google Scholar]
- 20.Barynin VV, Whittaker MM, Antonyuk SV, Lamzin VS, Harrison PM, Artymiuk PJ, Whittaker JW. Structure. 2001;9:725. doi: 10.1016/s0969-2126(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 21.Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak MA, Krebs C, Bollinger JM., Jr. Science. 2007;316:1188. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 22.Boal AK, Cotruvo JA, Jr., Stubbe J, Rosenzweig AC. Science. 2010;329:1526. doi: 10.1126/science.1190187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cotruvo JA, Jr., Stubbe J. Biochemistry. 2011;50:1672. doi: 10.1021/bi101881d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM., Jr. Biochemistry. 2005;44:8138. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 25.Bollinger JM, Jr., Price JC, Hoffart LM, Barr EW, Krebs C. Eur. J. Inorg. Chem. 2005:4245. [Google Scholar]
- 26.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr. Biochemistry. 2009;48:4331. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broadwater JA, Ai JY, Loehr TM, Sanders Loehr J, Fox BG. Biochemistry. 1998;37:14664. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 28.Paulsen KE, Liu Y, Fox BG, Lipscomb JD, Münck E, Stankovich MT. Biochemistry. 1994;33:713. doi: 10.1021/bi00169a013. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Nesheim JC, Lee SK, Lipscomb JD. J. Biol. Chem. 1995;270:24662. doi: 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- 30.Bailey LJ, Mccoy JG, Phillips GN, Fox BG. Proc. Natl. Acad. Sci. USA. 2008;105:19194. doi: 10.1073/pnas.0807948105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tinberg CE, Lippard SJ. Acc. Chem. Res. 2011;44:280. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SJ, McCormick MS, Lippard SJ, Cho US. Nature. 2013;494:380. doi: 10.1038/nature11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem. Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 34.Neidig ML, Brown CD, Light KM, Fujimori DG, Nolan EM, Price JC, Barr EW, Bollinger JM, Jr., Krebs C, Walsh CT, Solomon EI. J. Am. Chem. Soc. 2007;129:14224. doi: 10.1021/ja074557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pavel EG, Zhou J, Busby RW, Gunsior M, Townsend CA, Solomon EI. J. Am. Chem. Soc. 1998;120:743. [Google Scholar]
- 36.Hitchman ML. Measurement of dissolved oxygen. John Wiley and Sons; New York N. Y: 1978. [Google Scholar]
- 37.Connors KA. Chemical kinetics: the study of reaction rates in solution. Wiley-VCH; 1990. [Google Scholar]
- 38.Espenson JH. Chemical kinetics and reaction mechanisms. McGraw-Hill; New York: 1995. [Google Scholar]
- 39.Jiang W, Hoffart LM, Krebs C, Bollinger JM., Jr. Biochemistry. 2007;46:8709. doi: 10.1021/bi700906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitteck JT, Cicchillo RM, van der Donk WA. J. Am. Chem. Soc. 2009;131:16225. doi: 10.1021/ja906238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr., Krebs C. Biochemistry. 2003;42:7497. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 42.Fish WW. In: In Methods Enzymol. James F, Riordan BLV, editors. Vol. 158. Academic Press; 1988. p. 357. [Google Scholar]
- 43.Münck E. In: Aspects of 57Fe Mössbauer spectroscopy In Physical Methods in Bioinorganic Chemistry. Que L Jr., editor. University Science Books; Sausalito: 2000. pp. 287–319. [Google Scholar]
- 44.Kurtz DM. Chem. Rev. 1990;90:585. [Google Scholar]
- 45.Fox BG, Shanklin J, Somerville C, Münck E. Proc. Natl. Acad. Sci. USA. 1993;90:2486. doi: 10.1073/pnas.90.6.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown CA, Remar GJ, Musselman RL, Solomon EI. Inorg. Chem. 1995;34:688. [Google Scholar]
- 47.Eser BE, Das D, Han J, Jones PR, Marsh EN. G. Biochemistry. 2011;50:10743. doi: 10.1021/bi2012417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valentine AM, Stahl SS, Lippard SJ. J. Am. Chem. Soc. 1999;121:3876. [Google Scholar]
- 49.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr. Proc. Natl. Acad. Sci. USA. 2006;103:6130. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayashi Y, Kayatani T, Sugimoto H, Suzuki M, Inomata K, Uehara A, Mizutani Y, Kitagawa T, Maeda Y. J. Am. Chem. Soc. 1995;117:11220. [Google Scholar]
- 51.Broadwater JA, Achim C, Münck E, Fox BG. Biochemistry. 1999;38:12197. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 52.Dong YH, Zang Y, Shu LJ, Wilkinson EC, Que L;, Jr., Kauffmann K, Münck E. J. Am. Chem. Soc. 1997;119:12683. [Google Scholar]
- 53.Bollinger JM, Jr., Krebs C, Vicol A, Chen SX, Ley BA, Edmondson DE, Huynh BH. J. Am. Chem. Soc. 1998;120:1094. [Google Scholar]
- 54.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr., Krebs C. J. Am. Chem. Soc. 2009;131:13608. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tinberg CE, Lippard SJ. Biochemistry. 2009;48:12145. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Biochemistry. 1998;37:9871. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- 57.Zaugg WS. J. Biol. Chem. 1964;239:3964. [PubMed] [Google Scholar]
- 58.Paul B, Das D, Ellington B, Marsh ENG. J. Am. Chem. Soc. 2013 doi: 10.1021/ja3115949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eser BE, Das D, Han J, Jones PR, Marsh ENG. Biochemistry. 2012;51:5703. doi: 10.1021/bi300837j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das D, Eser BE, Han J, Sciore A, Marsh ENG. Angew. Chem. Int. Ed. 2012;51:7881. [Google Scholar]
- 61.Das D, Eser BE, Han J, Sciore A, Marsh ENG. Angew. Chem. Int. Ed. 2011;50:7148. doi: 10.1002/anie.201101552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. J. Am. Chem. Soc. 2001;123:7388. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 63.Brazeau BJ, Lipscomb JD. Biochemistry. 2000;39:13503. doi: 10.1021/bi001473l. [DOI] [PubMed] [Google Scholar]
- 64.Xue GQ, Pokutsa A, Que L., Jr. J. Am. Chem. Soc. 2011;133:16657. doi: 10.1021/ja207131g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang YS, Broadwater JA, Pulver SC, Fox BG, Solomon EI. J. Am. Chem. Soc. 1999;121:2770. [Google Scholar]
- 66.Liu KE, Valentine AM, Wang DL, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J. Am. Chem. Soc. 1995;117:10174. [Google Scholar]
- 67.Tong WH, Chen S, Lloyd SG, Edmondson DE, Huynh BH, Stubbe J. J. Am. Chem. Soc. 1996;118:2107. [Google Scholar]
- 68.Yun D, Garcìa-Serres R, Chicalese BM, An YH, Huynh BH, Bollinger JM., Jr. Biochemistry. 2007;46:1925. doi: 10.1021/bi061717n. [DOI] [PubMed] [Google Scholar]
- 69.Murray LJ, Naik SG, Ortillo DO, Garcìa Serres R, Lee JK, Huynh BH, Lippard SJ. J. Am. Chem. Soc. 2007;129:14500. doi: 10.1021/ja076121h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li N, Korboukh VK, Krebs C, Bollinger JM., Jr. Proc. Natl. Acad. Sci. USA. 2010;107:15722. doi: 10.1073/pnas.1002785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ballou DP, Entsch B, Cole LJ. Biochem. Biophys. Res. Commun. 2005;338:590. doi: 10.1016/j.bbrc.2005.09.081. [DOI] [PubMed] [Google Scholar]
- 72.LeCloux DD, Barrios AM, Lippard SJ. Biorg. Med. Chem. 1999;7:763. doi: 10.1016/s0968-0896(98)00270-3. [DOI] [PubMed] [Google Scholar]
- 73.Jameson GNL, Jin W, Krebs C, Perreira AS, Tavares P, Liu XF, Theil EC, Huynh BH. Biochemistry. 2002;41:13435. doi: 10.1021/bi026478s. [DOI] [PubMed] [Google Scholar]
- 74.Bollinger JM, Jr., Diao Y, Matthews ML, Xing G, Krebs C. Dalton Trans. 2009:905. doi: 10.1039/b811885j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirao H, Morokuma K. J. Am. Chem. Soc. 2009;131:17206. doi: 10.1021/ja905296w. [DOI] [PubMed] [Google Scholar]
- 76.Cicchillo RM, Zhang HJ, Blodgett JAV, Whitteck JT, Li GY, Nair SK, van der Donk WA, Metcalf WW. Nature. 2009;459:871. doi: 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hirao H, Morokuma K. J. Am. Chem. Soc. 2010;132:17901. doi: 10.1021/ja108174d. [DOI] [PubMed] [Google Scholar]
- 78.Metcalf WW, Griffin BM, Cicchillo RM, Gao JT, Janga SC, Cooke HA, Circello BT, Evans BS, Martens-Habbena W, Stahl DA, van der Donk WA. Science. 2012;337:1104. doi: 10.1126/science.1219875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roberts ES, Vaz AD, Coon MJ. Proc. Natl. Acad. Sci. U S A. 1991;88:8963. doi: 10.1073/pnas.88.20.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim K, Lippard SJ. J. Am. Chem. Soc. 1996;118:4914. [Google Scholar]
- 81.Brunold TC, Tamura N, Kitajima N, Moro-oka Y, Solomon EI. J. Am. Chem. Soc. 1998;120:5674. [Google Scholar]
- 82.Moënne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM., Jr. Biochemistry. 1998;37:14659. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]
- 83.Ménage S, Brennan BA, Juarez-Garcia C, Münck E, Que L., Jr. J. Am. Chem. Soc. 1990;112:6423. [Google Scholar]
- 84.Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. J. Am. Chem. Soc. 2005;127:826. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 85.Rinaldo D, Philipp DM, Lippard SJ, Friesner RA. J. Am. Chem. Soc. 2007;129:3135. doi: 10.1021/ja0654074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okamura MY, Klotz IM, Johnson CE, Winter MRC, Williams RJP. Biochemistry. 1969;8:1951. doi: 10.1021/bi00833a027. [DOI] [PubMed] [Google Scholar]
- 87.Ookubo T, Sugimoto H, Nagayama T, Masuda H, Sato T, Tanaka K, Maeda Y, Ōkawa H, Hayashi Y, Uehara A, Suzuki M. J. Am. Chem. Soc. 1996;118:701. [Google Scholar]
- 88.LeCloux DD, Barrios AM, Mizoguchi TJ, Lippard SJ. J. Am. Chem. Soc. 1998;120:9001. [Google Scholar]
- 89.Holmes MA, Letrong I, Turley S, Sieker LC, Stenkamp RE. J. Mol. Biol. 1991;218:583. doi: 10.1016/0022-2836(91)90703-9. [DOI] [PubMed] [Google Scholar]
- 90.Choi YS, Zhang HJ, Brunzelle JS, Nair SK, Zhao HM. Proc. Natl. Acad. Sci. USA. 2008;105:6858. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Srnec M, Rokob TA, Schwartz JK, Kwak Y, Rulisek L, Solomon EI. Inorg. Chem. 2012;51:2806. doi: 10.1021/ic2018067. [DOI] [PubMed] [Google Scholar]
- 92.Vu VV, Emerson JP, Martinho M, Kim YS, Münck E, Park MH, Que L., Jr. Proc. Natl. Acad. Sci. USA. 2009;106:14814. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.