

Abstract
Metalloenzymes that utilize molecular oxygen as a co-substrate catalyze a wide variety of chemically difficult oxidation reactions. Significant insight into the reaction mechanisms of these enzymes can be obtained by the application of a combination of rapid kinetic and spectroscopic methods to the direct structural characterization of intermediate states. A key limitation of this approach is the low aqueous solubility (< 2 mM) of the co-substrate, O2, which undergoes further dilution (typically by one-third or one-half) upon initiation of reactions by rapid-mixing. This situation imposes a practical upper limit on [O2] (and therefore on the concentration of reactive intermediate(s) that can be rapidly accumulated) of ∼1-1.3 mM in such experiments as they are routinely carried out. However, many spectroscopic methods benefit from or require significantly greater concentrations of the species to be studied. To overcome this problem, we have recently developed two new approaches for the preparation of samples of oxygenated intermediates: (1) direct oxygenation of reduced metalloenzymes using gaseous O2 and (2) the in situ generation of O2 from chlorite catalyzed by the enzyme chlorite dismutase (Cld). Whereas the former method is applicable only to intermediates with half lives of several minutes, owing to the sluggishness of transport of O2 across the gas-liquid interface, the latter approach has been successfully applied to trap several intermediates at high concentration and purity by the freeze-quench method. The in situ approach permits generation of a pulse of at least 5 mM O2 within ∼ 1 ms and accumulation of O2 to effective concentrations of up to ∼ 11 mM (i.e. ∼ 10-fold greater than by the conventional approach). The use of these new techniques for studies of oxygenases and oxidases is discussed.
Keywords: oxygen, intermediate, ferryl, superoxo, peroxo, iron, non-heme
1 Introduction
1.1 O2 activation by metalloenzymes
Oxygenases and oxidases are enzymes that couple the reduction of O2 to the oxidation of their substrates. They are ubiquitous in nature and serve many important biological functions, including respiration [1], biosynthesis of enzyme cofactors [2-4], neurotransmitters [5], and natural products [6, 7], key steps in primary and secondary metabolism [6, 8, 9], degradation of xenobiotics [8], and regulation of transcription [10-14] and metabolism [15]. In general, the activation of O2 occurs at a reduced cofactor, which typically contains either a reduced transition metal, such as Fe(II) [8, 16-19] or Cu(I) [20-22], or a reduced flavin [23], and results in the generation of potently oxidizing reaction intermediates that initiate substrate oxidation. Studies discussed in this review, which involve the direct trapping and structural characterization of these intermediates, have been extremely important in our developing understanding of the mechanisms of these enzymes.
1.2 Trapping and characterization of intermediates
The use of rapid kinetic and spectroscopic methods as tools to study (metallo)enzyme mechanisms has a long history in mechanistic biochemistry. The first stopped-flow (SF) absorption-spectroscopic and freeze-quench (FQ) electron paramagnetic resonance (EPR)-spectroscopic studies were described approximately half a century ago (see e.g. [24-26]). Although the first application of the FQ method for the preparation of samples for 57Fe-Mössbauer spectroscopy was reported ∼20 years later in the early 1980s [27], the power of combining SF-absorption, FQ-EPR, and FQ-Mössbauer spectroscopies to trap and characterize reaction intermediates was not fully exploited until the 1990s in several detailed studies on the O2 activation reactions of carboxylate-bridged, non-heme-Fe2(II/II) proteins carried out collaboratively by several different research groups (including Vincent Huynh's) [28-39].
A generally (albeit not uniformly) successful strategy to apply these techniques has been to initiate the reaction by mixing a solution of the reactant complex [the form of the reduced enzyme with all (co)substrate(s) except O2 present at saturating levels] with an O2-containing solution. Addition of O2 is often quite fast (second-order rate constants for the addition of O2 to the reactant complex of 104 - 107 M−1s−1 [40-43]) in the reactions of wild-type enzyme with the natural substrates and, in many cases, has proven sufficiently fast to permit accumulation of intermediates. Typically, we begin our studies with SF absorption-spectroscopic experiments, which allow changes in the UV/visible region of the electromagnetic spectrum to be monitored in real time during a reaction. In most cases, the spectroscopic changes do not provide sufficient information to deduce the identities of absorbing species. However, they do provide insight into the kinetics of the reaction and greatly aid in the selection of suitable reaction times for samples to be prepared by the discontinuous FQ method for further characterization of intermediates by other spectroscopic methods.
We generally use FQ-EPR and FQ-57Fe-Mössbauer spectroscopy in the next phase of our mechanistic studies, because these two methods are complementary and are ideally suited for the detection and characterization of all 57Fe-labeled species contained in a sample [44, 45]. The spectroscopic parameters reveal aspects of the electronic structure(s) of detected species, such as the total electron-spin ground state, zero-field splitting parameters of the electron-spin ground state, spin and oxidation state of individual Fe ions, nuclearity of a multi-nuclear iron cofactor, and hyperfine coupling interactions. By using distinguishing spectroscopic features of a given reactant, intermediate, or product state, it is possible to determine each state’s concentration as a function of reaction time.
Further insight into the geometric and electronic structures of reaction intermediates can be obtained by studying samples maximally enriched in the intermediate state by other spectroscopic methods [46, 47]. These methods include: (i) FQ electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM) spectroscopies, which provide insight into the location of nuclei with non-zero magnetic moment relative to the paramagnetic center via detection of the hyperfine interactions; (ii) X-ray absorption spectroscopy (XAS), particularly the extended X-ray absorption fine structure (EXAFS) region of the XAS spectrum, which yields information about the geometric structure of the metal center including distances to nearby nuclei; (iii) resonance Raman spectroscopy, which yields insight into vibrational properties associated with the chromophore (often the metal center); (iv) magnetic circular dichroism (MCD) spectroscopy, a method interrogating electronic transitions in the UV/visible/NIR region and providing insight into the electron-spin ground state of the absorbing species; and (v) nuclear resonance vibrational spectroscopy (NRVS), a rather new and specialized technique that yields information about the vibrational properties associated with the 57Fe center. The comparison of experimentally observed spectroscopic parameters to those calculated for hypothetical model structures using density functional theory (DFT) [9, 48, 49] may then afford local, high-resolution structural information on reaction intermediates and thereby provide important insight into a reaction mechanism [50-53].
1.3 Limitations to trapping and characterization of intermediates
There are general challenges and limitations that one faces in the application of several of the above spectroscopic methods to samples prepared by the FQ method. These factors can render the experiments very challenging, or even impossible. The three main limitations are (i) unfavorable reaction kinetics, which result in modest accumulation and thus low purity of the target intermediate, (ii) low absolute concentration of the intermediate owing to limitations in solubility of reaction components (vide infra), and (iii) physical properties of the sample that are incompatible with the spectroscopic method.
1.3.1 Limitations due to unfavorable reaction kinetics
The extent to which an intermediate may accumulate is governed by the reaction kinetics, which are intrinsic to the enzyme and reaction under investigation and can generally be modified only for relatively modest gains by varying the reaction conditions. The intermediate’s accumulation depends on its rate constants of formation (kform) and decay (kdecay). An intermediate accumulates more for increasing kform and for decreasing kdecay. kform is in most cases determined by the reaction between the reactant complex and O2, which will generally exhibit second-order rate constants of 104-107 M−1s−1 [40-43]. For rapid-mixing experiments with [O2] of ∼1 mM (after mixing), the intermediates form with effective first-order rates of 101-104 s−1. kdecay depends on the nature of the chemical step in the reaction mechanism and is typically a unimolecular step. When the effective kform is comparable to or greater than kdecay, then the intermediate accumulates to levels permitting its characterization [28, 43, 54-58]. If kdecay is greater than the effective kform, then the intermediate accumulates to a lesser extent, and its identification may be very difficult or even impossible.
It is often possible to change the reaction kinetics significantly by using chemically or isotopically modified substrates and/or variant enzymes. These studies generally aim at accumulating the intermediate to a greater extent by slowing its decay. In our experience, the most successful perturbation involves the use isotopologs of the natural substrate in studies of enzymes that cleave strong C-H bonds. The sterically and electrostatically innocuous substitution of the target hydrogen of the natural substrate with deuterium results in greater accumulation of the C-H/D-cleaving intermediate, because (i) the formation of the intermediate is not affected by the isotopic substitution and (ii) the decay of the intermediate generally exhibits a large deuterium kinetic isotope effect (D-KIEs). We have observed D-KIEs on C-H/D cleavage ranging from ∼10 to ∼60 [42, 55, 57, 59-61]. Among the C-H-cleaving enzymes that we have studied so far, the use of the deuterated natural substrate has proven to be a generally successful strategy for accumulating the C-H/D-cleaving intermediate to levels sufficient to provide mechanistic insight.
There are also many examples of the successful trapping and characterization of reaction intermediates using chemically modified substrate analogs and/or variant enzymes [28, 38, 62-65]. The effect of the perturbation is less predictable compared to the effect of isotopic substitution described above, because the chemical perturbation might not only slow down decay of the intermediate (the desired effect), but it might also slow down formation of the intermediate, in some cases owing to reduced substrate triggering (ST) efficacy. ST is a phenomenon commonly observed in oxygenases and oxidases, in which addition of O2 is accelerated by some change caused by binding of one or more substrate. ST ensures that potently oxidizing reaction intermediates are formed exclusively (or at least preferentially) in the presence of the substrate. A well understood case of ST occurs in the reactions of the mononuclear non- heme-iron oxidases and oxygenases. Seminal studies by Solomon and co-workers showed that the conformational change associated with binding of the prime substrate promotes dissociation of a water molecule from the octahedral, six-coordinate Fe(II) center to yield a square-pyramidal, five-coordinate Fe(II) center, poised to be attacked by O2 at the vacated site [9, 66-69]. It has frequently been found that chemically modified substrates and variant enzymes are less effective at ST, rendering this strategy for accumulation of intermediates less generally effective than one might anticipate.
Whether the accumulation of intermediates to greater extent in the presence of a chemically modified substrate is successful or not depends on the extent to which kform and kdecay are perturbed. An example for the successful application of this strategy is our work on the Fe(II)-and α-ketoglutarate (αKG)-dependent halogenase, SyrB2, which is described in greater detail in section 2.1.2 [57].
A stark illustration of the failure of using analogs to accumulate an intermediate is our work on the enzyme taurine:αKG dioxygenase (TauD) from Escherichia coli (Ec) with the substrate analog 2,2-F2-taurine [70]. These studies were carried out before our discovery of the Fe(IV)-oxo intermediate in the reaction with the native substrate, taurine [54]. At that time, it was thought that the C-H-cleaving intermediate might be too reactive to be trapped [9, 71]. The 2,2-F2-taurine analog was synthesized with the expectation that the replacement of the target hydrogen with fluorine would decrease kdecay and result in greater accumulation of the C-H-cleaving intermediate. We further expected that the formation of the intermediate might not be affected, because, in the working hypothesis for the mechanism of the Fe(II)- and αKG-dependent oxygenases, the formation of the C-H-cleaving intermediate involves addition of O2 and decarboxylation of αKG to yield succinate and CO2, but it does not involve the prime substrate, taurine [9, 71, 72]. Surprisingly, the reaction of O2 with the TauD·Fe(II) αKG·2,2-F2-taurine complex is significantly slower than with the native quaternary complex (with taurine), despite the fact that the spectroscopic properties of the reactant complex with 2,2-F2-taurine are nearly identical to those with the natural substrate, taurine [70]. As a result of the diminished kform, the ferryl intermediate accumulates much less (∼10%) compared to the reaction with the native substrate, taurine (∼50%) [70].
Another example of the failure of chemically modified substrate to permit accumulation of intermediates to useful levels is our work on phenylalanine hydroxylase (PheH) from Chromobacterium violaceum (Cv) [73], an Fe(II)- and tetrahydropterin (BH4)-dependent aromatic amino acid hydroxylase [5]. Ferryl intermediates have been observed in tyrosine hydroxylase [74] and Cv PheH [73] in the presence of their natural substrates (tyrosine and phenylalanine, respectively), but the intermediate accumulates only to ∼20-25% in each case. With the intent to augment its accumulation, we used the perdeuterated cyclohexylalanine analog. Despite the fact that previous studies have shown that the reaction of Cv PheH with this analog results in hydroxylation of one of its aliphatic C-H bonds, exhibits a deuterium kinetic isotope effect of ∼15, and is fully coupled [75], the ferryl intermediate does not accumulate to a detectable level, as judged by the absence of the well-resolved high-energy line of the ferryl complex in the Mössbauer spectra, in the reaction of Cv PheH with the perdeuterated analog, presumably because addition of O2 (and therefore formation of the intermediate) is significantly slowed by the diminished ST efficacy of the non-native substrate [73].
1.3.2 Limitations caused by the physical properties of freeze-quenched samples
Transmission-based spectroscopic methods, such as low-temperature MCD spectroscopy, cannot, in general, be carried out on samples prepared by the FQ method, because the samples are not transparent. In a collaborative study, the Solomon group successfully demonstrated that samples of the high-valent Fe2(III/IV) intermediate, X, of Ec ribonucleotide reductase (RNR) suitable for MCD spectroscopy could be prepared by a modified FQ procedure [52, 76]. The double variant of the Ec RNR-β2 protein, Y122F/Y356F-β2, was used for this study, because the lifetime of X is ∼5-fold greater in this variant than in wild-type Ec RNR β2. The increased lifetime of X allowed liquid N2, which is much less efficient than the commonly used iso-pentane and ethane, to be used as cryosolvent in this case. After evaporation of the liquid N2, the dried freeze-quenched material containing X was mulled at −30 °C with glycerol and placed in a MCD cell. EPR measurements carried out in parallel ensured that X did not decay noticeably during this procedure.
Although this procedure was applied successfully to the study of Ec X, it is important to note that it is not generally applicable. For example, the high-valent Fe(IV)-oxo intermediate, J, from Ec TauD [54, 77] could not be prepared by this method, because it decays rapidly during the mulling process at −30 °C, as demonstrated by the disappearance of the unique quadrupole doublet associated with J in the Mössbauer spectra. Attempts to prepare samples of Ec TauD J suitable for MCD spectroscopy using modified conditions (lower mulling temperature, different mulling agent, shorter mulling time) have not been unsuccessful to date.
1.3.3 Limitations imposed by the physical properties of O2 gas
Dioxygen, O2, is only modestly soluble in aqueous solutions at ambient pressure (∼ 2 mM) due to its apolar nature [78]. Because transient-state kinetic experiments generally involve the rapid mixing of two solutions (in our preferred setup, one syringe contains an O2-free solution of the reactant complex, and the other contains an O2-saturated buffer solution) the concentration of O2 after mixing is further diminished to ∼1 mM or ∼1.3 mM in experiments involving a 1:1 (v/v) or 1:2 (v/v) mixing ratio, respectively. This value provides the upper limit for the absolute concentration of an intermediate that can be prepared in such a rapid-mixing experiment, even in the most favorable case that the intermediate would accumulate to stoichiometric levels (i.e. kform >> kdecay). Although intermediate concentrations of ∼1-1.3 mM are sufficient for many spectroscopic methods, some of the aforementioned techniques benefit from, or even require, higher concentrations.
Beyond its stoichiometric requirement for intermediate formation, the rather low solubility of O2 can also kinetically limit the concentration to which an intermediate can be accumulated, because the first step in the reaction sequence, the addition of O2 to the reactant complex, is bimolecular. Therefore, the apparent first-order rate constant, kform, increases as [O2] increases, limited by its modest solubility. The enzyme myo-inositol oxygenase (MIOX) serves as an example of this issue, which is discussed in more detail in section 2.2.5.
The gaseous nature of O2 also impedes quantitative kinetic studies of oxygenases and oxidases, such as in experiments to define the dependence of reaction rates on the concentration of O2 (e.g. [40, 41, 43]) or studies that require a well-defined ratio of reactants (e.g. [79]). In general, it is much more difficult to control and vary [O2] in solutions to be used in rapid-mixing experiments. As a consequence, values of [O2] can have significantly greater experimental uncertainty compared to those of non-volatile substrates.
2 New approaches to the trapping of reactive intermediates at high concentration
2.1 Trapping intermediates using gaseous O2
One possibility for the preparation of samples of reactive intermediates at high absolute concentration involves the exposure of a concentrated solution of the reactant complex to O2 gas, delivered on a Schlenck line to a degassed solution of the reactant complex. The O2-exposed solution is stirred vigorously under ∼2 atm O2, subsequently transferred into a suitable sample holder, and frozen. A rather long reaction time with O2 is required, because diffusion across the gas-liquid interface is inefficient (compared to the mixing of two solutions during a FQ experiment) especially given the high viscosity of the concentrated protein reactant. Because this method of oxygenation is much slower and kinetically heterogeneous (i.e. asynchronous) compared to oxygenation by mixing with an O2-containing solution, it can be applied only to the accumulation of intermediates with half-lives of minutes. We have used the direct oxygenation method for preparation of three metalloenzyme intermediates in high yield for detailed spectroscopic studies, which are described in the following sections.
2.1.1 The peroxo-Fe2(III/III) intermediate of Streptomyces thioluteus AurF
The peroxo-Fe2(III/III) intermediate found in the ferritin-like non-heme diiron-carboxylate N-oxygenase, AurF, from Streptomyces thioluteus is a very rare example of a chemically competent oxygenated intermediate that has a half-life of > ∼ 5 min and can therefore be trapped by the direct exposure of the reactant complex to O2 gas [56]. AurF catalyzes the unusual 6e--oxidation of para-aminobenzoate (Ar-NH2) to para-nitrobenzoate (Ar-NO2), a key step in the biosynthesis of the nitroaryl-containing natural product, aureothin [80-82]. In the first step, AurF promotes the 2e--oxidation (N-oxygenation) of Ar-NH2 to yield the para-hydroxylaminobenzoate (Ar-NHOH) intermediate [56], while in the second step of the reaction, AurF catalyzes the 4e-- oxidation of Ar-NHOH to Ar-NO2 [79]. The reactive oxygenated intermediate in both reactions is a peroxo-Fe2(III/III) complex, which is formed rapidly (k ∼ 105 M−1s−1 at 5 °C) in the reaction of the Fe2(II/II) form of AurF with O2 [56, 79]. The intermediate is remarkably stable in the absence of substrate (t1/2 ≈ 7 min at 5 °C), but reacts rapidly with the substrate (Ar-NH2 or Ar-NHOH; t1/2 ≈ 5 ms at 5 °C) [56, 79]. Thus, the formation and decay of the intermediate are kinetically so remarkably well resolved that the intermediate accumulates to near-stoichiometric levels and can be trapped in high purity by the direct oxygenation method. Comparison of the Mössbauer features of the various Fe-species of AurF present in samples prepared by the conventional FQ method and direct oxygenation method revealed the presence of a small amount of a Fe2(III/III) cluster in the sample prepared by direct oxygenation; this species was not present in samples prepared by the FQ method [56, 79]. This cluster presumably forms upon slow decay of the intermediate (t1/2 ≈ 7 min) in the absence of substrate as a consequence of the rather long O2-exposure time required for the direct-oxygenation method. However, this species constituted only a very minor fraction of the iron in the sample (∼10%). The detailed characterization of peroxo-Fe2(III/III) intermediate by a combination of spectroscopic methods, in particular, the MCD- and NRVS-spectroscopic studies carried out by the Solomon group, are ongoing and expected to yield fundamental new insight into the intermediate's geometric and electronic structure.
2.1.2 The halo-Fe(IV)-oxo intermediates of the αKG-dependent halogenases
The Fe(II)- and αKG-dependent halogenases functionalize aliphatic carbon atoms with a halogen (chlorine or bromine) during in the biosyntheses of various halogenated natural products by non-ribosomal peptide synthetases [83, 84]. The prime substrates of the halogenases are amino acids tethered via a thioester linkage to a phosphopantetheinyl (PPant) arm that is covalently attached to a thiolation carrier protein (or domain) of the non-ribosomal peptide synthetase (NRPS) complex. These enzymes are a subgroup of the large family of Fe(II)- and αKG-dependent oxygenases [6, 9, 17, 19], which in most cases promote the hydroxylation of their prime substrates. These enzymes activate O2 at a Fe(II) center to couple the decarboxylation of the co-substrate, αKG, to the formation of a potently oxidizing Fe(IV)-oxo (ferryl) intermediate, which abstracts hydrogen from the strong, aliphatic target C-H bond of the substrate. The unique outcome of the halogenases results from recombination of the substrate radical with the halogen atom from the halo-Fe(III)-hydroxo complex (formed by the hydrogen abstraction step) and is governed by the position of the target C-H bond relative to the halogen and oxo/hydroxo ligands to the Fe cofactor [85]. In contrast to the αKG-dependent hydroxylases, for which one ferryl intermediate is observed [54, 60], the αKG-dependent halogenases have two distinct halo-ferryl complexes that are partially resolved by Mössbauer spectroscopy [55, 57, 86]. Elucidation of their structures by a combination of spectroscopic methods is an important step towards understanding the unique reactivity of the halogenases. In the halogenase SyrB2 from Streptomyces, which halogenates the Cγ-methyl group of L-threonine tethered to the thiolation domain of SyrB1, the lifetime of the halo-ferryl complexes could be significantly extended by deuteration of the target Cγ-H bond, but remained too short for the direct oxygenation method to be applied. However, the use of two substrate analogs extended the lifetime of the halo-ferryl complexes dramatically [57]. For the cyclopropylglycine (Cpg)-containing analog, the modified carbon (Cγ) is part of a cyclopropyl ring and the Cγ-H bond is, consequently, unusually strong (homolytic bond dissociation energy ≈ 106 kcal/mol [87]). As a consequence, the halo-ferryl complexes are greatly stabilized (t1/2 ∼ 1.8 h at 0 °C) [57]. Alternatively, the L-alanine- containing analog, in which the Cy methyl group containing the natural target C-H bond is removed, also extends the lifetime significantly (t1/2 ∼ 0.7 h at 0 °C) [57]. The remarkable stability of the halo-ferryl complexes of SyrB2 with the Cpg-containing analog allowed for preparation of samples in high purity and concentration by the direct oxygenation method, permitting characterization of the chloro-ferryl complexes by EXAFS spectroscopy [57]. The characterization of the chloro- and bromo-ferryl complexes by NRVS and MCD is currently ongoing in collaboration with the Solomon group. Importantly, these halo-ferryl complexes are currently the only enzyme ferryl complexes that can be interrogated by MCD spectroscopy. These studies are expected to complement similar studies on non-heme ferryl models [88, 89].
2.1.3 The Mn(IV)/Fe(IV) intermediate of Chlamydia trachomatis ribonucleotide reductase
The class Ic ribonucleotide reductase (RNR) from Chlamydia trachomatis (Ct) utilizes a high-valent Mn(IV)/Fe(III) cofactor in its β subunit [4, 90-92] to reversibly generate the catalytically essential cysteinyl radical in its α subunit. The cysteinyl radical forms by a long-range electron-transfer reaction that occurs by a radical-hopping mechanism and is mediated by three tyrosine residues that are conserved for all class I RNRs [93-98]. The Mn(IV)/Fe(III) cluster forms in the reaction of the Mn(II)/Fe(II) cluster with O2 [43]. This reaction proceeds via a Mn(IV)/Fe(IV) intermediate [43], which is reduced by one electron delivered to the Fe(IV) site of the cluster via two conserved residues, W51 and Y222, of the β subunit. These two residues form a radical-hopping pathway that is functionally orthogonal to the ET pathway that is operant in cysteinyl radical formation during the catalytic reaction [99]. The characterization of the geometric and electronic structures of the high-valent Mn(IV)/Fe(III) and Mn(IV)/Fe(IV) complexes is a major goal of our research. Whereas the Mn(IV)/Fe(III) product of the reaction is stable and has been prepared in high concentration and purity for characterization by Mn- and Fe-k-edge EXAFS spectroscopy [100] and ongoing collaborative studies with the Solomon group using MCD and NRVS spectroscopies, the transient nature of the Mn(IV)/Fe(IV) intermediate makes the preparation of samples of it more challenging. While the decay rate constant of the Mn(IV)/Fe(IV) intermediate in the wild-type protein precludes the use of the direct oxygenation method (kdecay = 0.02 s−1 at 5 °C), the decay is slowed 10-fold (kdecay = 0.002 s−1) in the Y222F variant [99], making the direct oxygenation method feasible for the preparation of samples of the Mn(IV)/Fe(IV) intermediate in this variant. While we have successfully prepared samples of the Mn(IV)/Fe(IV) in high concentration and purity by the direct oxygenation method (as determined by EPR and Mössbauer experiments on these samples), we note that many attempts yielded samples with significant amounts of the reactant Mn(II)/Fe(II) form, rendering these samples unsuitable for additional spectroscopic experiments. The presence of unreacted Mn(II)/Fe(II) can be rationalized by the kinetic heterogeneity of the O2 addition step initiated by direct exposure of the viscous solution of the reactant complex to O2 gas. These difficulties in sample preparation suggest that the lifetime of the Mn(IV)/Fe(IV) intermediate in the Y222F-β variant of Ct RNR (t1/2 ∼ 6 min) represents a lower limit for the applicability of this method. As described in section 2.2.4, samples of the Mn(IV)/Fe(IV) intermediate can now be prepared at even higher concentration and purity more reliably and conveniently by using O2 generated in situ by the Cld/ClO2- system. However, the in situ generation of O2 via Cld/ClO2- does not allow for preparation of samples suitable for MCD spectroscopy. The optimization of the direct oxygenation method for the preparation of MCD samples of the Mn(IV)/Fe(IV) intermediate for collaborative studies with the Solomon group are ongoing.
2.2 Trapping intermediates by using O2 generated in situ from ClO2- by chlorite dismutase
The heme-dependent enzyme, chlorite dismutase (Cld), catalyzes the conversion of ClO2- to Cl- and O2 [101]. This reaction is a key step in the detoxification of ClO2- in proteobacteria that respire on perchlorate, ClO4- [102]. The stepwise reduction of ClO4- to ClO3- and ClO2- at a molybdopterin-containing, membrane-bound enzyme, perchlorate reductase, is coupled to the generation of a proton gradient [103]. The product of this reaction, ClO2-, is toxic and must be efficiently removed. Dechloromonas aromatica (Da) Cld has been studied in detail [104-106]. It catalyzes the conversion of ClO2- to Cl- and O2 very efficiently (kcat = 2 × 106 s−1 at 4 °C and pH 5.2) and is capable of performing ∼1,700 turnovers before the enzyme begins to exhibit oxidative inactivation.
We have recently shown that Da Cld can be used to generate O2 in situ from ClO2- for rapid-kinetic studies of oxygenases and oxidases [107]. It is possible to generate a pulse of ∼5 mM O2 in ∼1 ms and to achieve effective O2 concentrations of up to ∼11 mM. Thus, the concentration of O2 generated in situ by the Cld/ClO2- system exceeds the O2 concentrations attainable from dissolved O2 by ∼ 10-fold.
This recently developed method, which we describe briefly below, has already been used to accumulate several intermediates in greater absolute concentration than was previously possible. In addition to the ability to increase the absolute concentration of key intermediates, the method offers two other advantages for mechanistic studies: (i) greater range of [O2] and greater precision with which [O2] can be controlled in kinetic experiments and (ii) the ability to accumulate reaction intermediates to greater levels by shifting the equilibrium of the reversible O2 addition to the reactant complex to the side of the oxygenated intermediate. These advantages are highlighted below for the reactions of the Mn(II)/Fe(II) form of Ct RNR-β2 with O2 (section 2.2.4) and the Fe2(II/III)·MI complex of MIOX with O2 (section 2.2.5).
2.2.1 Experimental considerations
Rapid kinetic studies of oxygenases or oxidases are conceptually simple and typically involve a single mix of an O2-free solution of the reactant enzyme complex with an O2-containing buffer solution. By contrast, experiments involving O2 generated in situ by the Cld/ClO2- system are more challenging and require the mixing of ClO2-, Cld, and the reactant complex in a fashion that favors the two desired reactions (O2 generation by the Cld/ClO2-system and the reaction of O2 with the reactant complex) and disfavors possible side reactions, including:
Reduction of the catalytically active Fe(III) heme center of Cld by one of the components of the reduced reactant complex of the target enzyme;
Inhibition of Cld by the coordination of one of the substrates of the target enzyme to the heme cofactor;
Oxidation of one of the components of the reactant complex by ClO2-; and
Metal-mediated decomposition of ClO2-.
In addition to the reactions arising from cross-reactivity of components of the Cld and target oxygenase/oxidase systems, the auto-inactivation of Cld, which is presumably due to oxidative destruction of the heme cofactor during turnover, after ∼1,700 turnovers must be taken into account in design of these experiments. This potential limitation can be overcome by adjusting the ClO2-:Cld ratio to < 1,500 to ensure that ClO2- is (almost) completely consumed.
Because these non-enzymatic side reactions (1) – (4) are much slower than the reaction of the target oxygenase/oxidase with O2, we designed a double-mixing experiment, which in the first step involves mixing of an O2-free solution of the reactant complex with an anaerobic solution of Cld for a time that is as short as the instrumentation allows. The intent is to prevent a reaction between these two components, such as reduction of the Fe(III)-heme center of Cld by the reduced reactant complex, by minimizing this first delay time. In the second step, the resultant solution is mixed with a ClO2--containing solution, which will initiate the in situ generation of O2 by Cld and the reaction of O2 with the reactant complex of the enzyme to be studied. Because ClO2- is highly soluble and because Cld is required only at catalytic concentrations (typically 5-30 μM final) that are well within its solubility limit (up to 0.5 mM), it is possible to use rather concentrated solutions of Cld and ClO2- and employ a mixing ratio of 4:1:1 (v/v/v) of the solutions containing target reactant enzyme complex, Cld, and ClO2-, respectively. This mixing ratio diminishes the concentration of the target enzyme by only 33%, and the upper limit of the absolute concentration of the target intermediate is no longer limited by the low solubility of O2, but rather by the solubility of the target enzyme itself, which in most cases significantly exceeds the limit of ∼1-1.3 mM imposed by the solubility of O2.
Another important experimental consideration is the unavoidable presence of Cld heme cofactor in the low μM concentration range in samples prepared by the Cld/ClO2- method and, as a consequence, the possible interference of its spectroscopic features with those of the target enzyme under investigation. Whereas for some methods there is very little interference (e.g. Mössbauer, EPR, XAS), the intensely absorbing Soret band of the heme cofactor is clearly detectable in stopped-flow absorption experiments and may significantly interfere with authentic signals associated with states of the target enzyme. It is likely that significant interference will also play a role in resonance Raman- and MCD-spectroscopic experiments. Careful control experiments, in which Cld is reacted with ClO2- in the absence of the target enzyme, will be required for such experiments.
In order to explore the efficacy of the in situ generation of O2 by the Cld/ClO2- system, we studied the generation of the stable oxy-myoglobin (oxy-Mb) adduct from reduced, Fe(II)-Mb and O2 [107]. We used the 4:1:1 (v/v/v) double-mixing protocol, in which a solution of Fe(II)-Mb was mixed with a Cld-containing solution for ∼3 ms in the first step, followed by mixing with a ClO2--containing solution and freeze-quenching after ∼15 ms. Using 57Fe-Mössbauer spectroscopy, we showed that the Cld/ClO2- system ([Cld]final = 20 μM and [ClO2-]final = 17 mM) is capable of generating sufficient O2 within ∼15 ms to convert 6.7 mM Fe(II)-Mb nearly quantitatively (∼98%) to the oxy-Mb adduct. Thus, the Cld/ClO2- system is capable of generating 6.7 mM O2 within ∼15 ms, and the O2 remains available for reaction with a metalloprotein.
2.2.2 The Fe(IV)-oxo intermediate, J, of Escherichia coli taurine:αKG dioxygenase
The Fe(II)- and αKG-dependent oxygenases are a family of enzymes with diverse biological functions [6, 7, 9, 17, 19]. The most extensively studied member of these enzymes is Ec taurine:αKG dioxygenase (TauD) [77]. The key intermediate of the catalytic cycle is a Fe(IV)-oxo intermediate, termed J, which cleaves the C1-H bond of the substrate, 2-aminoethane-1-sulfonate or taurine [45, 53, 54, 108-110]. The Fe center of J is coordinated by the three amino acid ligands of the His2/(Glu/Asp)1 facial triad [111, 112], the co-product, succinate, and the oxo ligand. One of the His ligands is trans to the oxo group, while the other His and the two carboxylate ligands provide the equatorial ligands. The Fe(IV) center is either in a trigonal bipyramidal (with both carboxylates serving as monodentate ligands) or distorted octahedral (with one of the carboxylates in a bidentate binding mode) coordination environment [45, 53]. As a consequence of the weak ligand field in the xy-plane, the energy of the dx2-y2 orbital is diminished, resulting in the unusual high-spin (S = 2) electronic configuration [53]. The strategy of lowering the energy of the dx2-y2 orbital has also been successfully accomplished in inorganic high-spin ferryl models [113-118]. Because the geometric and electronic structures of Ec TauD J are well understood, J can serve as a benchmark for the new and emerging technique NRVS. Although it is possible to obtain samples of J in high purity (∼80%), the absolute concentration is limited by solubility of O2 (1.3 mM) and less than the concentration range required for NRVS. Thus, samples prepared with dissolved O2 are not suitable for NRVS. We prepared samples of J using the Cld/ClO2- method with comparable purity (∼80%), but significantly greater absolute concentration (3.2 mM total Fe, 2.5 mM J) for NRVS studies with the Solomon group [107] that are expected to complement studies of non-heme ferryl models [89, 117].
2.2.3 The Fe2(III/IV) intermediate, X, of Escherichia coli ribonucleotide reductase
A class Ia RNR utilizes a stable tyrosyl radical, which is in close proximity to a Fe2(III/III) cluster, in its β2 subunit [2, 119, 120] to generate the catalytically essential cysteinyl radical in its α subunit, in the same manner that the class Ic Ct RNR uses the Mn(IV)/Fe(III) cluster [93, 121]. The tyrosyl radical/Fe2(III/III) cofactor is generated by activation of O2 at the Fe2(II/II) form [122]. This reaction has been extensively studies for Ec RNR [28, 30-32, 35, 40, 122]. The last step of this reaction is the one-electron oxidation of the radical tyrosine, Y122, by the Fe2(III/IV) intermediate, termed X, which itself is reduced to the Fe2(III/III) cluster [28]. Although X has been studied by a wide variety of spectroscopic techniques [36, 38, 52, 123, 124], no consensus structure for X has been obtained. Importantly, the Fe-Fe distance of X determined by EXAFS spectroscopy (dFe-Fe = 2.5 Å) [38] is significantly shorter than Fe-Fe separations for an inorganic model for X (dFe-Fe = 2.68 Å) [125] and models for X derived from other spectroscopies and calculations [51, 52, 124]. The re-determination of the Fe-Fe separation in X by EXAFS spectroscopy using significantly more concentrated samples could resolve this long-standing conundrum. We used the Cld/ClO2- method to prepare samples of X with comparable purity (∼70%) and significantly greater concentration (2.0 mM) than reported in the published EXAFS study [107]. This concentration is 2.5-fold greater than those samples, which were prepared by the conventional FQ method (∼0.8 mM) [38]. The EXAFS-spectroscopic characterization of these samples is ongoing.
2.2.4 The Mn(IV)/Fe(IV) intermediate of Chlamydia trachomatis ribonucleotide reductase
As detailed in section 2.1.3, the Mn(IV)/Fe(IV) intermediate observed during reaction of the Mn(II)/Fe(II) cluster of Ct RNR-β2 with O2 is a major target for spectroscopic characterization. The intermediate prepared with the Y222F variant of Ct RNR-β2 has a half life of ∼6 min. The preparation of samples in high concentration by the direct oxygenation method has, not surprisingly, proven to be subject to greater variation than rapid-mixing methods. We recently prepared the Mn(IV)/Fe(IV) intermediate at high concentration (1.2 mM) and purity (∼63% of total Fe) using the Cld/ClO2- method [107]. Such samples will be used for Mn- and Fe-k-edge EXAFS studies to determine the Mn-Fe distance independently. These studies are presently ongoing. While the characterization of the Mn(IV)/Fe(IV) intermediate by NRVS in collaboration with the Solomon group will provide additional insight into its structure, these studies are not feasible with the samples we can presently prepare, because the absolute concentration is less that required for NRVS. Optimization of the procedure to prepare samples with the greater absolute concentration of the intermediate required for the NRVS studies of the Solomon group is currently being pursued in our group.
The reaction of the Mn(II)/Fe(II) cofactor of Ct RNR-β2 with O2 was also used as a kinetic test for the capability of the Cld/ClO2- method. The dependence of the kinetics of Mn(IV)/Fe(IV) intermediate formation on [O2] was defined [107]. These experiments were carried out by SF absorption spectroscopy, made possible by strong absorption at 390 nm of the Mn(IV)/Fe(IV) intermediate, which forms in a bimolecular reaction between the reactant complex and O2 [43]. The product of the reaction, the Mn(IV)/Fe(III) cluster, also absorbs at this wavelength, but its molar absorptivity is only roughly half that of the Mn(IV)/Fe(IV) intermediate [43]. Thus, the A390-vs-time traces (Figure 1A) reflect formation and decay of the Mn(IV)/Fe(IV) intermediate. It is apparent that the intermediate forms more rapidly with increasing [ClO2-], and, at the lesser ClO2– concentrations, the apparent first-order rate constant for formation of the intermediate, kform depends linearly on [ClO2-] (Figure 1B). Importantly, the values of kform are identical to those obtained at the same [O2] from experiments with dissolved O2 (gray diamonds), thereby confirming that ClO2- is quantitatively converted to O2 by Cld so rapidly that no lag phase on formation of the Mn(IV)/Fe(IV) intermediate is observed. At greater [ClO2-], values of kform deviate from this first-order dependence. However, increasing [Cld] results in greater kform for [ClO2-] > 4 mM, without affecting kform for [ClO2-] ≤ 4 mM. This observation demonstrates that the discrepancy is due to inactivation of Cld at higher [ClO2-] [107]. These studies illustrate the virtue of the Cld/ClO2- system as a tool for kinetic studies in which [O2] must be varied. Compared to experiments using dissolved O2, the linear range is significantly greater and the O2 concentration can be controlled with greater precision.
Figure 1.
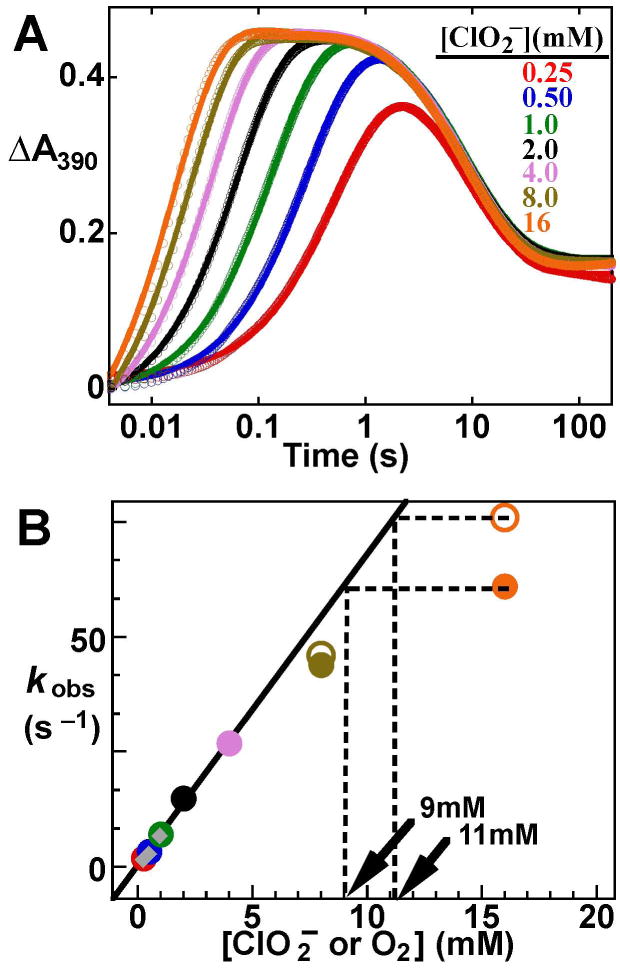
Reaction of the Mn(II)/Fe(II) form of Ct RNR-β2 with O2 generated in situ by the Cld/ClO2- system. (A) Delineation of the [O2] dependence of the reaction by variation of [ClO2-]. A390-vs-time traces following rapid mixing at 5 °C of a solution containing 0.2 mM RNR- 2, 0.6 mM Mn(II), 0.2 mM Fe(II), and 0.01 mM Cld with an equal volume of a ClO2--containing solution. The final values of [ClO2-] are 0.25 mM (red), 0.50 mM (blue), 1.0 mM (green), 2.0 mM (black), 4.0 mM (light purple), 8 mM (gold), and 16 mM (orange). Traces were analyzed by non-linear regression using the equation for two exponential phases (solid lines) in order to extract observed first-order rate constants for formation of the Mn(IV)/Fe(IV) intermediate (kobs). (B) Plot of kobs versus [ClO2-] or [O2]. The solid circles are values of kobs for the above experiment ([Cld]final = 5 μM). The open circles are values of kobs from an identical experiment, except for [Cld]final = 10 μM. The gray diamond points are values of kobs obtained after mixing with either O2-saturated buffer or buffer prepared by diluting O2-saturated buffer 2- or 4-fold with O2-free buffer, as has been done in the past to define the [O2]-dependence of the reaction. The points with [ClO2-] · 4 mM were fit by the equation for a line (solid line). Extrapolation of the kobs for the reaction with 16 mM ClO2- to the linear fit line (dashed lines) gives effective [O2] of 9 mM or 11 mM, respectively. Reprinted with permission from [107]. Copyright 2012 American Chemical Society.
2.2.5 The superoxo-Fe2(III/III) intermediate, G, of Mus musculus myo-inositol oxygenase
The enzyme myo-inositol oxygenase (MIOX) catalyzes the four-electron oxidation of myo-inositol (MI) to D-glucuronate using O2 as the oxidant [126-128]. The reaction begins with the reversible addition of O2 to the mixed-valent Fe2(II/III) cofactor [129, 130] having the substrate, myo-inositol (MI), coordinated at the Fe(III) site [131]. This reaction yields formally a superoxo-Fe2(III/III) complex, termed G [61]. Formation of G occurs with a second-order rate constant of 95 mM−1s−1, which corresponds to an apparent kform of ∼100 s−1 for [O2] ≈ 1 mM [61]. G can decay by two pathways: the reductive elimination of O2, which occurs with k ∼40 s−1, and cleavage of the C1-H(D) bond of MI, which occurs with rate constants of kH > 250 s−1 and kD = 48 s−1, respectively, resulting in a significant D-KIE of ∼ 8-15. Thus, even with the deuterated substrate, the rate of breakdown of G is comparable to the rate of formation at experimentally accessible [O2], thereby precluding accumulation of G to more than ∼40% of the concentration of the reactant complex. An important consequence of the reversibility of O2 addition is that samples quenched at the reaction time to appropriate to trap the maximum concentration of G still contain significant concentrations of the reactant complex.
We recently showed that the use of the Cld/ClO2- system [20 μM Cld and 16 mM ClO2-; under those condition the effective [O2] is ∼11 mM in experiments with Ct RNR (see section 2.2.4)] allows for ∼2-fold greater accumulation of G and nearly complete consumption of the reactant complex (∼15-fold less remaining) than in the published experiments conducted by rapid mixing with O2-containing solutions [107]. This observation is an important step toward the more thorough characterization of G with other spectroscopic methods. In particular, the combination of 57Fe ENDOR and Mössbauer experiments is expected to provide detailed insight into the electronic structure of G. The 57Fe hyperfine tensors determined from 57Fe ENDOR spectroscopy will be an important constraint for the determination of other parameters (in particular the isomer shifts, δ) required for simulation of the magnetically split Mössbauer spectra of G. This approach was used successfully to study the Fe2(III/IV) intermediate, X, of Ec RNR [36]. The ability to prepare samples enriched in G and with only small amounts of the reactant Fe2(II/III)•MI complex will facilitate this analysis considerably. These studies are now being pursued in our group.
3 Conclusion and Outlook
We have developed two new methods for the preparation of oxygenated intermediates in concentrations significantly exceeding those attainable in samples prepared by rapid-mixing of solutions with dissolved O2.
The direct oxygenation method involves exposure of an anaerobic solution of the reactant complex to gaseous O2 with vigorous stirring of the resultant solution, transfer of the sample into the appropriate spectroscopy cell, and freezing of the sample. Because the transfer of O2 from the gas phase to the viscous liquid phase is very inefficient, the oxygenation process takes several minutes. The method is therefore applicable only to intermediates that have half-lives greater than ∼6 min. This criterion is not met for the vast majority of intermediates and therefore the method has only limited applicability. However, for those intermediates with half lives >6 min, the method is extremely useful and allows for preparation of samples for virtually all spectroscopies, including transmission-based methods (e.g. MCD) and methods requiring very high-concentrations of material (e.g. NRVS).
The Cld/ClO2- method is fundamentally different and involves the in situ generation of dissolved O2. The experimental setup is more complex, because the method requires two reactions, the generation of O2 by Cld and the reaction of O2 with the target oxygenase/oxidase, to take place without interference. Although many side reactions can potentially occur between the various components of the two reactions, these are, for all cases studied by us so far, much slower than the desired reactions. The double-mixing protocol, in which the reactant complex and Cld are mixed in the first step and immediately thereafter mixed with ClO2-, has in our experience been successful in suppressing these potential side reactions. Thus, we anticipate that the Cld/ClO2- method will be applicable to most oxygenases and oxidases.
The method has many advantages for mechanistic studies that cannot be realized by other methods. The fact that the accessible concentration range of O2 is ∼10-fold greater has two important consequences. First, it allows for the preparation of intermediates in unprecedented, multi-millimolar concentrations for detailed characterization by various spectroscopies. Second, kinetic studies, in which the dependence of the reaction kinetics on [O2] is interrogated over a wide range of [O2], are rendered more convenient and accurate. We note that our study is not the first example of the interrogation of a reaction of O2 with a reduced metalloenzyme over a wide range of [O2]. Bailey, et al. reported on O2 binding to cytochrome c oxidase by SF absorption spectroscopy [132]. However, these studies utilized a custom-built, high-pressure SF apparatus that is not routinely available. Moreover, a similar apparatus for the application of the FQ method, i.e. high-pressure FQ, has not, to our knowledge, ever been described. By contrast, the Cld/ClO2- method requires only standard SF and FQ equipment.
Another important advantage of the Cld/ClO2- method is the ability to accumulate intermediates that form upon addition of O2 (i.e. the “O2-adducts”, which are generally believed to have an superoxo-Fe(III) electronic structure [64, 133-137]) to the reactant complex to greater levels by accelerating kform. For the superoxo-Fe2(III/III) intermediate, G, from MIOX, we illustrated that the ∼10-fold greater [O2] results in acceleration of kform, thereby shifting equilibrium of the reversible O2-addition step to favor G. The use of the Cld/ClO2- method may prove to be an important tool for the identification of the many proposed superoxo-Fe(III) intermediates that are thought to cleave C-H bonds [138, 139], in particular when using the specifically deuterated substrates. For many other enzymes, such as the Fe(II)- and αKG-dependent oxygenases and the Fe(II)- and BH4-dependent aromatic amino acid hydroxylases, superoxo-Fe(III) intermediates have been proposed as reaction intermediates en route to the experimentally observed ferryl intermediates, but the superoxo-Fe(III) complexes have never been directly detected, presumably because they do not accumulate to detectable levels under standard reaction conditions. It is conceivable that the ∼10-fold faster formation of these complexes may result in their accumulation. However, the reaction may be so fast that the maximum time of accumulation of these intermediates is less than the dead time of conventional rapid-mix instrumentation. The “freeze hyperquenching” technique [140, 141], which allows the reaction to be quenched after significantly shorter reaction times (∼20-50 μs) might then allow for trapping and characterization of these complexes.
Highlights.
spectroscopy on intermediate states has illuminated the mechanisms of O2-activating metalloenzymes
the modest solubility of O2 can limit the concentration and purity of target intermediate states
long-lived intermediates can be built up further by direct exposure of reactant complexes to O2(g)
chlorite dismutase (Cld) can rapidly generate a ∼ 10 mM pulse of O2 from ClO2-
O2 evolved by Cld can yield oxygenated intermediates at high concentration and purity
Acknowledgments
This work was supported by the National Institutes of Health (GM-55365, GM-69657, and DK-74641 to JMB and CK), the National Science Foundation (MCB-642058, CHE-724084, and CHE-1058931 to JMB and CK), and the Alfred P. Sloan Foundation Minority PhD Scholarship Program (to LMKD). We thank our co-workers and collaborators whose work is cited herein. This paper is dedicated to Ed Solomon on the occasion of his 65th birthday. The approaches summarized here set the stage to continue our joint work with Ed and to provide his group with milliliters of samples containing intermediates in the multi-millimolar range for more detailed characterization. We look forward to this work.
Abbreviations
- α-KG
α-ketoglutarate
- Ar-NH2
para-aminobenzoate
- Ar-NHOH
para-hydroxylaminobenzoate
- Ar-NO2
para-nitrobenzoate
- BH4
tetrahydropterin
- Cld
chlorite dismutase
- Cpg
cyclopropylglycine
- Ct
Chlamydia trachomatis
- Cv
Chromobacterium violaceum
- Da
Dechloromonas aromatica
- DFT
density functional theory
- D-KIE
deuterium kinetic isotope effect
- d4-taurine
1,1,2,2-[2H4]-2-aminoethane-1-sulfonic acid
- d6-MI
1,2,3,4,5,6-[2H6]-cyclohexan-(1,2,3,5/4,6)-hexa-ol
- Ec
Escherichia coli
- ENDOR
electron nuclear double resonance
- EPR
electron paramagnetic resonance
- ESEEM
electron spin echo envelope modulation
- EXAFS
extended X-ray absorption fine structure
- ferryl
Fe(IV)-oxo
- FQ
freeze-quench
- kD
rate constant for C-D cleavage
- kH
rate constant for C-H cleavage
- MCD
magnetic circular dichroism
- MI
myo-inositol or cyclohexan-(1,2,3,5/4,6)-hexa-ol
- MIOX
myo-inositol oxygenase
- NIR
near infrared
- NRPS
non-ribosomal peptide synthetase
- NRVS
nuclear resonance vibrational spectroscopy
- PheH
phenylalanine hydroxylase
- PPant
phosphopantetheine
- RNR
ribonucleotide reductase
- SF
stopped-flow
- ST
substrate triggering
- TauD
taurine:α-ketoglutarate dioxygenase
- t1/2
half life
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Carsten Krebs, Email: ckrebs@psu.edu.
J. Martin Bollinger, Jr, Email: jmb21@psu.edu.
References cited
- 1.Ferguson Miller S, Babcock GT. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Atkin CL, Thelander L, Reichard P, Lang G. J Biol Chem. 1973;248:7464–7472. [PubMed] [Google Scholar]
- 3.Klinman JP, Mu D. Annu Rev Biochem. 1994;63:299–344. doi: 10.1146/annurev.bi.63.070194.001503. [DOI] [PubMed] [Google Scholar]
- 4.Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak MA, Krebs C, Bollinger JM., Jr Science. 2007;316:1188–1191. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]
- 5.Fitzpatrick PF. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 7.Kershaw NJ, Caines MEC, Sleeman MC, Schofield CJ. Chem Comm. 2005:4251–4263. doi: 10.1039/b505964j. [DOI] [PubMed] [Google Scholar]
- 8.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 9.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 10.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 11.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 12.Tsukada Yi, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 13.Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 14.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 15.Gerken T, Girard CA, Tung YCL, Webby CJ, Saudek V, Hewitson KS, Yeo GSH, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O'Rahilly S, Schofield CJ. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baik MH, Martin N, Friesner RA, Lippard SJ. Chem Rev. 2003;103:2385–2419. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- 17.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 18.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 21.Chen P, Solomon EI. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 22.Klinman JP. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 23.Massey V. J Biol Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 24.Bray RC. Biochem J. 1961;81:189–195. doi: 10.1042/bj0810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chance B, Eisenhart RH, Gibson QH, Lonberg-Holm KK. Rapid Mixing and Sampling Techniques in Biochemistry. Academic Press; New York: 1964. [DOI] [PubMed] [Google Scholar]
- 26.Ballou DP, Palmer GA. Anal Chem. 1974;46:1248–1253. [Google Scholar]
- 27.Rutter R, Hager LP, Dhonau H, Hendrich MP, Valentine M, Debrunner PG. Biochemistry. 1984;23:6809–6816. doi: 10.1021/bi00321a082. [DOI] [PubMed] [Google Scholar]
- 28.Bollinger JM, Jr, Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 29.Lee SK, Fox BG, Froland WA, Lipscomb JD, Münck E. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 30.Ravi N, Bollinger JM, Jr, Huynh BH, Edmondson DE, Stubbe J. J Am Chem Soc. 1994;116:8007–8014. [Google Scholar]
- 31.Bollinger JM, Jr, Tong WH, Ravi N, Huynh BH, Edmondson DE, Stubbe J. J Am Chem Soc. 1994;116:8015–8023. [Google Scholar]
- 32.Bollinger JM, Jr, Tong WH, Ravi N, Huynh BH, Edmondson DE, Stubbe J. J Am Chem Soc. 1994;116:8024–8032. [Google Scholar]
- 33.Liu KE, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1994;116:7465–7466. [Google Scholar]
- 34.Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 35.Tong WH, Chen S, Lloyd SG, Edmondson DE, Huynh BH, Stubbe J. J Am Chem Soc. 1996;118:2107–2108. [Google Scholar]
- 36.Sturgeon BE, Burdi D, Chen S, Huynh BH, Edmondson DE, Stubbe J, Hoffman BM. J Am Chem Soc. 1996;118:7551–7557. [Google Scholar]
- 37.Shu L, Nesheim JC, Kauffmann KE, Münck E, Lipscomb JD, Que L., Jr Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 38.Riggs-Gelasco PJ, Shu L, Chen S, Burdi D, Huynh BH, Que L, Jr, Stubbe J. J Am Chem Soc. 1998;120:849–860. [Google Scholar]
- 39.Pereira AS, Small GW, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Biochemistry. 1998;37:9871–9876. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- 40.Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JM., Jr J Am Chem Soc. 2000;122:12195–12206. [Google Scholar]
- 41.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM., Jr Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 42.Bollinger JM, Jr, Krebs C, Inorg J. Biochem. 2006;100:586–605. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 43.Jiang W, Hoffart LM, Krebs C, Bollinger JM., Jr Biochemistry. 2007;46:8709–8716. doi: 10.1021/bi700906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Münck E. Aspects of 57Fe Mössbauer spectroscopy. In: Que L Jr, editor. Physical Methods in Bioinorganic Chemistry. University Science Books; Sausalito, CA: 2000. pp. 287–319. [Google Scholar]
- 45.Krebs C, Price JC, Baldwin J, Saleh L, Green MT, Bollinger JM., Jr Inorg Chem. 2005;44:742–757. doi: 10.1021/ic048523l. [DOI] [PubMed] [Google Scholar]
- 46.Solomon EI, Hansen MA. Bioinorganic Spectroscopy. In: Solomon EI, Lever ABP, editors. Inorganic electronic Structure and Spectroscopy, Volume II Applications and Case Studies. John Wiley & Sons; New York: 1999. [Google Scholar]
- 47.Que L., Jr . Physical Methods in Bioinorganic Chemistry. University Science Books; Sausalito, CA: 2000. [Google Scholar]
- 48.Neese F. Curr Opin Chem Biol. 2003;7:125–135. doi: 10.1016/s1367-5931(02)00006-6. [DOI] [PubMed] [Google Scholar]
- 49.Neese F. J Biol Inorg Chem. 2006;11:702–711. doi: 10.1007/s00775-006-0138-1. [DOI] [PubMed] [Google Scholar]
- 50.Skulan AJ, Brunold TC, Baldwin J, Saleh L, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2004;126:8842–8855. doi: 10.1021/ja049106a. [DOI] [PubMed] [Google Scholar]
- 51.Han WG, Liu TQ, Lovell T, Noodleman L. J Am Chem Soc. 2005;127:15778–15790. doi: 10.1021/ja050904q. [DOI] [PubMed] [Google Scholar]
- 52.Mitić N, Clay MD, Saleh L, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2007;129:9049–9065. doi: 10.1021/ja070909i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM, Jr, Neese F, Krebs C. J Am Chem Soc. 2007;129:6168–6179. doi: 10.1021/ja067899q. [DOI] [PubMed] [Google Scholar]
- 54.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 55.Galonić DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Nature Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 56.Korboukh VK, Li N, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 59.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 60.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Proc Natl Acad Sci USA. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Proc Natl Acad Sci USA. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bollinger JM, Jr, Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. J Am Chem Soc. 1998;120:1094–1095. [Google Scholar]
- 63.Saleh L, Krebs C, Ley BA, Naik S, Huynh BH, Bollinger JM., Jr Biochemistry. 2004;43:5953–5964. doi: 10.1021/bi036099e. [DOI] [PubMed] [Google Scholar]
- 64.Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD. Proc Natl Acad Sci USA. 2010;107:16788–16793. doi: 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murray LJ, García-Serres R, Naik S, Huynh BH, Lippard SJ. J Am Chem Soc. 2006;128:7458–7459. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kemsley JN, Mitić N, Zaleski KL, Caradonna JP, Solomon EI. J Am Chem Soc. 1999;121:1528–1536. [Google Scholar]
- 67.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 2001;123:7388–7398. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 68.Solomon EI. Inorg Chem. 2001;40:3656–3669. doi: 10.1021/ic010348a. [DOI] [PubMed] [Google Scholar]
- 69.Ohta T, Chakrabarty S, Lipscomb JD, Solomon EI. J Am Chem Soc. 2008;130:1601–1610. doi: 10.1021/ja074769o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Price JC. PhD Thesis. The Pennsylvania State University; 2005. [Google Scholar]
- 71.Que L, Ho RYN. Chem Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 72.Hanauske-Abel HM, Günzler V. J Theor Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 73.Panay AJ, Lee M, Krebs C, Bollinger JM, Jr, Fitzpatrick PF. Biochemistry. 2011;50:1928–1933. doi: 10.1021/bi1019868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eser BE, Barr EW, Frantom PA, Saleh L, Bollinger JM, Jr, Krebs C, Fitzpatrick PF. J Am Chem Soc. 2007;129:11334–11335. doi: 10.1021/ja074446s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Panay AJ, Fitzpatrick PF. J Am Chem Soc. 2010;132:5584–5585. doi: 10.1021/ja101563t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mitić N, Saleh L, Schenk G, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2003;125:11200–11201. doi: 10.1021/ja036556e. [DOI] [PubMed] [Google Scholar]
- 77.Bollinger JM, Jr, Price JC, Hoffart LM, Barr EW, Krebs C. Eur J Inorg Chem. 2005;2005:4245–4254. [Google Scholar]
- 78.Hitchman ML. Measurement of Dissolved Oxygen. Wiley; New York: 1978. [Google Scholar]
- 79.Li N, Korboukh VK, Krebs C, Bollinger JM., Jr Proc Natl Acad Sci USA. 2010;107:15722–15727. doi: 10.1073/pnas.1002785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hirata Y, Nakata H, Yamada K, Okuhara K, Naito T. Tetrahedron. 1961;14:252–274. [Google Scholar]
- 81.He J, Hertweck C. J Am Chem Soc. 2004;126:3694–3695. doi: 10.1021/ja039328t. [DOI] [PubMed] [Google Scholar]
- 82.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci USA. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vaillancourt FH, Yin J, Walsh CT. Proc Natl Acad Sci USA. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 85.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM., Jr Proc Natl Acad Sci USA. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galonić Fujimori D, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Bollinger JM, Jr, Krebs C, Riggs-Gelasco PJ. J Am Chem Soc. 2007;129:13408–13409. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 87.Luo YR. Comprehensive handbook of chemical bond energies. CRC Press Inc.; Boca Raton, FL: 2007. [Google Scholar]
- 88.Decker A, Rohde JU, Que L, Solomon EI. J Am Chem Soc. 2004;126:5378–5379. doi: 10.1021/ja0498033. [DOI] [PubMed] [Google Scholar]
- 89.Bell CB, Wong SD, Xiao YM, Klinker EJ, Tenderholt AL, Smith MC, Rohde JU, Que L, Cramer SP, Solomon EI. Angew Chem-Int Edit. 2008;47:9071–9074. doi: 10.1002/anie.200803740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang W, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2007;129:7504–7505. doi: 10.1021/ja072528a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang W, Yun D, Saleh L, Bollinger JM, Jr, Krebs C. Biochemistry. 2008;47:13736–13744. doi: 10.1021/bi8017625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bollinger JM, Jr, Jiang W, Green MT, Krebs C. Curr Opin Struct Biol. 2008;18:650–657. doi: 10.1016/j.sbi.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 93.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 94.Jiang W, Xie J, Varano PT, Krebs C, Bollinger JM., Jr Biochemistry. 2010;49:5340–5349. doi: 10.1021/bi100037b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seyedsayamdost MR, Xie J, Chan CTY, Schultz PG, Stubbe J. J Am Chem Soc. 2007;129:15060–15071. doi: 10.1021/ja076043y. [DOI] [PubMed] [Google Scholar]
- 96.Seyedsayamdost MR, Stubbe J. J Am Chem Soc. 2007;129:2226–2227. doi: 10.1021/ja0685607. [DOI] [PubMed] [Google Scholar]
- 97.Yokoyama K, Uhlin U, Stubbe J. J Am Chem Soc. 2010;132:15368–15379. doi: 10.1021/ja1069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yokoyama K, Smith AA, Corzilius B, Griffin RG, Stubbe J. J Am Chem Soc. 2011;133:18420–18432. doi: 10.1021/ja207455k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang W, Saleh L, Barr EW, Xie J, Gardner MM, Krebs C, Bollinger JM., Jr Biochemistry. 2008:8477–8484. doi: 10.1021/bi800881m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Younker JM, Krest CM, Jiang W, Krebs C, Bollinger JM, Jr, Green MT. J Am Chem Soc. 2008;130:15022–15027. doi: 10.1021/ja804365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van Glinkel CG, Rikken GB, Kroon AGM, Kengen SWM. Arch Microbiol. 1996;166:321–326. doi: 10.1007/s002030050390. [DOI] [PubMed] [Google Scholar]
- 102.Coates JD, Achenbach LA. Nature Rev Microbiol. 2004;2:569–580. doi: 10.1038/nrmicro926. [DOI] [PubMed] [Google Scholar]
- 103.Bender KS, Shang C, Chakraborty R, Belchik SM, Coates JD, Achenbach LA. J Bacteriol. 2005;187:5090–5096. doi: 10.1128/JB.187.15.5090-5096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Streit BR, DuBois JL. Biochemistry. 2008;47:5271–5280. doi: 10.1021/bi800163x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Streit BR, Blanc B, Lukat-Rodgers GS, Rodgers KR, DuBois JL. J Am Chem Soc. 2010;132:5711–5724. doi: 10.1021/ja9082182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goblirsch B, Kurker RC, Streit BR, Wilmot CM, DuBois JL. J Mol Biol. 2011;408:379–398. doi: 10.1016/j.jmb.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dassama LMK, Yosca TH, Conner DA, Lee MH, Blanc B, Streit BR, Green MT, DuBois JL, Krebs C, Bollinger JM., Jr Biochemistry. 2012;51:1607–1616. doi: 10.1021/bi201906x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Proshlyakov DA, Henshaw TF, Monterosso GR, Ryle MJ, Hausinger RP. J Am Chem Soc. 2004;126:1022–1023. doi: 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- 109.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 110.Grzyska PK, Appelman EH, Hausinger RP, Proshlyakov DA. Proc Natl Acad Sci USA. 2010;107:3982–3987. doi: 10.1073/pnas.0911565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Que L., Jr Nature Struct Biol. 2000;7:182–184. doi: 10.1038/73270. [DOI] [PubMed] [Google Scholar]
- 112.Koehntop KD, Emerson JP, Que L., Jr J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 113.England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L. Angew Chem-Int Edit. 2009;48:3622–3626. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.England J, Guo YS, Farquhar ER, Young VG, Münck E, Que L. J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2010;132:12188–12190. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.England J, Guo YS, Van Heuvelen KM, Cranswick MA, Rohde GT, Bominaar EL, Münck E, Que L. J Am Chem Soc. 2011;133:11880–11883. doi: 10.1021/ja2040909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wong SD, Bell CB, Liu LV, Kwak Y, England J, Alp EE, Zhao JY, Que L, Solomon EI. Angew Chem-Int Edit. 2011;50:3215–3218. doi: 10.1002/anie.201007692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bigi JP, Harman WH, Lassalle-Kaiser B, Robles DM, Stich TA, Yano J, Britt RD, Chang CJ. J Am Chem Soc. 2012;134:1536–1542. doi: 10.1021/ja207048h. [DOI] [PubMed] [Google Scholar]
- 119.Sjöberg BM, Reichard P, Gräslund A, Ehrenberg A. J Biol Chem. 1977;252:536–541. [PubMed] [Google Scholar]
- 120.Nordlund P, Sjöberg BM, Eklund H. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 121.Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Philos Trans Royal Soc B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stubbe J. Curr Opin Chem Biol. 2003;7:183–188. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 123.Burdi D, Sturgeon BE, Tong WH, Stubbe J, Hoffman BM. J Am Chem Soc. 1996;118:281–282. [Google Scholar]
- 124.Shanmugam M, Doan PE, Lees NS, Stubbe J, Hoffman BM. J Am Chem Soc. 2009;131:3370–3376. doi: 10.1021/ja809223s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hsu HF, Dong YH, Shu LJ, Young VG, Que L. J Am Chem Soc. 1999;121:5230–5237. [Google Scholar]
- 126.Charalampous FC, Lyras C. J Biol Chem. 1957;228:1–13. [PubMed] [Google Scholar]
- 127.Bollinger JM, Jr, Diao Y, Matthews ML, Xing G, Krebs C. Dalton Trans. 2009;6:905–914. doi: 10.1039/b811885j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hirao H, Morokuma K. J Am Chem Soc. 2009;131:17206–17214. doi: 10.1021/ja905296w. [DOI] [PubMed] [Google Scholar]
- 129.Xing G, Hoffart LM, Diao Y, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Biochemistry. 2006;45:5393–5401. doi: 10.1021/bi0519607. [DOI] [PubMed] [Google Scholar]
- 130.Xing G, Barr EW, Diao Y, Hoffart LM, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Biochemistry. 2006;45:5402–5412. doi: 10.1021/bi0526276. [DOI] [PubMed] [Google Scholar]
- 131.Brown PM, Caradoc-Davies TT, Dickson JMJ, Cooper GJS, Loomes KM, Baker EN. Proc Natl Acad Sci USA. 2006;103:15032–15037. doi: 10.1073/pnas.0605143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bailey JA, James CA, Woodruff WH. Biochem Biophys Res Commun. 1996;220:1055–1060. doi: 10.1006/bbrc.1996.0531. [DOI] [PubMed] [Google Scholar]
- 133.Schenk G, Pau MYM, Solomon EI. J Am Chem Soc. 2004;126:505–515. doi: 10.1021/ja036715u. [DOI] [PubMed] [Google Scholar]
- 134.Mukherjee A, Cranswick MA, Chakrabarti M, Paine TK, Fujisawa K, Münck E, Que L. Inorg Chem. 2010;49:3618–3628. doi: 10.1021/ic901891n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ye SF, Price JC, Barr EW, Green MT, Bollinger JM, Jr, Krebs C, Neese F. J Am Chem Soc. 2010;132:4739–4751. doi: 10.1021/ja909715g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee YM, Hong S, Morimoto Y, Shin W, Fukuzumi S, Nam W. J Am Chem Soc. 2010;132:10668–10670. doi: 10.1021/ja103903c. [DOI] [PubMed] [Google Scholar]
- 137.Cho KB, Chen H, Janardanan D, de Visser SP, Shaik S, Nam W. Chem Commun. 48:2189–2191. doi: 10.1039/c2cc17610f. [DOI] [PubMed] [Google Scholar]
- 138.Bollinger JM, Jr, Krebs C. Curr Opin Chem Biol. 2007;11:151–158. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 139.van der Donk WA, Krebs C, Bollinger JM., Jr Curr Opin Struct Biol. 2010;20:673–683. doi: 10.1016/j.sbi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lin Y, Gerfen GJ, Rousseau DL, Yeh SR. Anal Chem. 2003;75:5381–5386. doi: 10.1021/ac0346205. [DOI] [PubMed] [Google Scholar]
- 141.Cherepanov AV, de Vries S. Biochim Biophys Acta-Bioenergetics. 2004;1656:1–31. doi: 10.1016/j.bbabio.2004.02.006. [DOI] [PubMed] [Google Scholar]