

Abstract
The class I ribonucleotide reductase from Chlamydia trachomatis uses a stable MnIV/FeIII cofactor to initiate nucleotide reduction by a free-radical mechanism. The enzyme provides the first example both of a Mn-dependent ribonucleotide reductase and of a Mn/Fe redox cofactor. In this work, we have used variable-field Mössbauer spectroscopy to demonstrate that the active cofactor has an S = 1 ground state due to antiferromagnetic coupling between the MnIV (SMn = 3/2) and high-spin FeIII (SFe = 5/2) sites.
The reduction of nucleotides to 2′-deoxynucleotides by ribonucleotide reductases (RNRs)1 begins with the abstraction of the 3′ hydrogen atom of the substrate by a transient cysteine thiyl radical (C•).2–4 Class I, II, and III RNRs differ in subunit composition and the nature of the cofactor used to generate the 3′-H-abstracting C•.2 A conventional class I RNR uses a stable tyrosyl radical (Y•), which is introduced into the enzyme’s homodimeric R2 subunit (also denoted β2) by reaction of O2 with an adjacent carboxylate-bridged non-heme diiron cluster,5 to generate the C• in its R1 subunit (also denoted α2) via long-range (~ 35 Å) proton-coupled electron transfer (PCET).6–8 The identification in Chlamydia trachomatis (Ct) and other species of pathogenic bacteria of RNRs having the class I subunit architecture but the Y•-harboring tyrosine replaced by phenylalanine raised the question of how such an RNR might function without the initiating Y•.9,10 We recently showed that Ct RNR uses a stable MnIV/FeIII cofactor, generated by reaction of the MnII/FeII-R2 complex with O2, for radical initiation.11 Although heterobinuclear Mn/Fe complexes of various oxidation states,12–14 including one example of an inorganic MnIV/FeIII complex,15 have been reported, Ct RNR is, to our knowledge, the first case in which an enzyme has been shown to use a Mn/Fe cluster as a redox cofactor. Here we characterize this novel cofactor by Mössbauer spectroscopy, showing that it has a triplet (STotal = 1) ground state resulting from antiferromagnetic coupling of its MnIV (SMn = 3/2) and high-spin FeIII (SFe = 5/2) constituents and providing parameters to calibrate calculation of its geometric and electronic structure.
The 4.2-K/zero-field Mössbauer spectrum of a sample prepared by reaction of a solution of R2, MnII, and FeII with O216 (Figure 1, hashed marks) comprises two quadrupole doublets with similar isomer shifts (δ) but different quadrupole splittings (ΔEQ). The minor doublet (25% of the total intensity) matches the spectrum of the Fe2III/III cluster, which is characterized by δ = 0.50 mm/s and ΔEQ = 0.79 mm/s (Figure 1, solid line). The major doublet (75% intensity) arises from the functional MnIV/FeIII cofactor.11 For in-depth characterization of this species, we collected Mössbauer spectra at 4.2 K in variable applied magnetic fields. To analyze these spectra, it was necessary first to remove the 25% contribution attributed to the Fe2III/III complex (Figure S1). The derived spectra of the MnIV/FeIII complex are shown in Figure 2.
Figure 1.
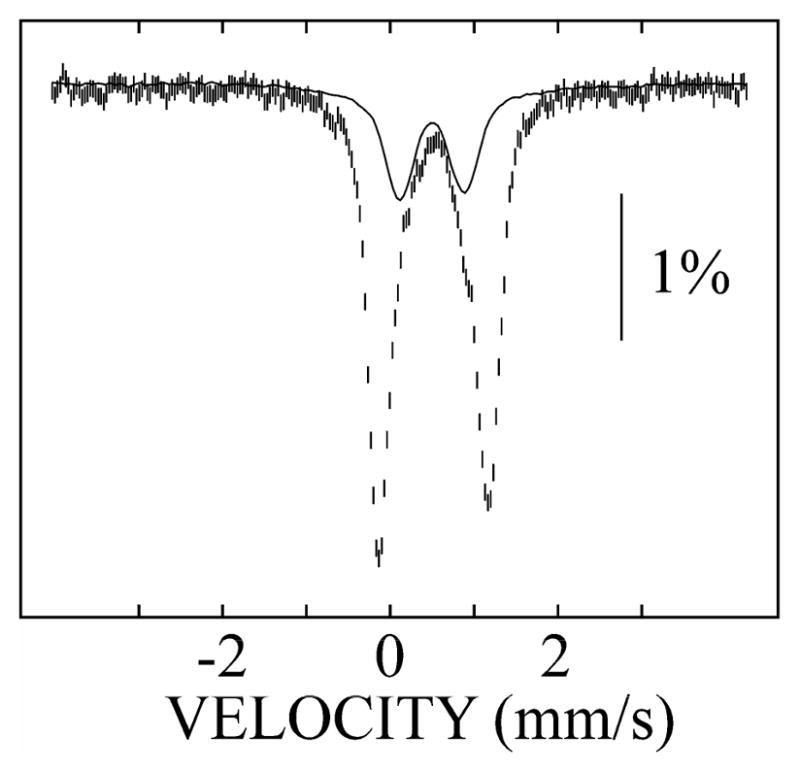
4.2-K/zero-field Mössbauer spectrum of the final product of the reaction of MnII/FeII Ct-R2 with O2 (hashed marks). The solid line is the spectrum of the final product of the reaction of FeII/FeII Ct-R2 with O2 (25% of the total intensity).
Figure 2.
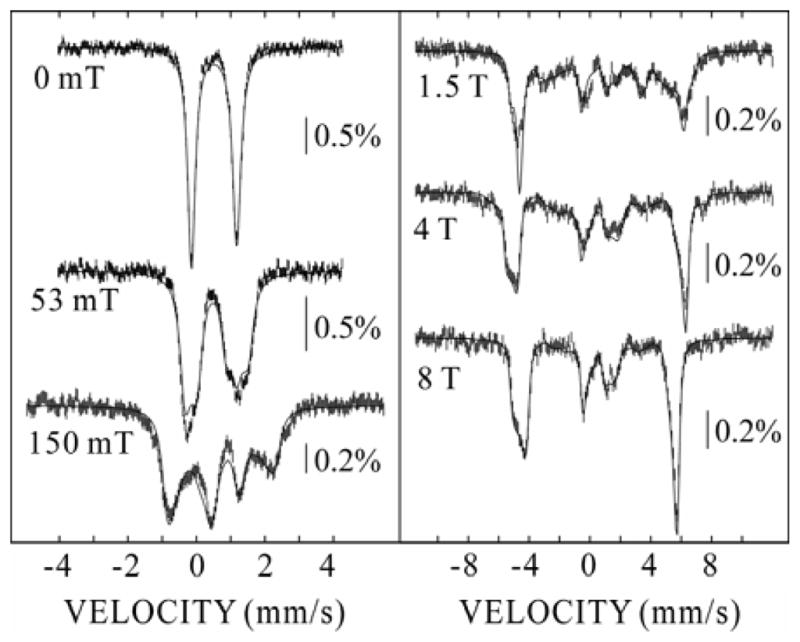
4.2-K Mössbauer spectra of MnIV/FeIII-R2 derived by component analysis of the experimental spectra. The solid lines are simulations according to the spin Hamiltonian given in the Supporting Information and the following parameters: STotal = 1, DS=1 = −1.9 cm−1, (E/D)S=1 = 0.33, δ = 0.52 mm/s, ΔEQ = 1.32 mm/s, η = −2.6, (A/gNβN)Fe = (−40.2, −38.9, −38.0) T.
The zero-field spectrum is a sharp quadrupole doublet with parameters (δ = 0.52 mm/s and ΔEQ = 1.32 mm/s) typical of high-spin FeIII. Spectra recorded in applied fields were analyzed with the assumption of slow relaxation and according to the spin Hamiltonian with respect to the total-spin ground state, STotal = 1, given in the Supporting Information. For this analysis, the parameters determined from the zero-field spectrum were fixed. The other parameters in the simulations are the asymmetry parameter, η, the axial and rhombic zero-field splitting (ZFS) parameters of the STotal = 1 ground state, DS=1 and (E/D)S=1, respectively, and the hyperfine tensor for the FeIII ion, (A/gNβN)Fe.
The fact that the overall splitting of the spectrum is larger at 4 T than at 8 T reveals that the electronic Zeeman term dominates the ZFS interaction at these magnetic field strengths. As a consequence, the spin expectation value of the ground state (〈S〉) and the internal magnetic field [Bint = −〈S〈·(A/gNβN)Fe] approach their limiting values (corresponding to 〈S〉 = −1) at these applied fields, and the hyperfine tensor can be determined accurately from the spectra. In a spin-coupled cluster, the A-tensor for the iron ion with respect to the STotal = 1 ground state (AFe) is given by equation 1, in which cFe and aFe represent the vector coupling coefficient and the intrinsic hyperfine tensor, respectively.17
| (1) |
For high-spin FeIII sites, (a/gNβN)Fe is dominated by the Fermi contact term and exhibits nearly isotropic values of ~ −21 T.18 For the STotal = 1 state, cFe = +7/4. Thus, (A/gNβN)Fe is expected to be nearly isotropic with values of ~ −37 T. Indeed, analysis of the spectra yielded a nearly isotropic hyperfine tensor, (A/gNβN)Fe = (−40.2, −38.9, −38.0) T, similar to the expected value.
With the AFe determined from the 4-T and 8-T spectra, the ZFS-parameters were then determined accurately from the spectra with lesser applied magnetic fields, under which conditions the ZFS is significant compared to the electronic Zeeman term. Simulation of the spectra provided for determination of the magnetic-field dependence of 〈S〉, which determines the magnitude of the internal magnetic field and the resultant splitting in each spectrum. From the field-dependent spectra, DS=1 = −1.9 cm−1 and (E/D)S=1 = 0.33 were found. DS=1 is related to the intrinsic D-values of the MnIV and FeIII ions via equation 2, with the assumption that the total spin states are well separated in energy (i.e., J ≫ D).17
| (2) |
DS=1 is dominated by the contribution from the FeIII site19 and can be rationalized with a value typical for high-spin FeIII (|DFe| of ~ 0.7 cm−1).18
The MnIV and FeIII ions could conceivably couple ferromagnetically to yield an STotal = 4 ground state. However, the inherent anisotropy of 〈S〉 of the STotal = 4 system precludes simulation of all the experimental spectra with a single set of spin-Hamiltonian parameters. Specifically, comparison of the 4-T and 8-T spectra establishes that 〈S〉 along all three molecular axes nearly reach saturation in an applied field of ~4 T. |DS=4| would therefore have to be small (< 0.5 cm−1, see Figure S2). Conversely, with a DS=4 of this small magnitude, 〈Sz〉 (for DS=4 < 0) saturates at ~30 mT (Figure S2), resulting in much greater magnetic hyperfine splitting in the weak-field spectra than is observed (Figure S3). Thus, ferromagnetic coupling to yield STotal = 4 can be ruled out.
The demonstration by Mössbauer spectroscopy that the MnIV/FeIII cluster in the active form of Ct R2 has a STotal = 1 ground state arising from antiferromagnetic coupling between the SMn = 3/2 and SFe = 5/2 ions represents an important first step toward defining its electronic structure. Moreover, parameters obtained in the analysis can now be used to calibrate density functional theory calculations to extract deeper insight.20,21 This insight will permit an important unresolved aspect of class I RNR function – how electron transfer to the oxidized cofactor in R2 is “gated” so as to occur only in the active holoenzyme complex – to be addressed. Direct interrogation of the radical initiation step in conventional class I RNRs has proven to be extremely challenging.7 The R2 product of radical transfer does not accumulate during turnover because of unfavorable kinetics.7,22 Accumulation of the reduced cofactor can be promoted by use of radical-trapping substrate analogues or R1 variants,23–26 but extracting atomic-level insight into the changes to the cofactor upon reduction is not straightforward. The reduced Y lacks a useful spectroscopic signature. The Fe2III/III cluster is either unchanged by the radical-transfer step or, at most, may transfer a proton to the Y from a coordinated water ligand, a change not obviously demonstrable by the spectroscopic methods to which the EPR-silent cluster is amenable. The use of the novel cofactor by Ct RNR affords a unique opportunity to dissect the initiation step. The metal cluster undergoes reduction from MnIV/FeIII to MnIII/FeIII, which is, by contrast to the reduced cofactor of a conventional class I RNR, EPR active.11 Moreover, although the reduced form might, as in E. coli RNR, not accumulate during turnover, it can be prepared in stable form and its structure is affected by binding of R1 and nucleotides, as demonstrated by marked changes to its EPR spectrum.11 The structural changes caused by formation of the complex are almost certainly elements of the gating mechanism. By defining what they are and extending the description of the geometric and electronic structure of the MnIV/FeIII cofactor initiated by this work, insight into the mechanisms of radical transfer and conformational gating thereof may be obtained.
Supplementary Material
Scheme 1.
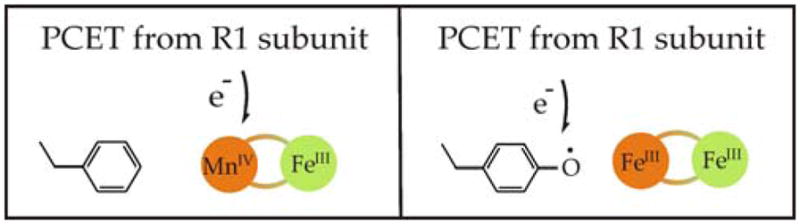
Radical-generating cofactor of Ct RNR (left) and conventional class I RNR (right).
Acknowledgments
This work was supported by the National Institutes of Health (GM-55365 to JMB), the Beckman Foundation (Young Investigator Award to CK), and the Dreyfus Foundation (Teacher Scholar Award to CK).
Footnotes
Supporting Information Available: Rationale for method of sample preparation, component analysis to resolve the spectra of MnIV/FeIII-R2 shown in Figure 2, analysis of these spectra for the hypothetical case of an STotal = 4 ground state, selected plots of the spin expectation values, and the spin Hamiltonian used for analysis of the Mössbauer spectra. This material is available free of charge from the journal website: http://pubs.acs.org.
References
- 1.Abbreviations used: Ct: Chlamydia trachomatis; PCET: proton-coupled electron transfer; RNR: ribonucleotide reductase; ZFS: zero field splitting.
- 2.Licht S, Gerfen GJ, Stubbe J. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- 3.Stubbe J. Curr Opin Struct Biol. 2000;10:731–736. doi: 10.1016/s0959-440x(00)00153-6. [DOI] [PubMed] [Google Scholar]
- 4.Nordlund P, Reichard P. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 5.Atkin CL, Thelander L, Reichard P. J Biol Chem. 1973;248:7464–7472. [PubMed] [Google Scholar]
- 6.Stubbe J, Riggs-Gelasco P. Trends Biochem Sci. 1998;23:438–443. doi: 10.1016/s0968-0004(98)01296-1. [DOI] [PubMed] [Google Scholar]
- 7.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 8.Stubbe J. Curr Opin Chem Biol. 2003;7:183–188. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 9.Roshick C, Iliffe-Lee ER, McClarty G. J Biol Chem. 2000;275:38111–38119. doi: 10.1074/jbc.M006367200. [DOI] [PubMed] [Google Scholar]
- 10.Högbom M, Stenmark P, Voevodskaya N, McClarty G, Gräslund A, Nordlund P. Science. 2004;305:245–248. doi: 10.1126/science.1098419. [DOI] [PubMed] [Google Scholar]
- 11.Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak M-A, Krebs C, Bollinger JM., Jr Science. 2007 doi: 10.1126/science.1141179. accepted. [DOI] [PubMed] [Google Scholar]
- 12.Bossek U, Weyhermüller T, Wieghardt K, Bonvoisin J, Girerd JJ. J Chem Soc, Chem Comm. 1989:633–636. [Google Scholar]
- 13.Schenk G, Boutchard CL, Carrington LE, Noble CJ, Moubaraki B, Murray KS, De Jersey J, Hanson GR, Hamilton S. J Biol Chem. 2001;276:19084–19088. doi: 10.1074/jbc.M009778200. [DOI] [PubMed] [Google Scholar]
- 14.Pierce BS, Hendrich MP. J Am Chem Soc. 2005;127:3613–3623. doi: 10.1021/ja0491937. [DOI] [PubMed] [Google Scholar]
- 15.Hotzelmann R, Wieghardt K, Flörke U, Haupt HJ, Weatherburn DC, Bonvoisin J, Blondin G, Girerd JJ. J Am Chem Soc. 1992;114:1681–1696. [Google Scholar]
- 16.An anaerobic solution of R2 (3.0 mM β), MnII (3.0 mM), and FeII (1.5 mM) in 100 mM HEPES (pH = 7.6) containing 10% (v/v) glycerol was mixed at 5 °C with an equal volume of an O2-saturated solution of the same buffer, and the reaction was allowed to proceed for 10 min before the sample was frozen. The control sample was prepared identically, except the MnII was omitted and the concentration of FeII was doubled in the protein reactant solution. See the Supporting Information for a discussion of the logic underlying the method of sample preparation.
- 17.Bencini A, Gatteschi D. EPR of exchange coupled systems. Springer; Berlin, Germany: 1990. [Google Scholar]
- 18.Münck E. In: Physical Methods in Bioinorganic Chemistry. Que L Jr, editor. University Science Books; Sausalito, CA: 2000. pp. 287–319. [Google Scholar]
- 19.High-spin FeIII and MnIV have well-isolated, non-degenerate orbital ground states and typically exhibit small intrinsic D-values.
- 20.Han WG, Liu T, Lovell T, Noodleman L. J Inorg Biochem. 2006;100:771–779. doi: 10.1016/j.jinorgbio.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 21.Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM, Jr, Neese F, Krebs C. J Am Chem Soc. 2007 doi: 10.1021/ja067899q. accepted. [DOI] [PubMed] [Google Scholar]
- 22.Ge J, Yu G, Ator MA, Stubbe J. Biochemistry. 2003;42:10071–10083. doi: 10.1021/bi034374r. [DOI] [PubMed] [Google Scholar]
- 23.Thelander L, Larsson B, Hobbs J, Eckstein F. J Biol Chem. 1976;251:1398–1405. [PubMed] [Google Scholar]
- 24.Mao SS, Johnston MI, Bollinger JM, Stubbe J. Proc Natl Acad Sci U S A. 1989;86:1485–1489. doi: 10.1073/pnas.86.5.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Persson AL, Eriksson M, Katterle B, Pötsch S, Sahlin M, Sjöberg BM. J Biol Chem. 1997;272:31533–31541. doi: 10.1074/jbc.272.50.31533. [DOI] [PubMed] [Google Scholar]
- 26.Stubbe J, van der Donk WA. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.