

Abstract
The absorption, distribution, metabolism and excretion (ADME) and pharmacokinetic (PK) properties of inorganic nanoparticles with hydrodynamic diameters between 2 and 20 nm are presently unpredictable. It is unclear whether unpredictable in vivo properties and effects arise from a subset of molecules in a nanomaterials preparation, or if the ADME/PK properties are ensemble properties of an entire preparation. Here we characterize the ADME/PK properties of atomically precise preparations of ligand protected gold nanoclusters in a murine model system. We constructed atomistic models and tested in vivo properties for five well defined compounds, based on crystallographically resolved Au25(SR)18 and Au102(SR)44 nanoclusters with different (SR) ligand shells. To rationalize unexpected distribution and excretion properties observed for several clusters in this study and others, we defined a set of atomistic structure–activity relationships (SAR) for nanoparticles, which includes previously investigated parameters such as particle hydrodynamic diameter and net charge, and new parameters such as hydrophobic surface area and surface charge density. Overall we find that small changes in particle formulation can provoke dramatic yet potentially predictable changes in ADME/PK.
Introduction
Ligand passivated metal and semiconductor nanoparticles can encode several physical properties of clinical interest for diagnostic imaging and therapy. Investigated diagnostic imaging properties include X-ray contrast,1–3 luminescence,4–6 and PET/SPECT.7 Preclinical therapeutics based on these nanomaterials can be analogous to antibody mimetics,8 or be targeted hyperthermal therapeutics,9–11 where the inorganic core of the nanomaterial interacts with extrinsic radiation (typically IR or RF) to produce localized therapeutic heat.
Predictive and complete understanding of biological absorption, distribution, metabolism, excretion (ADME) and pharmacokinetics (PK) of metal and semiconductor nanoparticles is important for realizing a comprehensive set of design criteria for determining which nanomaterials may ultimately be useful in clinic. Ideally a set of structure–activity relationships (SAR) will emerge, enabling design, synthesis and application of nanoparticles with accurately predicted circulation lifetimes, metabolism and clearance mechanisms, tissue penetrations, cellular and subcellular trafficking, and high localization efficiency to desired corporeal targets.
Current predictive understanding of the ADME/PK of inorganic nanoparticles is incomplete, in part because these experiments combine complex mixtures of nanoparticles (i.e., a 10.0 ± 1.0 nm gold colloid preparation contains on the order of 40 000 discrete molecular formulae) with blood which is also a complex mixture itself. The uncertainties inherent in nanoparticle preparation complicate the analysis of their biological activity.
Some very general conclusions about ADME/PK can be drawn. For instance, nanoparticles substantially larger than 20 nm in hydrodynamic diameter (i.e., the size of the smallest viruses) are relatively predictable in their PK and biodistribution.12–14 Such particles are generally cleared by the reticuloendothelial system (RES), with some control in circulation time imparted by the surface layer, where poly- and oligo-ethylene glycols are shown to dramatically increase blood circulation half life.15,16 The metabolism and excretion of inorganic particles in the RES is unpredictable.4,5 Some reports begin to establish metabolism and excretion with mechanism,17,18 while other reports conclude in long-term RES accumulation with unknown consequence,13 and RES toxicity13,19 all present and all arising from particle preparations of different nature.
The 2–20 nm hydrodynamic diameter range (smaller than viruses and other pathogens for which mammals have well established mechanisms for blood clearance) is a less predictable size regime, and one in which ADME/PK properties may be tunable for desired clinical applications. Predictive circulation lifetime is important as diagnostic imaging applications generally specify short circulation lifetimes while therapeutic applications generally benefit from extended circulation times.20
Within this size regime, three hydrodynamic diameter thresholds are important determinants of ADME/PK. These are the ~8 nmhydrodynamic threshold of glomerular wall filtration in the kidney, the 5 nm threshold for rapid extravascular equilibration, and the ~20 nm threshold for Enhanced Permeability and Retention (EPR).16,21 The glomerural wall of the kidney has a nominal pore size of 8 nm and as a negatively charged structure is more selective for filtration of positively charged particles. Zwitterionic particles may need to be as small as 5.5 nm to pass efficiently,4 and negatively charged particles can have unpredictable behavior including apparently extended circulation times18 and kidney accumulation,1 sometimes with notable associated toxicity.22 Still renal clearance of nanoparticles is often viewed as a favorable result,4,5 since it results in predictable clearance of nanoparticles relative to RES clearance. Toxicity in this size range also appears unpredictable, with 1.4 nm and 13 nm particles observed as causing acute toxicity,19,23,24 while other sizes appear essentially non-toxic in short term studies.
The study of ADME/PK of nanomaterials is complicated by the complex natures of both nanomaterial preparations and biological systems. While obvious that blood is a complicated mixture, less frequently highlighted is that most nanomaterials preparations are also complex mixtures. For instance, implicit in even ‘monodisperse’ preparations of nanomaterials is a 10% standard deviation in dimension measurement, suggesting that a ‘monodisperse’ 10 ± 1 nm spherical gold nanoparticle preparation corresponds to a statistical gold composition of 244 000 ± 70 000 atoms. The purity of the exactly 10 nm diameter Au244000 core is less than 1% in this preparation. Further complicating these mixtures is frequently an additional distribution in ligand shell composition, especially for mixed ligand shells.
In the present study, we make the first examination of the ADME/PK properties of ‘magic number’ gold clusters,25,26 specifically those scaffolded by the crystallographically determined Au25(SR)18 and Au102(SR)44 nanoclusters. We prepared each compound with three different ligand shells: an as-synthesized ligand shell (p-mer-captobenzoic acid and glutathione for Au102 and Au25, respectively) and two partially ligand exchanged shells with tetraethylene glycol functionality introduced in varying amounts. We modeled each of the five compounds as idealized atomistic models, and suggest that the structural features of these models give novel insight into the surprising ADME/PK activity observed for some of these preparations.
Materials and methods
Generation of models
Models of the ligands on Au25(GSH)18 (1), Au25(GSH)9-[S(CH2)6(EG)4OH]9 (2), Au25(GSH)6[S(CH2)6(EG)4OH]12 (3), Au102(pMBA)44 (4), Au102(pMBA)25[S(CH2)11(EG)4OH]19 (5) were generated with PRODRG27 and manually modeled onto clusters in PyMOL.28
Calculation of hydrodynamic radius
Models were input into Hydropro10 (ref. 29) with 0.0035 Pa s to simulate the viscosity of serum at 37 °C.30 Settings were left as recommended by Ortega et al.,29 except molecular weight and partial specific volume. Partial specific volume was calculated according to Durchschlag et al.31 for each of the ligands then multiplied by the number of ligands and added to the partial specific volume of the gold cluster divided by the molecular weight to obtain ν̄c.
Calculation of solvent accessible area
Solvent accessible area was calculated using PyMOL’s ability to calculate area. Hydrophilic atoms were set as nitrogen and oxygen and hydrophobic areas were set as carbon and sulfur. Solvent radius was set at 1.4 Å and modeled and the total area around each of the areas was calculated.
Calculation of surface charge
Models of ligands were input into PDB2PQR32,33 to convert models from PDB format to PQR, which was then input into Adaptive Poisson-Boltzmann Solver (APBS)34 to calculate total surface charge. The force field chosen for the calculations was SWANSON, the model was also allowed to optimize the hydrogen bonding network.
Reagents
All commercially available reagents were used without further purification. Tetrachloroauric (III) acid (HAuCl4·3H2O 99.99% metal basis, Alfa Aesar), p-mer-captobenzoic acid (>95.0%, TCI America), NaBH4 (98–99%, MP Biomedicals), MeOH (99.9%, Fisher Scientific), L-glutathione reduced (≥98%, Sigma-Aldrich), Diethylene glycol dimethyl ether (99%, Sigma-Aldrich), 2-[2-(1-mercaptoundec-6-yloxy)ethoxy]ethoxy-ethoxy-ethanol (HS–(CH2)6–EG4–OH, prochimia surfaces), [11-(methylcarbonylthio) undecyl]tetra(ethylene glycol) (AcS–(CH2)11–EG4–OH, 95%, Sigma-Aldrich). Nanopure water (resistivity 18.2 MΩ cm) was produced with a Barnstead NANOpure water system.
Synthesis and characterization of Au@GSH (compound 1)
A 50mL conical was charged with 6mL 100mMglutathione (0.6 mmol, 3 equiv.) in 0.3 M NaOH solution. 2 mL of HAuCl4·3H2O in diethylene glycol dimethyl ether solution (0.2 mmol, 1 equiv., a non-metal spatula should be used to weigh out HAuCl4·3H2O) was also added to the conical. The reaction was shaken at rt for 30 min. 5 min prior to the end of the 30 min, a suspension of 0.5 mM NaBH4 in dry diethylene glycol dimethyl ether (about 19 mL) was sonicated at rt for 5 min. 17 mL of the NaBH4 suspension of (0.0085 mmol, 0.043 equiv.) was added to the reaction which turned orange over about 20 seconds and was quenched by the addition of methanol (to a final volume of about 50 mL). The content of the conical was then mixed and then centrifuged in a swinging bucket rotor at 4000 rpm and 4 °C for 10 min. The clear and colorless supernatant was then decanted and the orange precipitate was air dried. Gel electrophoresis visualization was run on a 30% polyacrylamide gel (19 : 1, acrylamide : bisacrylamide) at 175 V for 3 h. The nanoparticle bands were visible by eye and with a UV trans-illuminator, thus no staining steps were performed for visualization.
Ligand exchange reaction of Au@GSH (1) cluster with 18-mercapto-3,6,9,12-tetraoxaoctadecan-1-ol (HS–(CH2)6–EG4–OH) (compounds 2 and 3)
A 500 μM solution of Au@GSH (0.001 mmol, 10 mg in 2 mL H2O) and a 0.1 M solution of the HS–(CH2)6–EG4–OH (0.04 mmol, 12.4 mg in 0.4 mL THF) were prepared. For the 1 : 1 incoming ligand : outgoing ligand reaction (compound 2): 1mL of Au@GSH solution and 0.125 mL of HS–(CH2)6–EG4–OH solution were mixed then diluted with H2O to a final volume of 5 mL, shaken at rt for 1 h. The crude product was purified by ultrafiltration spin columns (5000 Da cutoff) and washed with 3 × 10 mL 1 : 1H2O : MeOH. The remaining orange liquid was placed into a 15 mL conical and was lyophilized until dry. For the 2 : 1 incoming ligand : outgoing ligand reaction (compound 3): 0.250 mL of HS–(CH2)6–EG4–OH solution was used instead of 0.125 mL (see above).
Synthesis and characterization of Au102pMBA44 (compound 4)
Au102pMBA44 was synthesized according to published procedure. 46 HAuCl4·3H2O was dissolved (0.209 g, 0.50 mmol, a nonmetal spatula should be used to weigh out HAuCl4·3H2O) in nanopure H2O (19.0 mL, 0.028 M based on Au) in a 50 mL conical. In a separate 50 mL conical, p-mercaptobenzoic acid (0.292 g, 1.89 mmol) was dissolved in a solution composed of nanopure H2O (18.43 mL) and 10 M NaOH (0.57 mL, 5.70 mmol). The resulting p-mercaptobenzoic acid/NaOH solution was 0.10M based on p-mercaptobenzoic acid, 0.30M based on NaOH, and the pH was determined to be >9. A 1 L Erlenmeyer flask was equipped with a stir bar and nanopure H2O was added to it (51.5 mL). In three separate beakers, the following solutions were dispensed: (1) 0.028 M HAuCl4 solution (17.8 mL, 0.5 mmol, 1.0 equiv.), (2) 0.10 M p-mercaptobenzoic acid/0.30 M NaOH (15.5 mL, 1.5 mmol, 3.0 equiv. of p-mercaptobenzoic acid and 5.7 mmol, 11.4 equiv. of NaOH) solution, (3) MeOH (75 mL). Under stirring, the HAuCl4 solution was poured into the 1 L Erlenmeyer flask (containing H2O), this was immediately followed by the addition of the p-mercaptobenzoic acid/NaOH solution. The reaction turned from yellow to orange upon the addition of the p-mercaptobenzoic acid/NaOH solution. Immediately afterwards the beaker of MeOH was also added to the 1 L flask. The reaction was allowed to stir at room temperature for 1 h. During that time, the reaction turned from dark orange to light orange. After 1 hour, pulverized solid NaBH4 (20.8 mg, 0.55 mmol, 1.1 equiv.) was added to the stirring reaction to reduce the polymer; the reaction continued to stir at room temperature for 17 h. The reaction turned black upon the addition of solid NaBH4. After 17 h MeOH was added to the 1 L flask until the total volume was approximately 800 mL, then 5 M NH4OAc (40 mL) was also added. The reaction was then split into about twenty 50 mL conicals, which were capped and then centrifuged in a swinging bucket rotor at 4000 rpm and 4 °C for 10 min. The supernatant was then decanted and the precipitate was allowed to dry by inverting the conical on a paper towel for about 1 hour. The precipitate in each conical was then dissolved in about 200 μL of nanopure water. The nanoparticle solutions were then combined into 4 conicals. Next, the particles were washed by performing the following: 500 μL of 2 M NH4OAc was added to each of the four conicals, then MeOH was added until the total volume in each conical was about 45 mL. The conicals were shaken to mix and were centrifuged at 4000 rpm and 4 °C for 10 min in a swinging bucket rotor. The resulting supernatant was decanted and the precipitates were dried in vacuo at room temperature for at least 2 h. Gel electrophoresis visualization was run on a 20% poly-acrylamide gel (19 : 1, acrylamide : bisacrylamide) at 110 V for 2 h. The nanoparticle bands were visible by eye, thus no staining steps were performed for visualization. The synthesized particles were run against a standard Au102pMBA44 sample of which the formula/structure has been confirmed by X-ray crystallography.
Fractional precipitation of Au102(pMBA)44
The reaction outcome varies depending on the quality of the solid NaBH4. In the case where many large and/or insoluble products were formed, a simple fractional precipitation removed the majority of the larger products. First, all particles from the reaction were dissolved in nanopure water (9.24 mL) in a 50 mL conical, then 2 M NH4OAc (0.76 mL, 1.52 mmol, 0.076 M final concentration) was added. The solution was thoroughly mixed followed by the addition of MeOH (10 mL, 50%). The suspension was shaken again to mix and the conical was centrifuged at 4000 rpm and 4 °C. The supernatant was decanted into a new 50 mL conical and then re-spun until pellets (larger impurities) no longer formed. The remaining black solution (purified Au102(pMBA)44) was again transferred to a new 50 mL conical. MeOH was then added to the conical until the total volume was about 45 mL. The conical was shaken to mix and was then centrifuged at 4 °C for 10 min. The resulting supernatant was decanted and the precipitate (purified Au102(pMBA)44) was dried in vacuo at room temperature for at least 2 h.
Deprotection of [11-(methylcarbonylthio)undecyl] tetra(ethylene glycol) (AcS–(CH2)11–EG4–OH)35
The acyl-protected thiol was refluxed at 100 °C in 10% HCl/MeOH for 18 h. The reaction mixture was cooled and dichloromethane was added. The separated organic layer was washed twice with H2O, twice with saturated NaHCO3, and dried with Na2SO4. The solvent was removed in vacuo to give the product 23-mercapto-3,6,9,12-tetraoxatricosan-1-ol [HS–(CH2)11–EG4–OH] as a clear oil. 1H NMR (300 MHz, CDCl3) δ 3.80–3.76 (m, 2H), 3.75–3.61 (m, 14H), 3.50 (t, J = 6.0 Hz, 2H), 2.58 (q, J = 6.0 Hz, 1H), 2.23 (brs, 1H), 1.70–1.58 (m, 4H), 1.47–1.29 (m, 14H).
Ligand exchange reaction of Au102(pMBA)44 with 23-mercapto-3,6,9,12-tetraoxatricosan-1-ol [HS–(CH2)11–EG4–OH] (compounds 5)
A 500 μM solution of Au102pMBA44 (6.6 μmol, 178 mg in 13.37 mL H2O) and a 0.1 M solution of HS–(CH2)11–EG4–OH (0.79 mmol, 304 mg in 7.90 mL THF) were prepared. For the 2 : 1 incoming ligand : outgoing ligand reaction (compound 5): 1.32 mL of HS–(CH2)11–EG4–OH solution were mixed and diluted with H2O to a final volume of 15 mL. Then the reaction was shaken at rt for 1 h, then the crude product was purified by ultrafiltration spin columns (5000 Da cutoff) and was washed with 3 × 10 mL 1 : 1H2O : MeOH. The remaining orange liquid was placed into a 15 mL conical and lyophilized until dry.
Animal models
Animals were housed in polycarbonate cages and kept on a 12 h light/dark cycle with water and food given ad libitum. C57BL/6NCr male mice, 8–14 weeks old, weighing 20–30 g, were purchased from the National Cancer Institute (Frederick, MD, USA). Compounds 1–4 were dissolved in nanopure water and compound 5 was dissolved in 5% DMSO, 5% Tween-80 in 90% D5W solution. All the particle solutions were filtered through a 0.45 micron filter, and the dosage concentrations were determined after the filtration step. Approximate dosing solution concentration for compound 1 was 2.0 mM, both compounds 2 and 3 were 0.81 mM, compound 4 was 2.71 × 10−5 M, and compound 5 was 5.94 × 10−5 M. Tail vein injection was carried out and the injection volume was 100 μL of solution/25 g of mouse body weight. The mice were euthanized at 6, 12, 24, 48, 72, and 96 h time points via cardiac exsanguination under anesthesia (isoflurane). Urine and feces from 0–6 h were collected as they were produced in the cage. Feces from 6–12 h and 12–24 h were collected at the end of the time point from the cage. Urine from 6–12 h and 12–24 h were collected at the end of the time point by washing the cage with 20–30 mL nanopure water followed by lyophilization of the sample. The organs were collected at the end of each time point. All experimental procedures were approved by the Institutional Animal Care and Use Committee at Colorado State University.
ICP-MS analysis
Blood
Blood sample (0.5 mL) was measured out using a pipette in a 15 mL conical. Aqua regia (4 mL, approximately 3 : 1 HCl : HNO3) was added to the conical and allowed to be stored at room temperature for two days with the conical gently capped. Water was then added to a final volume of 5 mL. Organs/feces: The organs/feces were weighed out in 15 mL conical. Samples that weighted more than 0.3 g were digested with 4 mL of aqua regia at room temperature over two days then diluted with water up to 5 mL total volume. Samples that weighted less than 0.3 g were digested with 2 mL of aqua regia at room temperature over 2 days then diluted with water up to 2.5 or 3 mL total volume. Urine: liquid urine sample (typically 0.5 mL) was measured out using a pipette and lyophilized urine sample was treated as a dry powder. Aqua regia was added to the conical and incubated at room temperature for two days with the conical gently capped. Water was then added to a final volume of 5 mL. All the samples were sent to MidWest Laboratories, Inc. (Omaha, NE) for ICP-MS analysis.
Luminescence images
Following collection of tissues for biodistribution, hepatic luminescence in the liver was determined using a cryogenically cooled IVIS 100 imaging system coupled to a data acquisition computer running LivingImage software (PerkinElmer, Waltham, MA). A digital grayscale image was acquired followed by acquisition and overlay of a pseudocolor image representing the spatial distribution of detected photons emerging from within the liver after subtracting background luminescence. Signal intensity was quantified as the sum of all detected photons within the region of interest per second. The excitation (640 nm ± 25 nm) and emission (732.5 nm ± 37.5 nm) used the Cy 5.5 filters.
Results & discussion
A grand challenge in the adaptation of nanoparticles for clinical purpose is the development of robust, predictive structure–activity relationships for in vivo nanoparticle behaviors such as ADME and PK. To advance toward this goal, we establish in vivo properties for the structurally characterized nanoclusters Au25(SR)18 and Au102(SR)44 where SR is either the thiolate ligand used in the native synthesis (glutathione for Au25 and p-mercaptobenzoic acid for Au102) or ligand exchanged preparations of these clusters with mixtures of original ligand and tetraethylene glycol. We proceeded with these oligoethylene glycols because of literature showing that these molecules may suppress protein absorption thereby improving predictive biological properties, and also extend or allow the manipulation of blood PK.4,15,19,36–38
Predictive modeling
For each of the compounds listed in Fig. 1, we generated atomisitic models (Table 1). The inorganic portion of each atomistic model was taken from the X-ray crystal structure of either Au25 (ref. 39 and 40) or Au102.41 Ligand placement on exchanged models draws partially from our and other studies of structural ligand exchange, enabling some speculation as to the location of exchanged ligands.42 For instance, in Au25, the more exchangeable ligand sites are on those sulfur atoms that are closest to Au(0), and on Au102, ligand exchange occurs at solvent exposed Au or adjacent Au atoms. From these atomistic models, which are accurate for the as-synthesized product and a reasonable approximation as to the structure of ligand exchanged products, we calculated expected hydrodynamic diameter (with HYDROPRO29), net surface charge (by simple count), surface charge density (by PDB2PQR32,33), and hydrophobicity (by PyMOL).28 The results of these calculations are summarized in Table 1. While these parameters are widely understood as biochemically significant, previous studies have focused primarily on net charge and hydrodynamic diameter as determinants of PK and excretion mechanism.
Fig. 1.
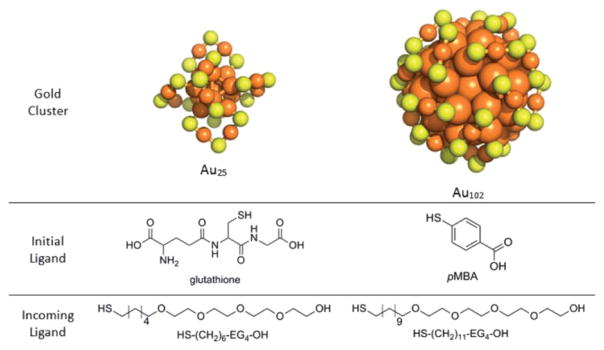
Structures of each molecular component used in the study. Orange spheres depict gold atoms and yellow spheres depict sulfur atoms. The carbon and hydrogen atoms of the ligand layer have been excluded for clarity.
Table 1.
Theoretical properties of compounds 1–5b
| Compound | Proposed targeted molecular formula | Calculated stokes diameter (nm)a | Net surface charge (e−) | Surface charge density (charge Å−2) | Hydrophobic surface area (%) | Expected clearance mechanism % renal/% RES |
|---|---|---|---|---|---|---|
| 1 | Au25(GSH)18 | 2.44 | 18− | 1.3 × 10−3 (−) | 63% | 100/0 |
| 2 | Au25(GSH)9 [S(CH2)6(EG)4OH]9 | 4.55 | 9− | 6.7 × 10−4 (−) | 72% | 60/40 |
| 3 | Au25(GSH)6 [S(CH2)6(EG)4OH]12 | 4.72 | 6− | 4.4 × 10−4 (−) | 74% | 60/40 |
| 4 | Au102(pMBA)44 | 3.28 | 44− | 6.6 × 10−3 (−) | 77% | 90/10 |
| 5 | Au102(pMBA)25 [S(CH2)11(EG)4OH]19 | 5.71 | 25− | 8.4 × 10−4 (−) | 79% | 40/60 |
Calculated stokes diameter is in serum.
Information on the Au102 1 : 1 exchanged compound can be found in the ESI.
Atomistic models are especially useful in the case of small nanoclusters because methods for characterizing hydrodynamic diameter and surface charge, such as dynamic light scattering and particle tracking, have large errors for particles substantially smaller than 5–10 nm hydrodynamic diameter. Other properties calculated from these atomistic models, such as surface charge density, and hydrophobicity, are more difficult to measure directly and may allow post-experimental explanation of observed in vivo properties and development of more sophisticated structure–activity relationships.
Synthesis and characterization of Au25, Au102 & exchange products
The Au25 and Au102 nanoclusters are now well characterized by multiple groups.39,43–45 The synthesis is described in greater detail in the experimental section. Au25(GSH)18 synthesis was by a novel method (manuscript in preparation) and Au102(pMBA)44 as previously described.46 Initial characterization of the Au25(GSH)18 by polyacrylamide gel electrophoresis used in this study suggested the presence of a single discrete product, but subsequent characterization of this product after mouse experimentation had already begun showed the presence of multiple products of which Au25(GSH)18 is suggested as the largest product as determined by the visible color of this and other products.47,48 The Au102(pMBA)44 product appears approximately pure as assessed by polyacrylamide gel electrophoresis (ESI, Fig. S1†). There is some uncertainty in the exact nature of this preparation, because the subset of any Au102(pMBA)44 preparation that crystallizes is small, and mass spec analysis suggests the presence of neighboring products.46,49
Oligoethylene glycol moiety containing (OEG, Fig. 1) modifications of the as-synthesized Au25 and Au102 were made with of 1 : 1 and 2 : 1 incoming : outgoing ligand exchanges of OEG compounds shown in Fig. 1 onto Au25 and Au102 cores as described in the Experimental section. The molecular formula assignment of the Au102-based compound (5) was based on the ratios of the aromatic 1H NMR signal (4H) versus the aliphatic 1H NMR signal (18H) (Fig. S16†). The Au102-based compound (exchanged with HS–(CH2)11–EG4–OH at 1 : 1 incoming : outgoing ratio) was also prepared for this study. Incomplete ADME/PK data and the theoretical properties of this compound can be found in the ESI (Fig. S3, S17, and Table S1†).
Average molecular formula assignments for the Au25-based compounds 2 and 3 were difficult to determine by NMR, because of overlapping 1H signals from glutathione and OEG. Therefore, the molecular formula of the Au25-based ligand exchanged compounds (2 and 3) are proposed formulae based on the assumption that the ligand exchange assumes equilibrium exchange conditions. This assumption may overestimate the amount of OEG on these ligand exchange products relative to the idealized models.
ADME/PK of Au25-based compounds
The ADME/PK activities of each Au25-based product (compounds 1, 2 and 3) were tested in 8–14 weeks old C57BL/6NCr male mice. Time dependent absorption, biodistribution, and excretion was determined by ICP-MS analysis of gold content in lung, liver, kidney, blood, urine and feces from three mice per time point.
Fig. 2 shows the blood drug levels of the Au25-based compounds 1, 2 and 3. The pharmacokinetic parameters calculated by noncompartmental analyses are shown in Table 2. The calculated circulation half lives of compounds 1, 2 and 3 are relatively similar at 20.1, 16.0, and 15.6 hours respectively. Interestingly, modification with increasing coverage of OEG ligand (which also leads to the reduction of GSH coverage), there is an increase in area under the curve (AUC), and a reduction in half-life, clearance and apparent steady state volume of distribution (Table 2). These findings help support the ability to predict the whole blood PK parameters by “tuning” these nanoparticles.
Fig. 2.
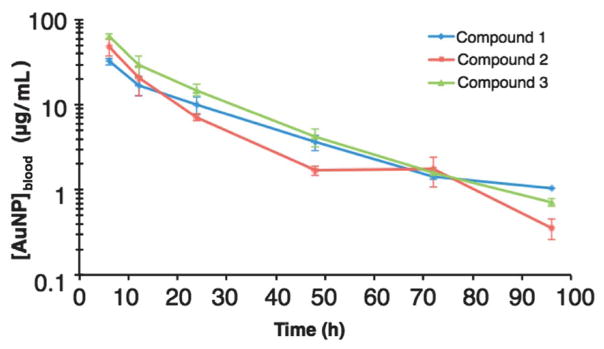
Blood drug concentration vs. time curves of Au25-based compounds. Data represents the mean of three animals per time point per compound.
Table 2.
Whole blood pharmacokinetic values of compounds 1, 2 and 3a
| Compound | Doseave (mg) | AUC0→96 h (μg mL−1)h | t1/22λ (h) | CL (mL h−1) | Vss (mL) |
|---|---|---|---|---|---|
| 1 | 2.16 | 847 | 20.1 | 2.47 | 53.4 |
| 2 | 0.923 | 1020 | 16.0 | 0.797 | 10.4 |
| 3 | 0.989 | 1480 | 15.6 | 0.542 | 8.02 |
Noncompartmental modeling was used for the calculation of pharmacokinetic parameters based on the composite data in Fig. 2.
Time dependent distribution of Au25-based compounds 1, 2 and 3 over a 96 hours time course are shown in Fig. 3. Notably compound 1 appears to accumulate in the kidneys over the course of the experiment, while compounds 2 and 3 (those with OEG) – also have accumulations in the kidney but even more accumulate in the liver. This change in distribution predicts the dominance of excretion into urine for compound 1 and in feces for compounds 2 and 3 observed (Fig. S2†). Analysis of excretion products, shown in Fig. 4, confirms this, meaning that a small change in particle surface composition can dramatically change excretion mechanism.
Fig. 3.
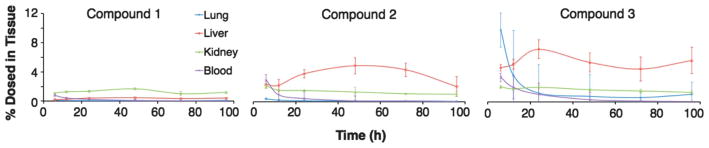
Percent of Au nanoparticles found (of total amount dosed) in tissue of Au25-based compounds in various tissues at noted post-injection times. See Fig. S11–S13† for the graphs zoomed in at the lower percentages.
Fig. 4.
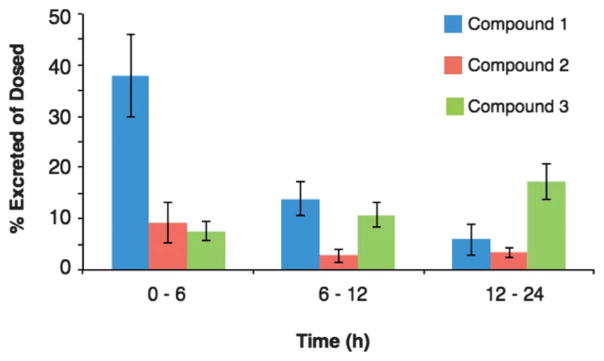
Percent of Au nanoparticles found (of total amount dosed) in excretion (urine and feces combined) for Au25-based compounds.
Long term, potentially irreversible accumulation in filtration associated organs such as the lungs, liver and spleen is a major concern emerging investigation of metal nanoparticles in preclinical models.50 Current studies suggest that smaller gold nanoparticles may be metabolized and excreted by hepatic mechanisms,18 while larger gold nanoparticles persist in the liver indefinitely.13 Metabolism of smaller particles may be by biologically sourced oxidative thiolate etching,51 perhaps by intrinsic GSH, which results in (–Au(I)–SR–)n oligomers similar to Au(I) based FDA approved rheumatoid arthritis drugs such as aurothiomalate (trade name Myochrisine.) The intrinsic luminescence of the Au25(SR)18 compound52,53 allowed observation of the hepatic uptake and clearance of the hepatically cleared Au25-based compound 3 over time. Fig. 5 shows the luminescence of compound 3 in livers at the noted time points after administration. We suggest that etching precedes excretion, but speculate that since the analogous rheumatoid arthritis drugs are not associated with long term metal accumulation, that the Au(I)-SR etching products will be excreted. This does appear consistent with analysis of feces over the course of the experiment which shows increasing concentrations of Au as the experiment progressed toward its 96 hours termination point.
Fig. 5.
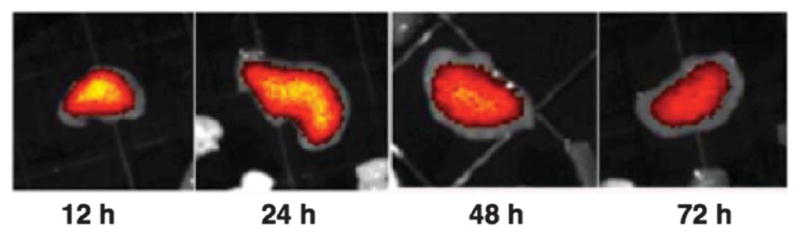
The luminescence within representative livers ex vivo at noted post-injection time points for compound 3.
With no OEG, particles locate primarily to the kidneys and urine – consistent with the other observations of GSH–Au, although we surprisingly observe more kidney residence than other studies.18 With an average of half a ligand shell of OEG (compound 2), particles locate primarily in the liver, and with an average of two thirds of a ligand shell of OEG (compound 3) the particles locate initially in the lungs (Fig. 3). While particles of similar size are known to locate to similar organ systems,12,14,54 we believe this is the first observation of particle modification destination based on a mixed ligand shell composition.
ADME/PK of Au102
Establishing the ADME/PK properties of the Au102(pMBA)44 based compounds 4 and 5 was attempted in the same manner as for the Au25(GSH)18 compounds 1, 2 and 3. Analysis of time-dependent biodistribution and excretion suggests that the Au102 compounds locate primarily to the liver and spleen, regardless of the composition of the ligand shell (Fig. 6 and 7). In each case, the hydrodynamic diameter of the compound predicts substantial renal clearance, and the accumulation in RES suggests substantial binding of proteins to these particles, increasing their hydrodynamic diameter resulting in RES accumulation. Surprisingly, the incorporation of OEG in these compounds did not change their distribution properties, indicating that in this case OEG did not suppress protein binding.
Fig. 6.
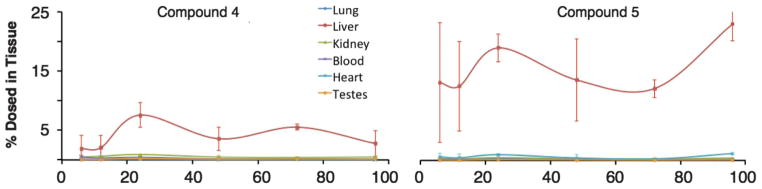
Percent of Au nanoparticles found (of total amount dosed) in tissue of Au102-based compounds in various tissues at noted post-injection times. See Fig. S14–S15† for the graph zoomed in at the lower percentages.
Fig. 7.
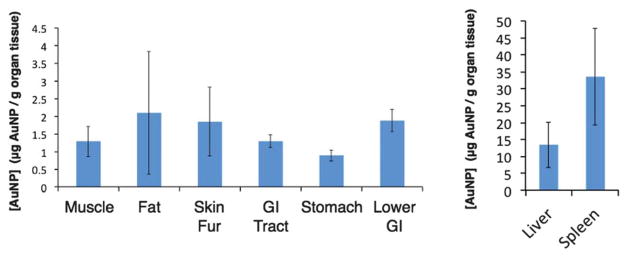
Concentration of compound 4 in various tissues at 24 h. The graph on the right shows the concentration of compound 4 in the liver and spleen on a different scale.
Analysis of excretion products for compounds 4 and 5 show excretion of 4.8% and 0.9% of the starting material within 12 hours. Fig. S3† shows the relative amounts of each in urine and feces at 6, 12 and 24 hours post injection. Notably, excretion mechanism does not appear to change with ligand shell modification.
Predictive SAR may enable design of nanoparticles with predictable excretion mechanism, biodistribution, metabolism and blood PK. Precise and predictable tuning of these properties is desirable. Nanoparticles are investigated as diagnostic imaging agents, therapeutic agents, and combined theranostic agents. Each of these modalities has a different optimal corporeal half life.20
The seminal work of Choi et al.4,5 suggested that the ADME/PK properties of inorganic nanoparticles and explicitly their excretion mechanism might be predicted by comparison to proteins with comparable hydrodynamic diameter, as long as the particles had a net surface charge of zero achieved either through zwitterionic ligands such as cysteine or neutral ligands such as oligoethylene glycols. Subsequent work from many groups demonstrate that nanoparticle ADME/PK is much more complex. For instance, particles with net-negative charge can avoid opsonization and protein corona formation which can dramatically increase the effective hydrodynamic diameter of nanoparticles, as judged by renal clearance. Thus, in some instances particles with a net negative charge behave as predicted by their hydrodynamic diameter,7,18,37,55 but in other instances do not.22 Moreover, the models for predictive ADME/PK do not yet account at all for the result of targeting molecules (RGD, folate, antibodies, etc) on ADME/PK, which is a serious limitation.
In this study five compounds were tested, all with hydrodynamic diameter less than 6.25 nm. Since these diameters are smaller than the 8 nm cutoff commonly cited for filtration through the glomerural wall of the kidney, and have net negative charge, substantial renal excretion is predicted. This is observed only for compound 1. To explain the excretion properties of compounds 2–5, we offer insights into SAR that arise from the atomistic models.
The distribution of compounds 2–5 into the RES is consistent with an increase in their effective hydrodynamic diameter above the ~8 nm cutoff for renal filtration that can result from opsonization (absorption of immuno-proteins) and protein corona formation (absorption of other proteins.) The net negative charge of compounds 2–5 and the addition of OEG are hypothesized to suppress these effects. Furthermore the apparent absorption of proteins to compounds 2 and 3 is especially surprising, as the OEG added to these compounds is hypothesized to decrease protein absorption compared to compound 1 because of several other studies that show decrease in protein binding as a result of OEG addition15,56
A possible explanation for this unpredicted behavior arises from analysis of hydrophobic surface area in the atomistic models. Hydrophobic interactions are well known to drive substantial specific and non-specific biological interactions. Larger relative amounts of hydrophobic surface area on a nanoparticle might provoke both specific (immuno-) and nonspecific protein nanoparticle aggregation. Table 1 shows the calculated percentage of hydrophobic surface area for each compound, calculated in PyMOL as described in the methods section. The amount of hydrophobic surface area increases after OEG addition because the OEG used in this study incorporates a C6 or C11 linker, separating the tetraethylene glycol functionality from the metal core. When packed into a monolayer, for instance on a sufficiently large {111} facet, this aliphatic region is buried and not solvent accessible. In the case of the small nanoclusters used in this study, two aspects may expose the aliphatic region: first, the ligand shell is a mixed monolayer of GSH and OEG, and this may allow solvent exposure of the aliphatic region. Second, the high radius of curvature of the particles may expose the aliphatic region even in areas where the surface is essentially OEG saturated. Thus, especially in the case of compounds 2 and 3, the Au25-based nanoclusters, with an inorganic diameter of 1.1 nm, are the smallest OEG passivated particles studied in vivo so far, and small size and mixed ligand shell may account in large part for the unpredicted results.
The blood pharmacokinetics of the GSH protected particles indicates a substantially longer blood half-life than suggested by a simple hydrodynamic diameter calculation and comparison to naturally occurring proteins of similar size. For instance, the 2.44 nm hydrodynamic diameter of compound 1 is comparable to that of the inulin with a 3.0 nm hydrodynamic diameter.4,57 These molecules have respective circulation half life of 20.1 hours and 9 minutes, respectively. The high density of negative charge on compound 1 may account for the long circulation half life, because glomerular filtration is faster for neutral or positively charged compounds.58 Other published examinations of the properties of injected GSH protected nanoparticles show similarly extended circulation times with substantial renal clearance, indicating that this somewhat surprising result is not artifactual.18
Conclusions
For the first time, the circulation properties of nanomaterials characterized in terms of molecular formula are examined. We show that the characteristics (i.e., circulation half life and especially biodistribution) of such particles depend critically upon their surface structure, as inferred from putative molecular formula atomistic models.
In aggregate, our results suggest that blood PK, distribution and excretion mechanism depend not only on the widely recognized parameters of net charge and hydrodynamic diameter, but also on exposed hydrophobic surfaces and surface charge density. Additionally, the specific 3-D structure of the organic portion of the cluster may also play an important role. Furthermore, we establish SAR properties for atomistically modeled particles, showing that renal clearance, transient lung accumulation, and RES accumulation are all highly sensitive to surface structure of nanoclusters, if not actual 3-D relationships of ligands. The increasingly precise surface modifications that now appear available42 for some clusters may ultimately enable quantitative understanding.
We further show that the partial exchange with OEG in this instance increased the hydrophobicity of the nanoparticles, resulting in either opsonization or protein corona formation, resulting in particle entrapment in the reticuloendothelial system. These results in whole are superficially contradictory to many others that suggest that OEG increases circulation lifetime and biocompatibility, primarily by suppressing (rather than increasing as we observe here) protein absorption. The superficial contradiction may be explained in part by the larger nanoparticle sizes (with decreased radius of curvature) used in other studies.
Supplementary Material
Acknowledgments
We thank Xi Chen and Hannu Häkkinen for sharing unpublished coordinates of DFT-optimized Au25(GSH)18 – cluster. The authors thank the CSU Cancer Supercluster for supporting this work. This work was also supported by startup funds from Colorado State University. This work was also supported by a University of Colorado Shared Resource Grant. This research was conducted while CJA was a New Investigator in Alzheimer’s Disease Grant recipient from the American Federation for Aging Research. Funding Sources: CSU Chemistry startup, CSU Cancer Supercluster, Univ CO Shared Resource Grant (P30CA046934).
Footnotes
Electronic supplementary information (ESI) available: The polyacrylamide gel that shows the purity of Au102pMBA44, excretion graphs for compounds 1–5, atomistic models of the Au25 and Au102-based compounds, the zoomed in versions of Fig. 3 and 6, 1H NMR of compound 5, information on the Au102 1 : 1 exchange compound, and blood drug concentration vs. time curves of Au102-based compounds. See DOI: 10.1039/c3nr03121g
Contributor Information
Daniel L. Gustafson, Email: daniel.gustafson@colostate.edu.
Christopher J. Ackerson, Email: ackerson@mail.colostate.edu.
Notes and references
- 1.Hainfeld JF, Slatkin DN, Focella TM, Smilowitz HM. Br J Radiol. 2006;79:248–253. doi: 10.1259/bjr/13169882. [DOI] [PubMed] [Google Scholar]
- 2.Hainfeld JF, O’Connor MJ, Dilmanian FA, Slatkin DN, Adams DJ, Smilowitz HM. Br J Radiol. 2011;84:526–533. doi: 10.1259/bjr/42612922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghann WE, Aras O, Fleiter T, Daniel MC. Langmuir. 2012;28:10398–10408. doi: 10.1021/la301694q. [DOI] [PubMed] [Google Scholar]
- 4.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, Frangioni JV. Nat Nanotechnol. 2010;5:42–47. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang T, Murray R. J Phys Chem B. 2001;105:12498–12502. [Google Scholar]
- 7.Zhou C, Long M, Qin Y, Sun X, Zheng J. Angew Chem, Int Ed. 2011;50:3168–3172. doi: 10.1002/anie.201007321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bresee J, Maier KE, Melander C, Feldheim DL. Chem Commun. 2010;46:7516–7518. doi: 10.1039/c0cc02663h. [DOI] [PubMed] [Google Scholar]
- 9.Cherukuri P, Glazer ES, Curley SA. Adv Drug Delivery Rev. 2010;62:339–345. doi: 10.1016/j.addr.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, Bardhan R, Bartels M, Perez-Torres C, Pautler RG, Halas NJ, Joshi A. Mol Cancer Ther. 2010;9:1028–1038. doi: 10.1158/1535-7163.MCT-09-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bardhan R, Lal S, Joshi A, Halas NJ. Acc Chem Res. 2011;44:936–946. doi: 10.1021/ar200023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonavane G, Tomoda K, Makino K. Colloids Surf, B. 2008;66:274–280. doi: 10.1016/j.colsurfb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Lasagna-Reeves C, Gonzalez-Romero D, Barria MA, Olmedo I, Clos A, Sadagopa Ramanujam VM, Urayama A, Vergara L, Kogan MJ, Soto C. Biochem Biophys Res Commun. 2010;393:649–655. doi: 10.1016/j.bbrc.2010.02.046. [DOI] [PubMed] [Google Scholar]
- 14.De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJ, Geertsma RE. Biomaterials. 2008;29:1912–1919. doi: 10.1016/j.biomaterials.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 15.Huynh NT, Roger E, Lautram N, Benoît J-P, Passirani C. Nanomedicine. 5:1415–1433. doi: 10.2217/nnm.10.113. [DOI] [PubMed] [Google Scholar]
- 16.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Nano Lett. 2010;9:1909–1915. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 17.Sadauskas E, Wallin H, Stoltenberg M, Vogel U, Doering P, Larsen A, Danscher G. Part Fibre Toxicol. 2007;4:10. doi: 10.1186/1743-8977-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson CA, Salleng KJ, Cliffel DE, Feldheim DL. Nanomedicine. 2013;9:257–263. doi: 10.1016/j.nano.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Cho WS, Cho M, Jeong J, Choi M, Cho HY, Han BS, Kim SH, Kim HO, Lim YT, Chung BH, Jeong J. Toxicol Appl Pharmacol. 2009;236:16–24. doi: 10.1016/j.taap.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 20.Cheng Z, Zaki AA, Hui JZ, Muzykantov VR, Tsourkas A. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumura Y, Maeda H. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 22.Simpson CA, Huffman BJ, Gerdon AE, Cliffel DE. Chem Res Toxicol. 2010;23:1608–1616. doi: 10.1021/tx100209t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Y, Neuss S, Leifert A, Fischler M, Wen F, Simon U, Schmid G, Brandau W, Jahnen-Dechent W. Small. 2007;3:1941–1949. doi: 10.1002/smll.200700378. [DOI] [PubMed] [Google Scholar]
- 24.Pan Y, Leifert A, Ruau D, Neuss S, Bornemann J, Schmid G, Brandau W, Simon U, Jahnen-Dechent W. Small. 2009;5:2067–2076. doi: 10.1002/smll.200900466. [DOI] [PubMed] [Google Scholar]
- 25.Walter M, Akola J, Lopez-Acevedo O, Jadzinsky PD, Calero G, Ackerson CJ, Whetten RL, Grönbeck H, Häkkinen H. Proc Natl Acad Sci U S A. 2008;105:9157–9162. doi: 10.1073/pnas.0801001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin R. Nanoscale. 2010;2:343–362. doi: 10.1039/b9nr00160c. [DOI] [PubMed] [Google Scholar]
- 27.Schüttelkopf AW, van Aalten DMF. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 28.deLano W. PyMol User Manual [Google Scholar]
- 29.Ortega A, Amoròs D, Garcîa de la Torre J. Biophys J. 2011;101:892–898. doi: 10.1016/j.bpj.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chien S, Usami S, Taylor HM, Lundberg JL, Gregersen MI. J Appl Physiol. 1966;21:81–87. doi: 10.1152/jappl.1966.21.1.81. [DOI] [PubMed] [Google Scholar]
- 31.Durchschlag H, Zipper P. In: Ultracentrifugation. Lechner MD, editor. Springer; Darmstadt: 1994. pp. 20–39. [Google Scholar]
- 32.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Klebe G, Baker NA. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi I, Kim YK, Min DH, Lee S, Yeo WS. J Am Chem Soc. 2011;133:16718–16721. doi: 10.1021/ja2083587. [DOI] [PubMed] [Google Scholar]
- 36.Tong L, Wei Q, Wei A, Cheng JX. Photochem Photobiol. 2009;85:21–32. doi: 10.1111/j.1751-1097.2008.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson CA, Agrawal AC, Balinski A, Harkness KM, Cliffel DE. ACS Nano. 2011;5:3577–3584. doi: 10.1021/nn103148x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D, Park S, Lee JH, Jeong YY, Jon S. J Am Chem Soc. 2007;129:7661–7665. doi: 10.1021/ja071471p. [DOI] [PubMed] [Google Scholar]
- 39.Heaven MW, Dass A, White PS, Holt KM, Murray RW. J Am Chem Soc. 2008;130:3754–3755. doi: 10.1021/ja800561b. [DOI] [PubMed] [Google Scholar]
- 40.Zhu M, Aikens CM, Hollander FJ, Schatz GC, Jin R. J Am Chem Soc. 2008;130:5883–5885. doi: 10.1021/ja801173r. [DOI] [PubMed] [Google Scholar]
- 41.Jadzinsky PD, Calero G, Ackerson CJ, Bushnell DA, Kornberg RD. Science. 2007;318:430–433. doi: 10.1126/science.1148624. [DOI] [PubMed] [Google Scholar]
- 42.Heinecke CL, Ni TW, Malola S, Mäkinen V, Wong OA, Häkkinen H, Ackerson CJ. J Am Chem Soc. 2012;134:13316–13322. doi: 10.1021/ja3032339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaaff T, Knight G, Shafigullin M, Borkman R, Whetten RL. J Phys Chem B. 1998;102:10643–10646. [Google Scholar]
- 44.Muhammed MA, Verma PK, Pal SK, Kumar RC, Omkumar RV, Pradeep T. Eur J Chem. 2009;15:10110–10120. doi: 10.1002/chem.200901425. [DOI] [PubMed] [Google Scholar]
- 45.Tofanelli MA, Ackerson CJ. J Am Chem Soc. 2012;134:16937–16940. doi: 10.1021/ja3072644. [DOI] [PubMed] [Google Scholar]
- 46.Wong OA, Heinecke CL, Simone AR, Whetten RL, Ackerson CJ. Nanoscale. 2012;4:4099. doi: 10.1039/c2nr30259d. [DOI] [PubMed] [Google Scholar]
- 47.Schaaff T, Whetten RL. J Phys Chem B. 2000;104:2630–2641. [Google Scholar]
- 48.Negishi Y, Nobusada K, Tsukuda T. J Am Chem Soc. 2005;127:5261–5270. doi: 10.1021/ja042218h. [DOI] [PubMed] [Google Scholar]
- 49.Levi-Kalisman Y, Jadzinsky PD, Kalisman N, Tsunoyama H, Tsukuda T, Bushnell DA, Kornberg RD. J Am Chem Soc. 2011;133:2976–2982. doi: 10.1021/ja109131w. [DOI] [PubMed] [Google Scholar]
- 50.Arvizo R, Bhattacharya R, Mukherjee P. Expert Opin Drug Delivery. 2010;7:753–763. doi: 10.1517/17425241003777010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schaaff T, Whetten RL. J Phys Chem B. 1999;103:9394–9396. [Google Scholar]
- 52.Wu Z, Jin R. Nano Lett. 2010;10:2568–2573. doi: 10.1021/nl101225f. [DOI] [PubMed] [Google Scholar]
- 53.Templeton A, Chen S, Gross S, Murray RW. Langmuir. 1999;15:66–76. [Google Scholar]
- 54.Semmler-Behnke M, Kreyling WG, Lipka J, Fertsch S, Wenk A, Takenaka S, Schmid G, Brandau W. Small. 2008;4:2108–2111. doi: 10.1002/smll.200800922. [DOI] [PubMed] [Google Scholar]
- 55.Mattoussi H, Rotello VM. Adv Drug Delivery Rev. 2013;65:605–606. doi: 10.1016/j.addr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 56.Monopoli MP, Aberg C, Salvati A, Dawson KA. Nat Nanotechnol. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 57.Prescott LF, McAuslane JAN, Freestone S. Eur J Clin Pharmacol. 1991;40:619–624. doi: 10.1007/BF00279982. [DOI] [PubMed] [Google Scholar]
- 58.Brenner BM, Hostetter TH, Humes HD. Am J Physiol. 1978;234:F455–F460. doi: 10.1152/ajprenal.1978.234.6.F455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.