

Abstract
Sequestration of Plasmodium falciparum-infected erythrocytes in host blood vessels is a key triggering event in the pathogenesis of severe childhood malaria, which is responsible for about one million deaths every year1. Sequestration is mediated by specific interactions between members of the P. falciparum erythrocyte membrane protein 1 (PfEMP1) family and receptors on the endothelial lining2. Severe malaria is associated with expression of specific PfEMP1 subtypes containing domain cassettes (DC) 8 and 133, but the endothelial receptor for parasites expressing these proteins was unknown4,5. Here, we identify endothelial protein C receptor (EPCR), which mediates cytoprotective effects of activated protein C6, as the endothelial receptor for DC8 and DC13 PfEMP1. We show that EPCR binding is mediated through the N-terminal cysteine-rich interdomain region (CIDRα1) of DC8 and group A PfEMP1 subfamilies and that CIDRα1 interferes with protein C binding to EPCR. This PfEMP1 adhesive property links P. falciparum cytoadhesion to a host receptor involved in anticoagulation and endothelial cytoprotective pathways and has implications for understanding malaria pathology and the development of new malaria interventions.
Each P. falciparum parasite genome harbors about 60 var genes encoding different PfEMP1 types, enabling the parasite to attach infected erythrocytes to different receptors on the vascular lining2. This attachment drives malaria pathologies, but also prevents passage of mature-stage infected erythrocytes through the spleen where they are destroyed. Based on the var 5′ upstream regions (UPS), PfEMP1 can be divided into groups A–E. In addition, the Duffy binding-like (DBL) and CIDR adhesion domains are subdivided into 147 sub-classes (e.g. CIDRα1.1)7. Despite a high rate of var gene recombination many tandem domain arrangements, called domain cassettes (DC), have been maintained through evolution, and are therefore thought to be of functional importance. The best example is DC2 aka VAR2CSA, which mediates binding in the placenta8, and it is of key importance to pathogenesis of pregnancy malaria. Severe malaria in children is associated with expression of a subset of PfEMP1 molecules characterized by DC8 (a unique group B/A chimeric gene) and DC13 (group A)3, but the endothelial receptor for these proteins has remained undefined4,5.
To identify the DC8-PfEMP1 receptor, we produced a full-length DC8-containing PfEMP1 using the var gene IT4var20 from the FCR3/IT4 parasite. This 288 kDa His-tagged recombinant protein (rIT4VAR20) was screened against an array of 2505 full-length human plasma membrane proteins expressed on HEK293 cells (Table S1) using the Retrogenix Cell Microarray. One specific hit for rIT4VAR20 identified endothelial protein C receptor (EPCR) as a potential binding partner9. EPCR is encoded by PROCR and is expressed on endothelial cells in most tissues10. Protein C (PC) binds EPCR promoting conversion to activated PC (APC)11. On endothelial cells, APC cleaves Protease Activated Receptor 1 (PAR1) resulting in broad endothelial cytoprotective12 and anti-inflammatory effects13. In the absence of EPCR/APC engagement, PAR1 activation can result in barrier-disruptive effects and activation of pro-inflammatory pathways13. In plasma, soluble APC exerts anticoagulative effects by proteolytic inactivation of blood coagulation Factors Va and VIIIa13. To identify the EPCR-binding region in DC8-PfEMP1 variants, we expressed individual recombinant protein domains from IT4VAR20 and two other DC8 variants (IT4VAR19 and 3D7-PFD0020c) and evaluated binding to rEPCR by ELISA (Fig. 1A). For all three proteins, EPCR binding mapped to the CIDRα1.1 domain within DC8 and not other extracellular domains. Using surface plasmon resonance (SPR) (Figs. 1C and S2), the binding kinetics of the IT4VAR20_CIDRα1.1::EPCR interaction (KD ~29 nM) was similar to that of the full-length protein (KD ~10 nM). This affinity is comparable to the binding of APC to EPCR for which a KD of 32nM has been reported14. These data confirmed that the DC8 CIDRα1.1 domain binds ECPR with a high and physiologically relevant affinity.
Fig. 1. Binding between recombinant PfEMP1 and EPCR.

a) Annotation of proteins (coloured boxes) and their ability to bind EPCR by ELISA (+/−).
b) EPCR binding (ELISA OD, ±s.d.) of 30 recombinant PfEMP1 proteins.
c) SPR sensorgrams of PfEMP1 binding to EPCR and CD36. Binding was tested in two-fold dilutions of PfEMP1 from 125nM for rIT4VAR20, 0.5μM for DD2VAR32 and DD2VAR01 and 1μM for the rest. Green: protein infusion.
d) Sensorgrams for two chips coated with EPCR. Red: EPCR coated chip sequentially flushed with DC8 CIDRα1.1 (2μM), buffer, APC (2μM) and buffer. Blue: EPCR coated chip flushed with APC only.
Previous work has shown that the N-terminal CIDR domain of PfEMP1 has diverged in sequence and functional properties. While group B and C PfEMP1 variants bind CD36, group A and B/A PfEMP1 variants do not15. To investigate EPCR-binding, a panel of 28 different CIDR variants representing both CD36-binding (CIDRα2-6) and non-CD36 binding sybtypes (CIDRα1, δ subtypes) were tested. Also included were full-length VAR2CSA and VAR3 (DC3) (Fig. 1B). All of the CIDRα1.1 and CIDRα1.4 proteins, representing DC8 and DC13 from different parasite genomes bound EPCR (Figs. 1 and S2). In addition, the group A CIDRα1.5 (DC15) and CIDRα1.7 proteins also bound EPCR. By contrast, other group A CIDR domains representing DC1 (CIDRα1.2/1.3), DC4 (CIDRα1.6), and PfEMP1 variants that facilitate rosetting between infected and uninfected erythrocytes (CIDRδ)16 did not bind EPCR; neither did group B CIDRα domains, VAR2CSA or VAR3 (Fig. 1B). As expected, the two group B CIDRα domains bound with high affinity to CD36 (KD ~12 nM) (Figs. 1C and S2). These results show that most PfEMP1 proteins have diverged into CD36 binding (group B and C) and EPCR binding types (group A and DC8), while the binding properties of a small subset of group A variants containing CIDRα1.2, 1.3, or 1.6, or atypical CIDR sequence types (CIDRβ,γ,δ) remain unknown.
Next we tested whether parasites expressing native DC8 PfEMP1 bound EPCR on endothelial cells. An FCR3/IT parasite line expressing IT4VAR20 was generated (Fig. S3) and found to bind brain derived endothelial cells via the PfEMP1::EPCR interaction, as demonstrated by reversal of binding by recombinant EPCR (rEPCR), anti-EPCR-antibodies and antibodies against either the full-length rIT4VAR20 or the IT4VAR20-CIDRα1.1 domain (Figs. 2 and S6). Anti-ICAM-1 antibodies, recombinant ICAM-1 or antibodies against heterologous PfEMP1 domains did not inhibit binding (Figs. 2 and S5). The CSA binding of VAR2CSA-expressing parasites or transformed human bone marrow endothelial cell/PECAM1 binding of IT4VAR02 (DC16-DC5 PfEMP1) expressing FCR3/IT parasites was not inhibited by rEPCR, anti-EPCR-antibodies or the antibodies against recombinant IT4VAR20 (Fig. 2C). To confirm these findings, additional three DC8-expressing parasite lines (FCR3/IT IT4VAR19, FCR3/IT IT4VAR06 and 3D7 PFD0020c) were generated (Fig. S3) and all found to bind brain-derived endothelial cells via EPCR (Table S3). Previous work has shown that DC8- and DC13-variants selected on brain endothelial cells also bind to non-brain microvascular endothelial cells from heart and lung4,5. Binding of the FCR3 IT4VAR19b parasite line (described in 4) to brain, heart, lung and bone marrow endothelial cells was evaluated and found to be mediated by EPCR (Table S3). Altogether, these results demonstrate cytoadhesion of DC8 PfEMP1 expressing parasites via EPCR on endothelial cells of diverse tissue origin.
Fig. 2. Binding of DC8 expressing parasites to human brain microvascular endothelial cells (HBMEC).
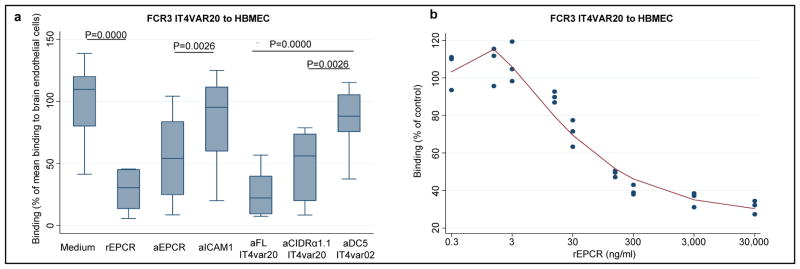
Binding characteristics of IT4VAR20 expressing FCR3 parasites shown as a) median binding (percentiles: 10,25,75,90) of n=10 independent experiments in relation to the mean control binding in medium and b) triplicate measures in one representative experiment of three. Binding was measured with rEPCR (60 μg/ml or as indicated) and anti-receptor IgG (20 μg/ml) or anti-PfEMP1 IgG (500 μg/ml). P values for comparisons were evaluated using wilcoxon test for paired differences.
Next, we explored the EPCR binding phenotype of two parasite isolates (1983 and 1965) from African children with severe malaria. As reported previously3, the ex vivo var transcript profiles of these isolates were dominated by DC8-coding transcripts. The isolates cryopreserved at diagnosis were thawed and selected on rEPCR before the var transcript profile and binding phenotypes were established. The analyses showed that the parasites predominantly expressed the DC8-encoding genes also expressed at high levels when the children were diagnosed (Fig. S4), and that the parasite lines bound to brain endothelial cells via EPCR (Fig. 2C). This suggests that the parasites causing severe malaria in these children expressed EPCR-binding PfEMP1. To extend this observation, the binding phenotypes of 15 severe malaria, 5 uncomplicated and 10 mild malaria isolates were compared (Tables 1 & S4). Parasites were snap frozen at diagnosis, thawed and exposed to short-term in vitro culture (2–12 parasite divisions, without rEPCR selection) to give parasites an opportunity to recover and expand in sufficient numbers for the binding assay. As previously reported17, the binding to ICAM-1 was higher in parasites causing severe malaria than in the control parasites (P<0.03 and P=0.0016 for parasites from children with uncomplicated and mild malaria, respectively). Among parasites from patients with severe malaria, EPCR-binding was significantly higher than ICAM-1 binding as measured in two independent assays (P=0.01 and P<0.05, respectively), and EPCR-binding was significantly higher among parasites causing severe malaria than in control parasites (P=0.0078 and P=0.0009 for parasites from children with uncomplicated and mild malaria, respectively). These data show that EPCR-binding is a common phenotype of parasites causing severe malaria, albeit studies including a larger number of children with different clinical manifestations will be needed to disentangle the role of EPCR binding in different forms of severe malaria.
Table 1.
Binding (median units* and 95CI) to human endothelial cells (HBMEC) and transfected CHO cells of parasite lines from patients with severe or mild malaria
| Parasites | HBMEC binding | HBMEC via EPCR§ | HBMEC via ICAM1§ | CHO-ICAM1$ | CHO-CD36$ |
|---|---|---|---|---|---|
| Severe malaria (N=15) | 58 [40;80] | 40# [20;50] | 27 [8;48] | 26 [10;42] | 19 [8;34] |
| Uncomplicated (N=5) Pvalue& | 25 [14;34] (P=0.021) | 7 [4;19] (P=0.0078) | 4 [1;20] (P=0.027) | 4 [0;6] (P=0.020) | 21 [12;25] (P=0.75) |
| Mild malaria (N=10) Pvalue% | 24 [12;45] (P=0.0039) | 8 [4;17] (P=0.0009) | 2 [0;6] (P=0.0016) | 0 [0;1] (P=0.0008) | 10 [2;37] (P=0.39) |
100 units represent optimal binding defined as the HBMEC binding observed with the IT4 parasite line selected to express IT4VAR20 (Figure 2).
HBMEC binding inhibited by incubating parasites in the presence of soluble EPCR (30 ug/ml) or anti-ICAM-1 monoclonal antibody (50 ug/ml) given as delta binding units (HBMEC binding subtracted binding in the presence of soluble EPCR or anti-ICAM1).
Binding to Chinese hamster ovary cells (CHO) transfected with ICAM-1 or CD36.
Among parasites from patients with severe disease EPCR binding statistically significantly higher (Wilcoxon test for paired data) than HBMEC ICAM-1 binding (P=0.01) or binding to CHO-ICAM1 (P<0.05).
Wilcoxon rank sum test for comparison between parasites from the two patients groups.
The pathogenesis of complicated malaria is far from completely understood and it is likely that there are several paths leading to severe outcomes. However, most of the pathology seems to be linked to an unrestrained inflammatory reaction in response to unchecked parasite growth. EPCR has been described as the “cell surface conductor of cytoprotective coagulation factor signaling”13 and interestingly, APC:EPCR/PAR1-induced signaling down-regulates five of the 13 previously described PfEMP1 ligands, including ICAM-16. We used SPR to investigate whether PfEMP1 might interfere with EPCR-mediated activation of APC. Pre-incubation of EPCR with either DC8 CIDRα1.1 or DC13 CIDRα1.4 domains prevented binding of APC (Figs. 1D and S2B). This indicated that the DC8 domains and APC competed for the same binding site. This notion was confirmed by showing that monoclonal antibody RCR-25218, an antibody that blocks APC binding to EPCR, also prevented parasite binding to endothelial cells (Fig. S5). However, the affinity (Kd = 1μM) of the commercial human purified APC used here was lower than reported earlier for native APC14. Thus, the extent to which native soluble APC can inhibit parasite binding remains to be explored. Taken together, these findings indicate that DC8-expressing parasite adhesion to brain endothelial cells is mediated by PfEMP1 interacting with EPCR at or close to the RCR-252 antibody binding site. It further suggests that PfEMP1 bind EPCR near or at the same region as APC. Thus, EPCR-mediated parasite cytoadhesion could interfere with activation of cytoprotective and anti-inflammatory pathways, which in turn may contribute to severe malaria pathology (Fig. S1). Soluble EPCR is shed from endothelial cells by metalloproteinase TACE/ADAM17 and circulates in plasma19. The levels of soluble EPCR differ between individuals and high plasma levels of EPCR (up to 500 ng/ml) are associated with specific PROCR haplotypes20, which in turn are associated with a higher risk of contracting venous thrombosis21. Of interest, addition of recombinant soluble EPCR at levels between 15–300 ng/ml showed a progressively higher inhibition of the binding between DC8 expressing parasites and endothelial cells (Fig. 2B). It is a possibility worthy of further exploration that human polymorphism in EPCR is a balanced polymorphism protecting individuals against severe malaria at the expense of a higher risk of thrombotic disease.
Our findings identify EPCR-binding as a property of PfEMP1. Previous studies aiming to link parasite binding phenotypes to disease outcome have not tested for binding to EPCR and results have been conflicting. Most consistently, ICAM-1 binding and rosetting have been identified as virulence factors22. However, the facts that DC8 and other EPCR binding variants are preferentially expressed young children with limited malaria immunity23,24 and the ubiquity of endothelial cells expressing EPCR, suggest that CIDRα1::EPCR-mediated P. falciparum cytoadhesion is the major virulence phenotype for severe malaria. Intriguingly, in a small number of case reports a recombinant form of APC (Xigris)25–27 was used to successfully treat severe malaria infections with remarkable recoveries. The results presented here open new avenues for studies of malaria pathogenesis and possibilities for development of new adjunct therapy and vaccines to treat or protect children from malaria death.
Methods
Recombinant PfEMP1 proteins and anti-PfEMP1 antibodies
The full-length exon 1 sequences of FCR3 IT4var20 and IT4var13 genes were codon-optimized for expression in Trichoplusia ni cells using the software GeneOptimizer§. DNA constructs containing a His-tag at the C-terminal end were synthesized by Geneart (Regensburg, Germany) and subcloned into the baculovirus expression vector pAcGP67-A (BD Biosciences) modified to contain a His-tag at the C-terminal end of the construct, by Geneart (Regensburg, Germany). Other insect cell produced proteins were generated by PCR from genomic DNA using primers shown in Table S2, as described in 31 5,4. Recombinant Esherichia coli proteins were generated as described previously4. Rat and rabbit immunizations and IgG preparations were as described in 5. Animal immunizations were conducted according to and approved by the Danish national Animal Experiments Inspectorate.
Human receptor interaction microarray screening
Screening for human protein ligands of DC8 PfEMP1 was performed using the Retrogenix Cell Microarray technology. Detection of recombinant His-tagged PfEMP1 interactions with the vector transfected HEK293 cell glass slide microarray was first optimized using an ICAM binding recombinant full-length IT4VAR13 protein (rIT4VAR13).
Retrogenix’s expression vectors encoding ICAM-1 or CD28 (negative control) were each spotted onto glass slides, and a human HEK293 monolayer was grown on top and reverse-transfected using Retrogenix’s optimized methodology. Slides were incubated with 10.5 ug/ml His-tagged rIT4VAR13, and interactions were detected using a mouse anti-His antibody (Millipore) followed by an AlexaFluor 647 anti-mouse antibody (Life Technologies). Slides were then imaged for fluorescence and analysed using ImageQuant software (GE).
For rIT4VAR20 receptor screening, 2505 expression vectors, each encoding a full-length human plasma membrane protein, were arrayed across 7 microarray slides, and human HEK293 cells were reverse-transfected. See Table S1 for the list of proteins screened. Each slide was incubated with 10.5 μg/ml rIT4VAR20. Detection of binding was performed using the mouse anti-HIS antibody followed by the AlexaFluor 647 anti-mouse antibody. Protein ‘hits’ were identified by visual inspection using ImageQuant software (GE). This primary screen produced a total of 16 hits to rIT4VAR20. The vectors encoding each of the 16 hits were sequenced, confirming their identities. A confirmation/specificity screen was done, with each of the 16 vectors re-spotted and re-probed with rIT4VAR20-anti-HIS, rIT4VAR13-anti-His or anti-His antibody alone, in order to determine which hits were specific for rIT4VAR20. In this screen only one hit, EPCR (Gene Id: PROCR) was specific for rIT4VAR20.
ELISA
MaxiSorp immunoplates (Nunc) were coated with recombinant human EPCR (250 ng/well, Sino Biological, 13320-H08H, consisting of the extracellular domain of human EPCR (PROCR) Met1-Thr209 fused with C-terminal poly-histidine tag) in PBS buffer pH 7,4 and blocked with PBS 3% skimmed milk. PfEMP1 proteins were added at a concentration of 10 μg/ml per DBL or CIDR domain in PBS, 1% skimmed milk and incubated for one hour at 37°C with gentle shaking. Peroxidase conjugated anti-V5 antibodies (Invitrogen, R961-25) were added at a dilution of 1:3000 for one hour at 37°C (baculovirus produced proteins) or 0.375 μg/mL rabbit anti-StrepII antibody (Genscript #A00626 (E. coli produced proteins). Secondary incubation with 1:400 anti-rabbit IgG/HRP (Alpha Diagnostics #20321) was carried out for E. coli proteins. Plates were developed with phosphate solution with 0.012% H2O2 substrate and o-phenylenediamine (baculo) or TMB Substrate (Alpha Diagnostics # 80091)(E. coli). The colorimetric reaction was stopped with 3 M H2SO4 after 10 min and the optical density (OD) was measured at 492 nm (baculo) or 1X stop solution (Alpha Diagnostics #80100) and read at 450 nM (E. coli). Reactivity with EPCR was tested in triplicates twice for E. Coli produced proteins and at least twice in duplicates for insect cell produced proteins, results from one representative experiment is given.
Surface Plasmon Resonance (SPR)
SPR experiments were carried out using a BIAcore T100 instrument (GE Healthcare). All experiments were performed in 10 mM HEPES pH 7.5, 150 mM NaCl at 25°C. Recombinant EPCR was immobilized on a CM5 chip (GE Healthcare) by amine coupling to a total loading of 1200RU. Binding partners were buffer exchanged into 10 mM HEPES pH 7.5, 150 mM NaCl and dilution series were injected over the rEPCR coated chip. Injections were 240 seconds with 300 second dissociation. The chip surface was regenerated in between injections with a 60 second pulse of 10 mM NaOH. Repeat injections showed that this treatment left the rEPCR coated surface undamaged. The specific binding response to rEPCR was obtained by subtracting the response given by analytes to an uncoupled surface. The kinetic sensorgrams were fitted to a global 1:1 interaction model to allow calculation of ka, kd and KD using BIAevaluation software 2.0.3 (GE Healthcare).
Clinical parasite isolates
The clinical parasite isolates were collected during studies conducted in Korogwe District in northeastern Tanzania about 100 km from the Indian Ocean3. Children admitted to Korogwe District Hospital with malaria symptoms were enrolled after obtaining informed consent from a parent or guardian. Children were immediately subjected to a clinical investigation, and a blood sample was collected for diagnostic and research purposes, after which treatment was instigated according to the national guidelines. Samples were also collected from patients recruited in two study villages during malaria surveys and diagnosed by a rapid diagnostic test. None of these patients had severe symptoms, and they were all treated as outpatients at the village clinic. The study received ethical clearance from the Ethical review board of National Institute for Medical Research, Tanzania ((National Institute of Medical Research/HQ/R.8a/Vol.IX/559) and informed consent was obtained from all study participant and/or parents or guardians. The patient samples representing severe malaria parasites were selected from a database among patients having a Blantyre coma score < 3 and/or a blood hemoglobin < 5 g/dl (Table S4). The samples from patients with mild malaria were selected on the basis of having a reasonable high parasite density to ensure a high culture success rate. Knowledge about ex vivo var gene transcription was not available when selecting the parasites to be tested for EPCR binding. At diagnosis pelleted erythrocytes (25–50 μL) from venous blood were preserved for later parasite culturing by adding an equal volume of freezing medium (28% glycerol and 3% D-sorbitol in a 0.65% saline solution) and then snap frozen in liquid nitrogen. Cultures of live parasite isolates were re-established by thawing in an equal volume of 3.5% saline solution, washing in RPMI and cultured with fresh uninfected red blood cells to allow reinvasion. Parasites were cultured for 4–22 days without any selection for binding phenotype until sufficient material was available for the binding assays (Table S4).
Malaria parasite culture, phenotype determination and manipulation
Malaria parasites were cultured and PfEMP1 expression phenotypes were determined by quantitative PCR and var gene or var gene type specific primers to determine the transcript level of var genes in relation to two internal control genes seryl-tRNA synthetase and aldolase as well as by flow cytometry using PfEMP1 specific polyclonal antibodies as previously described 3,28. The var transcript distribution of 1965 and 1983 parasite isolates were determined by counting unique DBLα var tags from 48 clones obtained by capillary sequencing (Macrogen Incobtained) of PCR products generated as in 3 cloned into pCR2.1 vectors.
Parasites were selected for either specific PfEMP1 expression using antibodies to recombinant PfEMP1 constructs as described previously 28 and/or by selection for binding to human brain endothelial cells and rEPCR. 50 μg/ml rEPCR was added in three spots (10 μl) onto the bottom of a 25 cm2 culture flask and allowed to adsorp to the plastic surface (two hours, RT). The spotting-liquid was gently aspirated and the bottom of the flask washed twice with PBS and blocked with skimmed milk (3%) in PBS (ON, 4°C). The flask was washed twice with parasite medium and 3 ml infected erythrocytes (parasitaemia ≥2%, hematocrit 2%) was added. The flask was then gassed with parasite gas mixture for 30 sec, tightly capped and incubated on a rocking platform (10 RPM, one hour, 37°C). Unbound erythrocytes were washed away using parasite medium and binding to spots was assessed using an inverted microscope. 10 ml parasite medium and 200 μl fresh human type O+ erythrocytes was added to the flask, which was gassed and incubated ON (37°C). The parasitaemia was recorded the following day.
History of parasite lines
The FCR3 IT4VAR20 line was generated by alternate antibody selection using rat anti-rIT4VAR20 antibodies and panning on HBMEC. The FCR3 IT4var19a line was generated by rEPCR selection from an IT4VAR03 expressing FCR3 (FCR3 IT4var03) line 29 by three continuous selections for rEPCR binding as described above. The FCR3 IT4var19b and FCR3 IT4var06 lines were generated by limiting dilution from the ItG-ICAM-1 line panned on THBMEC. The FCR3 IT4VAR06 line was further enriched by selection for binding to rEPCR as described above. The 3D7 PFD0020c line was generated by alternating antibody (rat anti-DBLγ6_PFD0020c antibodies) and rEPCR binding selections. The FCR3 IT4var02 parasites were generated by antibody selection (rabbit anti-DC5-IT4var20 serum), and FCR3 VAR2CSA lines were generated by panning on BeWo cells. Parasite isolates 1965 and 1983 were obtained from two patients diagnosed with severe malaria (described in 3). The 1965 and 1983 parasite isolates were selected for rEPCR binding as described above.
Parasite adhesion to human endothelial cells and CHO-cells
Copenhagen assay (Figures 2 & S5 and Tables 1 and S3)
Human brain microvascular endothelial cells (HBMEC)29 were grown in Endothelial Cell Medium (1001, ScienCell) and Chinese Hamster Ovary (CHO) cells (CHO-wild type, CHO-CD36 and CHO-ICAM-1 from ATCC) were grown in RPMI 1640 supplemented with 10% Fetal Bovine Serum (Lonza), 0.125 μg/ml gentamycin (Lonza) and 2 mmol/l L-glutamine (Sigma-Aldrich). Wells in flat-bottomed 96-well plates destined for HBMEC were pre-coated with 100 μl fibronectin (2 μg/cm2) (1918-FN-02M, R&D Systems) (ON, 37°C). One hundred μl endothelial cells or CHO-cells with 80000 cells/ml were seeded per well and grown to a monolayer over two days. The day before the adhesion assay, ring-stage infected erythrocytes were radioactively labelled with tritiated hypoxanthine (3H) (Amersham; 8.75 MBq/ml packed erythrocytes) in hypoxanthine free parasite medium and cultured over night. Radioactively labelled late trophozoite and schizont stages were purified using Magnetic Cell Sorting (MACS, Miltenyi Biotec) and adjusted to 1.25×107 cells/ml in 2% FCS (in RPMI 1640). Prior to the adhesion assay, HBMECs were washed once with PBS followed by addition of 20 μl 2% FCS in RPMI/well. For binding inhibition, 20 μl of antibodies or proteins diluted in PBS were added to HBMEC in triplicates and PBS alone was added as a control. IgG-purified anti-PfEMP1 rat or rabbit antibodies were added to a final concentration of 500 μg/ml. Polyclonal human EPCR antibodies (AF2245, R&D) and monoclonal antibody to ICAM-1 (MCA1615EL, AbDSerotec) were added at a final concentration of 50 μg/ml. Monoclonal antibody (RCR-252) to human EPCR (HM2145, Hycult biotech) was added at final concentrations of 5, 10, 20 and 30 μg/ml. Recombinant EPCR and ICAM-1 (ADP4 R&D Systems) were added at a final concentration of 30000 ng/ml (titration with human EPCR: 0.3, 3, 30, 300, 3000 or 30000 ng/ml). 20 μl late-stage infected erythrocytes were added to HBMECs and co-incubated on a rocking table for 0.25 hour to 1.5 hours at 37°C. Unbound infected erythrocytes were removed with a washing robot (Biomek 2000, Beckman Coulter). Radioactive material was harvested (Filtermate Harvester, PerkinElmer) and scintillation liquid added (Microscint 20, PerkinElmer). Radioactivity was measured in counts per minute (CPM) (Topcount NXT, PerkinElmer). Adhesion to wells blocked with 2% human albumin (A-1887, Sigma-Aldrich) was also measured in addition to the total amount of radioactivity added per well (max-value). Adhesion was calculated as the proportion (%) of bound radioactively labelled infected erythrocytes out of the total amount of radioactively labelled infected erythrocytes added per well. The binding was then normalised into units by assigning the value 100 to the percentage radioactivity bound and recovered under optimal conditions. The binding of FCR3 IT4VAR02 and FCR3 VAR2CSA parasite lines to Transformed Human Bone Marrow Endothelial Cells (TrHBMEC) or BeWo cells, respectively, was performed as described above for the HBMEC binding assay. Anti-PECAM-1 antibodies (BBA7, R&D Systems) were used at a final concentration of 20 μg/ml.
Seattle assay (Figure 2d)
Endothelial cell culture and adhesion of the FCR3 IT4VAR19b parasites was performed as described 4. Endothelial cells were primary human pulmonary microvascular endothelial cells (HPMEC) and human cardiac microvascular endothelial cells (HCMEC) (ScienCell) and immortalized transformed human brain microvascular endothelial cells (THBMEC) and bonemarrow endothelial cells (CDC-BMEC) provided by Dr. Kathryn Kellar (CDC, USA). In short, endothelial cells were grown to confluent layer on either collagen ((THBMEC & CDC-BMEC) or fibronectin (HPMEC & HCMEC) coated flasks or 8-wells slides were incubated statically with gelantin purified late stage parasite. Unbound parasites were removed by inverting slides upside down in binding medium, and bound parasites were quantified counting. All binding assays were done at least in duplicate. For antibody inhibition assays, cells were pre-incubated for 15 min with either 5 μg/ml of mouse monoclonal anti-human ICAM-1 (clone 15.2, Abcam ab20), 0.1 mg/ml of polyclonal goat anti-EPCR (AF2245, R&D) or 50 μg/ml of activated protein C (Sigma P2200). For inhibition with rEPCR, gelatin-enriched IEs were pre-incubated for 15 min with 50 μl/ml of recombinant EPCR prior to add IEs to the cells.
Supplementary Material
Acknowledgments
We thank Dr. Ali Salanti for supplying the full-length VAR2CSA protein and Susanne Lücking Nielsen for excellent technical assistance. We thank Lene Holberg Blicher, DMAC, Technical University of Denmark for DNA sequencing assistance. The work was supported by University of Copenhagen Program of Excellence, Lundbeck Foundation, Danish International Development Agency (DANIDA), Augustinus Fonden, Malaria Capacity Development Consortium (MCDC) and The Danish Medical Research Council as well as the National Institutes of Health (R01 AI47953 and U19 AI089688 to J.D.S.)
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions: LT, TL, JDS and AJB produced recombinant proteins; JF performed the protein array experiments; SSB, CWW, JEVP, MAN, MA, JSJ and JDS performed the work with malaria parasites; PM, JL and TGT organized clinical work and processed clinical samples; MKK performed the surface plasmon resonance studies; LT performed the ELISA studies. The study was conceived and planned by LT, TL and TGT. The manuscript was written by TL, TGT, LT, JDS, and MH. All authors read and commented on the manuscript. LT and TL contributed equally to the work.
Author Informaton: Reprints and permissions information is available at www.nature.com/reprints.
The authors have no competing financial interests.
References
- 1.Murray CJ, et al. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- 2.Kraemer SM, Smith JD. A family affair: var genes, PfEMP1 binding, and malaria disease. Curr Opin Microbiol. 2006;9:374–380. doi: 10.1016/J.Mib.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Lavstsen T, et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1791–1800. doi: 10.1073/pnas.1120455109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avril M, et al. A restricted subset of var genes mediates adherence of Plasmodium falciparum-infected erythrocytes to brain endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1782–1790. doi: 10.1073/pnas.1120534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claessens A, et al. A subset of group A-like var genes encodes the malaria parasite ligands for binding to human brain endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1772–1781. doi: 10.1073/pnas.1120461109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 7.Rask TS, Hansen DA, Theander TG, Gorm Pedersen A, Lavstsen T. Plasmodium falciparum erythrocyte membrane protein 1 diversity in seven genomes--divide and conquer. PLoS computational biology. 2010;6 doi: 10.1371/journal.pcbi.1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salanti A, et al. Evidence for the involvement of VAR2CSA in pregnancy-associated malaria. J Exp Med. 2004;200:1197–1203. doi: 10.1084/jem.20041579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukudome K, Esmon CT. Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. The Journal of biological chemistry. 1994;269:26486–26491. [PubMed] [Google Scholar]
- 10.Simmonds RE, Lane DA. Structural and functional implications of the intron/exon organization of the human endothelial cell protein C/activated protein C receptor (EPCR) gene: comparison with the structure of CD1/major histocompatibility complex alpha1 and alpha2 domains. Blood. 1999;94:632–641. [PubMed] [Google Scholar]
- 11.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10212–10216. doi: 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuepbach RA, Feistritzer C, Fernandez JA, Griffin JH, Riewald M. Protection of vascular barrier integrity by activated protein C in murine models depends on protease-activated receptor-1. Thrombosis and haemostasis. 2009;101:724–733. doi: 10.1160/th08-10-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gleeson EM, O’Donnell JS, Preston RJ. The endothelial cell protein C receptor: cell surface conductor of cytoprotective coagulation factor signaling. Cellular and molecular life sciences: CMLS. 2012;69:717–726. doi: 10.1007/s00018-011-0825-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukudome K, et al. The endothelial cell protein C receptor. Cell surface expression and direct ligand binding by the soluble receptor. The Journal of biological chemistry. 1996;271:17491–17498. doi: 10.1074/jbc.271.29.17491. [DOI] [PubMed] [Google Scholar]
- 15.Robinson BA, Welch TL, Smith JD. Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Molecular microbiology. 2003;47:1265–1278. doi: 10.1046/j.1365-2958.2003.03378.x. [DOI] [PubMed] [Google Scholar]
- 16.Ghumra A, et al. Induction of strain-transcending antibodies against Group A PfEMP1 surface antigens from virulent malaria parasites. PLoS pathogens. 2012;8:e1002665. doi: 10.1371/journal.ppat.1002665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochola LB, et al. Specific receptor usage in Plasmodium falciparum cytoadherence is associated with disease outcome. PloS one. 2011;6:e14741. doi: 10.1371/journal.pone.0014741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sturn DH, et al. Expression and function of the endothelial protein C receptor in human neutrophils. Blood. 2003;102:1499–1505. doi: 10.1182/blood-2002-12-3880. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, Qu D, Esmon NL, Esmon CT. Metalloproteolytic release of endothelial cell protein C receptor. The Journal of biological chemistry. 2000;275:6038–6044. doi: 10.1074/jbc.275.8.6038. [DOI] [PubMed] [Google Scholar]
- 20.Pintao MC, et al. High levels of protein C are determined by PROCR haplotype 3. Journal of thrombosis and haemostasis: JTH. 2011;9:969–976. doi: 10.1111/j.1538-7836.2011.04256.x. [DOI] [PubMed] [Google Scholar]
- 21.Dennis J, et al. The endothelial protein C receptor (PROCR) Ser219Gly variant and risk of common thrombotic disorders: a HuGE review and meta-analysis of evidence from observational studies. Blood. 2012;119:2392–2400. doi: 10.1182/blood-2011-10-383448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert reviews in molecular medicine. 2009;11:e16. doi: 10.1017/S1462399409001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cham GK, et al. Sequential, ordered acquisition of antibodies to Plasmodium falciparum erythrocyte membrane protein 1 domains. J Immunol. 2009;183:3356–3363. doi: 10.4049/jimmunol.0901331. [DOI] [PubMed] [Google Scholar]
- 24.Cham GK, et al. Hierarchical, domain type-specific acquisition of antibodies to Plasmodium falciparum erythrocyte membrane protein 1 in Tanzanian children. Infection and immunity. 2010;78:4653–4659. doi: 10.1128/IAI.00593-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kendrick BJ, Gray AG, Pickworth A, Watters MP. Drotrecogin alfa (activated) in severe falciparum malaria. Anaesthesia. 2006;61:899–902. doi: 10.1111/j.1365-2044.2006.04752.x. [DOI] [PubMed] [Google Scholar]
- 26.Rankin LG, Austin DL. The use of activated protein C in severe Plasmodium falciparum malaria. Anaesthesia and intensive care. 2007;35:428–432. doi: 10.1177/0310057X0703500320. [DOI] [PubMed] [Google Scholar]
- 27.Robak O, et al. The use of drotrecogin alfa in severe falciparum malaria. Anaesthesia and intensive care. 2010;38:751–754. doi: 10.1177/0310057X1003800421. [DOI] [PubMed] [Google Scholar]
- 28.Wang CW, et al. Evidence for in vitro and in vivo expression of the conserved VAR3 (type 3) plasmodium falciparum erythrocyte membrane protein 1. Malar J. 2012;11:129. doi: 10.1186/1475-2875-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stins MF, Gilles F, Kim KS. Selective expression of adhesion molecules on human brain microvascular endothelial cells. Journal of neuroimmunology. 1997;76:81–90. doi: 10.1016/s0165-5728(97)00036-2. [DOI] [PubMed] [Google Scholar]
- 30.Turner L, et al. Antibodies against PfEMP1, RIFIN, MSP3 and GLURP are acquired during controlled Plasmodium falciparum malaria infections in naive volunteers. PLoS One. 2011;6:e29025. doi: 10.1371/journal.pone.0029025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.