

Abstract
Analysis of the Polyp Prevention Trial showed an association between isorhamnetin-rich diet and a reduced risk of advanced adenoma recurrence; however, the mechanism of isorhamnetin’s chemoprotective effects remains unclear. Here we demonstrate that isorhamnetin prevents colorectal tumorigenesis of FVB/N mice treated with the chemical carcinogen azoxymethane (AOM) and subsequently exposed to colonic irritant dextran sodium sulfate (DSS). Dietary isorhamnetin decreased mortality, tumor number, and tumor burden by 62%, 35%, and 59%, respectively. Magnetic resonance imaging, histopathology, and immunohistochemical analysis revealed that dietary isorhamnetin resolved the DSS-induced inflammatory response faster than control diet. Isorhamnetin inhibited AOM/DSS induced oncogenic c-Src activation and β-catenin nuclear translocation, while promoting the expression of C-terminal Src Kinase (CSK), a negative regulator of Src family of tyrosine kinases. Similarly, in HT-29 colon cancer cells, isorhamnetin inhibited oncogenic Src activity and β-catenin nuclear translocation by inducing expression of CSK, as verified by RNAi knock down of CSK. Our observations suggest the chemoprotective effects of isorhamnetin in colon cancer are linked to its anti-inflammatory activities and its inhibition of oncogenic Src activity and consequential loss of nuclear β-catenin, activities that are dependent on CSK expression.
Keywords: isorhamnetin, colorectal Cancer, inflammation, flavonoid, prevention
Introduction
Colorectal cancer (CRC) is the fourth most commonly diagnosed type of cancer in the United States and it is the second leading cause of cancer related deaths (1). Chronic inflammation, such as ulcerative colitis and Crohn’s disease, are associated with increased risk of CRC (2–5).
The Src Family of tyrosine kinases (SFK) are nonreceptor protein tyrosine kinases (TK) that are activated in multiple cancers, including CRC (6). Increased Src activity in primary CRC is an indicator of poor prognosis (7). C-terminal Src kinase, also known as c-Src kinase (CSK), negatively regulates SFK by phosphorylation of a C-terminal tyrosine (Y530 in c-Src) (8). C-terminal phosphorylation of SFK stabilizes the protein in an inhibitory confirmation that prevents auto-phosphorylation of the activation loop tyrosine (Y419 in c-Src). The receptor-like tyrosine phosphatase CD45 and similar phosphastases impose a reciprocal regulation of Src by removing the C-terminal tyrosine phosphate (9).
Activated Src can regulate many downstream pathways including phosphatidylinositol 3-kinase (PI3-K), Ras-Mek-ERK, STAT3, and p130 to increase survival, proliferation, angiogenesis, motility, and invasion (6). Activated Src can also phosphorylate β-catenin, causing its release from E-cadherin sequestration at the plasma membrane and enhancing its nuclear localization (10). Colorectal cancer appears to be sensitive to loss of Src regulation. Both the loss of expression of the negative regulator CSK and the overexpression of Src have been implicated in colorectal carcinogenesis (8).
The cost of treating CRC is estimated to be $6.5 billion per year (11). Dietary change and use of dietary supplements, both feasible and safe, represents a viable and important strategy for preventing CRC (12). Recent analysis of the Polyp Prevention Trial (PPT), a clinical trial that investigated the role of diet modulation in the prevention of colorectal adenoma recurrence, suggested that consuming an isorhamnetin-rich diet was associated with a decreased risk of advanced adenoma recurrence (13).
Recently, it was shown that isorhamnetin can suppress skin cancer by binding to and inhibiting MAP (mitogen-activated protein)/ERK (extracellular signal regulated kinase) kinase (MEK) 1 and PI3-K (phosphoinositide 3-kinase) (14). To further investigate the potential role of flavonols in CRC prevention we assessed the effects of 4 flavonols, commonly consumed by humans, isorhamnetin, quercetin, rutin, and myricetin, in a mouse model for CRC. We found that supplementing the diet with isorhamnetin significantly reduced colorectal tumorigenesis in these mice. The isorhamnetin diet appeared to resolve the DSS induced inflammation as measured by magnetic resonance imaging (MRI), histopathology, and immunohistochemistry (IHC). Isorhamnetin also inhibited Src activity and nuclear localization of β-catenin and increased expression of CSK both in vivo and in colorectal carcinoma cells. Finally, we showed that inhibition of Src and nuclear β-catenin was dependent on the up-regulation of CSK in colon cancer cells and associated with CSK expression in isorhamnetin fed mice.
Materials and Methods
Animal studies
All mouse experiments were agreed to and regulated by the Animal Care and Use Committee of the National Cancer Institute (Frederick, MD). Pathogen-free male FVB/N mice were purchased from the NCI-Frederick Animal Production Area at 5 weeks of age and were exposed to a 12:12–hour light/dark cycle. Mice were given an AIN-96G purified diet from Harlan Teklad (Madison, WI) and drinking water ad libitum. At 6 weeks of age (Day 0, Fig. 1), mice were injected intraperitoneally with AOM (Sigma, St. Louis, MO) at a dose of 10 mg/kg body weight in 0.1 ml of saline (Fig. 1). Seven days later, mice were treated with 2% DSS, 36,000 to 50,000 kDa (MP Biomedicals LL, Solon, OH), dissolved in normal drinking water (reverse osmosis–purified water) for 7 days and then switch back to normal drinking water. Body weights were measured during the DSS treatment. Three days after DSS treatment ended, the mice were evenly sorted by change in body weight into 5 diet groups to ensure effects of the DSS were evenly represented in each of the different diets. Mice were single caged and started on the indicated diet (Day 17) (Supplementary Table 1). Isorhamnetin and myricetin were obtained from Jinan Haohua Industry Co., Ltd (Jinan Shandong, China) and quercetin and rutin were obtained from Sigma-Aldrich Co., LLC (St. Louis, MO). Purified diets were prepared by Harlan Laboratories (Madison, WI). Mice were allowed to eat ad libitum. One day before euthanasia (day 20, 25, and 33), 12 mice per group and time point were imaged by MRI as described previously (15). Mice were euthanized at indicated times and colon tissue collected.
Figure 1.
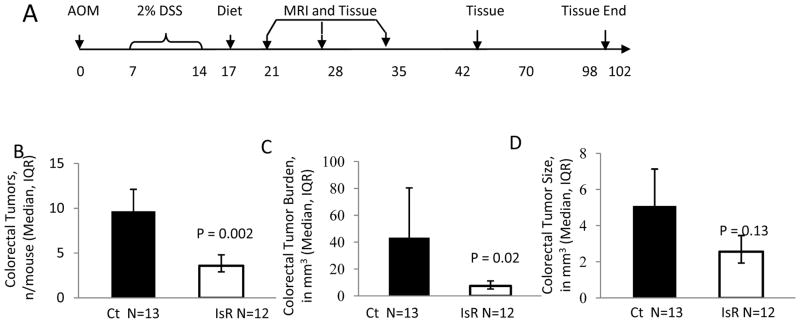
Isorhamnetin decreased AOM/DSS-induced colorectal tumorigenesis. A, Time line showing that mice were treated with AOM on day 0 and with 2% DSS from day 7 to 14. On day 17, mice were blocked by weight loss and randomly started within weight blocks on experimental diets. On day 20, 25 and 33 mice were imaged using MRI and one day later they were euthanized and tissue collected. At later times (day 59 and 102) mice were euthanized and tissue collected without MRI. B-D, Analysis of colorectums collected at day 102. B, Tumors numbers per mouse (geometric mean ± standard errror). C, Tumor burden in mm3/mouse (geometric mean ± standard errror). D, Tumor size in mm3/tumor (geometric mean ± standard error). Ct indicates control diet N=13, IsR indicts isorhamnetin diet N=12.
Histopathology
Colonic tissue was harvested from mice at various times after AOM or AOM DSS treatment and fixed in 4% formaldehyde overnight and stored in 70% ethanol. Fixed sections of colonic tissues were embedded in paraffin, cut into 6-μm sections, and put onto microscopic slides by Histoserv Inc. (Germantown, MD, USA). Slides were either stained with hematoxylin–eosin (H&E) for histological analysis via light microscopy or deparaffinized in xylene and rehydrated in graded ethanol for IHC. For histopathology analysis, an approximately 5-mm in length section of the distal colon was evaluated for each animal. The tissues were blinded and scored by the pathologist semiquantitatively from 0 to 15 as described (Supplementary Table 2). Colons were evaluated beginning at the colorectal junction and proceeding adorally 5-mm to the middle colon. Briefly, the severity of the leukocytic infiltrate in the mucosa was subjectively assessed as mild, moderate, or severe (1, 2, 3, respectively); the distribution was evaluated and denoted as focal/locally extensive, multifocal, or diffuse (1, 2, 3, respectively); and the depth was evaluated and denoted as mucosa only, extends to submucosa, and extends to muscularis mucosa or serosa (1, 2, 3, respectively). The distribution of atypical glandular hyperplasia (AGH)/squamous (sq) metaplasia was assessed as focal, multifocal or diffuse (1, 2, 3, respectively). The distribution of ulceration was assessed as focal, multifocal, or diffuse (1, 2, 3, respectively). If necrosis was present it was subjectively assessed as mild, moderate, or severe and scored accordingly (1, 2, 3, respectively). A score of 0 was assigned for each criterion not represented in the section, which was the case for the majority of mice for ulceration and necrosis. Total disease score per mouse was calculated by summation of the six parameter for each mouse.
Immunohistochemistry
For Immunohistochemistry, the sections were placed in low pH modified citrate buffer (Dako, Carpinteria, CA, USA) and subjected to antigen retrieval via pressure chamber. The sections were preincubated in 3% H2O2 for 20 minutes and blocked in normal goat serum for 1 hour. Sections were incubated overnight in primary antibody solution diluted as follows; Ki-67, phospho β-catenin654 (Abcam laboratories, Cambridge, MA, USA) 1:50, CD45 (BD biosciences, San Jose, CA, USA) 1:50, pSrcY417 and β-catenin (Cell signaling, Danvers, MA, USA) 1:100, and 1:1000, respectively. Slides were incubated for 1 hour in a biotin-conjugated secondary antibody diluted 1:200. The sections were treated with Vectorstain ABC kit (Vector laboratories, Burlingame, CA, USA); the resulting activity detected with Diaminobenzidine (Sigma, St. Louis, MI, USA) and counterstained in Hematoxylin. For CSK (Abcam Laboratories, Cambridge, MA), tissues were incubated 1:50 for 1 hr and detected with Ventana iView DAB detection kit (Ventana Medical Systems, Oro Valley, AZ, USA) on a Ventana BenchMark.
Cell Culture
Human colon cancer HT29 cell line was obtained from American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured as described previously (16).
Western blot
HT29 cells were seeded on 100mm plates and treated with either DMSO vehicle (<1%) or isorhamnetin (10μmol/L, 20μmol/L, and 40μmol/L) for 24 hours. The media was removed and cells were washed with cold PBS. Cells were lysed with complete RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein concentration was calculated using standard BSA curve. Approximately 30–80 μg of protein was loaded and separated by SDS-PAGE, and transferred to nitrocellulose membrane and probed with the following antibodies at indicated dilutions; CSK (1:1000), pSRC417 (1:1000), pSRC529 (1:1000), β-catenin (1:2000), pAKT (1:1000), pERK (1:2000), E-Cadherin (1:1000), and pGSK3αβ (1:1000) (Cell Signaling, Danvers, MA, USA) and pβ-catenin654 (Abcam laboratories, Cambridge, MA, USA) overnight at 4°C, followed by incubation in HRP labeled Secondary antibodies for 1 hour. Cells were developed with SuperSignal West Femto Chemiluminescent Substrate (Thermo scientific, Rockford, IL, USA). Each membrane was probed with β-Actin (1:5000, Sigma, St. Louis, MI, USA) to ensure consistent protein loading.
Real time-PCR
Total RNA from HT29 cultured cells was isolated using Trizol RNA extraction method. Briefly, chloroform was added to sample, and after shaking and centrifugation the aqueous layer was isolated. Isopropanol was next added to sample to precipitate the RNA. Following centrifugation the RNA pellet was washed with 70% ethanol. RNA was further purified using RNeasy kit (QIAGEN, Germantown, MD, USA). The cDNA was synthesized using Bio-Rad iScript reverse transcriptase and the PCR reactions were performed using Bio-Rad SYBR Green Master Mix carried out in Bio-Rad iCycler (Bio-Rad Laboratories, Hercules, CA, USA). The human CSK splicing variant 1 and 2 were purchased from IDT (San Jose, CA, USA) and sequence is as follows: sense, 5′-TCCGGCCCCGTCTCTCTTGG and antisense, 5′-ACCCTCACGGGCAGGACAGG. The PCR included non-template control and GAPDH as control primer set. The CSK transcript activity level was calculated using 2−Δ (ΔCT), where ΔCT=CTCSK-CTGAPDH and Δ (ΔCTstimulated -ΔCTcontrol).
CSK siRNA construct
HT29 cells were stably transfected with 3 unique 27mer siRNA duplexes - 2 nmol each against CSK siRNA (Trilencer-27, Origene technologies, Rockville, MD, USA) or a scrambled control siRNA using manufacturers transfection reagent and recommendations. After transfection, cells were incubated for 36 hours. The total protein was isolated and subjected to Western blot analysis.
Soft Agar Assay
HT29 cells were seeded onto 100 mm plates and pre-treated with either DMSO vehicle (<1%) or isorhamnetin (10 μmol/L, 20 μmol/L, and 40 μmol/L) for 24 hours. Cells were trypsinized and seeded at 30,000 cells in 2× DMEM media. Cell suspension was added 1:1 with 0.5% agarose (2 ml/well in a 6 well plate) and constitutes the top layer. The bottom layer consisted of 2 ml of 1.2% agarose. The cells were maintained in an incubator for 14 days and the colonies were scanned and counted with GelCount, (Oxford Optronix Ltd, Oxford United Kingdom).
Statistical Analyses
Statistical analyses were done using SAS, version 9.2 (SAS, Inc., Cary, NC) software. A Students’ unpaired t-test was used to compare treatment group averages for histopathology and cell culture data. Generalized linear models were used to compare treatment group averages for mouse morbidity (binomial; PROC GLIMMIX) and tumor data (PROC GLM). Beside diet, number of DSS rounds was included in the linear models. To achieve normality, tumor number was natural log-transformed and tumor burden and size was twice natural log-transformed, after adding a 1 to prevent values below zero. Histopathology and cell culture data are shown as raw means and their standard errors; tumor data are shown as least-squares means and their standard errors, which were back transformed to their original scale. All statistical tests were two-sided. The P values were not adjusted for multiple comparisons. Significance was declared at P ≤ 0.05 and a tendency at 0.05 to 0.10.
Results
Isorhamnetin prevents colon tumorigenesis in AOM/DSS treated mice
We previously showed that a high intake of flavonol-enriched foods and beverages, specifically isorhamnetin, kaempferol, and quercetin, was associated with decreased risk of advanced adenoma recurrence (13, 17). In order to verify the chemoprotective effects of individual flavonols on colorectal tumorigenesis, we fed FVB/N male mice previously treated with AOM and DSS, diets enriched with four primary flavonols consumed by humans (Table 1 and Supplementary Table 1). FVB/N mice are sensitive to the AOM/DSS and only require one cycle of DSS to promote tumorigenesis. Three days after the DSS treatment, mice were started on an AIN93-G control diet (18) or one supplemented with the indicated flavonol. Sensitive strains of mice treated with AOM and 1 week of DSS will develop colon dysplastic lesions, adenomas and adenocarcinomas; unfortunately some of these mice will have to be euthanized prematurely due to the large tumor burden and colorectal prolapsed (15, 19). In our study, feeding mice isorhamnetin after the DSS exposure produce a significantly higher survival rate (80%) compared to the mice fed the control diet (48%, p=0.02) (Table 1). The isorhamnetin-fed mice also developed 35% (p=0.03) fewer tumors and a 59% (p=0.04) smaller tumor burden than mice fed the control diet (Table 1). Because the decreased mortality and tumor number in the quercetin intervention was not significant and we saw no beneficial effects with rutin or myricetin diets, we conducted an in-depth study of the isorhamnetin intervention. A repeat of the isorhamnetin intervention showed 63% decrease in tumor number (p=0.002) and an 83% decrease in tumor burden (p=0.02) (Fig. 1). In this study, tumors did not progress to adenocarcinoma on either of the diet interventions. This data indicates a diet supplemented with isorhamnetin prevents colorectal tumorigenesis in a mouse model of colitis associated CRC.
Table 1.
Effect of feeding flavonols at equimolar concentrations (ppm) for 85 days to AOM/DSS-induced male FVB mice on morbidity, tumor number, and tumor burden (N = 32 per group)
| Dietary | Morbidity (%)1 | Tumor Number (n/mouse) | Tumor Burden (mm3/mouse) | ||||
|---|---|---|---|---|---|---|---|
| Flavonol | Avg ± SE2 | P vs. C3 | Avg ± SE | P vs. C | Avg ± SE | P vs. C | |
| Control | 0 | 52 ± 10 | 19.6 ± 1.2 | 172.7 ± 2.9 | |||
| Isorhamnetin | 552 | 20 ± 8 | 0.02 | 12.7 ± 1.1 | 0.03 | 74.5 ± 2.9 | 0.04 |
| Myricetin | 556 | 37 ±10 | 0.28 | 17.3 ± 1.1 | 0.52 | 178.2 ± 2.9 | 0.94 |
| Quercetin | 591 | 23 ± 8 | 0.04 | 14.8 ± 1.1 | 0.15 | 132.5 ± 2.9 | 0.54 |
| Rutin | 1,099 | 49 ± 10 | 0.88 | 17.6 ± 1.2 | 0.58 | 122.3 ± 2.9 | 0.42 |
Morbid mice had to be sacrificed because they either lost more than 10% of their maximum body weight or their intestines had prolapsed
Values are shown as geometric means and their standard errors. To achieve normality, tumor number had to be once natural log-transformed and tumor burden had to be twice natural log-transformed.
P vs C indicates P-values of natural log-transformed data when diet groups are compared to control diet.
Isorhamnetin resolves colitis faster in AOM/DSS treated mice
FVB/N mice exposed to 1 cycle of DSS will develop colitis that can be detected by MRI (15). Mice treated with AOM/DSS were imaged on day 20, 25 and 33 (Fig. 1) using MRI before and after IV administration of 0.2 mmol/kg Gd-DTPA contrast agent, (Fig. 2). In normal colons, little to no contrast was detected in the colonic epithelium (15), whereas in DSS treated mice, the contrast agent clearly reached the epithelial lining of the colon, indicating colitis at day 20 in both diet groups (Fig. 2A left panels). By day 25, colitis increased in mice fed both diets; however colitis was less severe in mice fed isorhamnetin (Fig. 2A middle panels). By day 33, isorhamnetin fed mice had less uptake of contrast agent in the colonic epithelium as compared to mice fed the control diet, indicating colitis had begun to resolve in these mice (Fig. 2A right panels). Histopathology analysis of the colonic tissue collected one day after MRI (day 21, 26 and 34) confirmed the results of the imaging (Fig. 2B). Regardless of day of sacrifice (no time × diet interaction at P < 0.20), dietary isorhamnetin decreased leukocyte distribution (P = 0.003), depth (P = 0.07) and severity (P = 0.11) of inflammation, and the distribution of atypical glandular hyperplasia (AGH) or squamous metaplasia (P=0.02), resulting in an overall lower histopathology score for isorhamnetin-fed mice (P = 0.02). The severity and distribution of inflammation was similar in the mice on day 21 and 26, irrespective of the diet. By day 34, however, the mice fed the isorhamnetin diet showed only localized or multifocal distribution of leukocytes with mild to moderate severity, while the mice fed the control diet still exhibited moderate to severe inflammation which was distributed diffusely within the section of colon evaluated (Figure 2B). Colons collected at days 21 and 26 from mice fed both the control and isorhamnetin diet showed atypical glandular hyperplasia and squamous metaplasia confined to the colorectal area in most sections. By day 34 the colons of the mice that were fed the control diet had a higher rate of microadenoma formation and a greater degree of hyperplasia and squamous metaplasia, which are considered premalignant lesions, as compared to the mice fed the isorhamnetin diet. Unlike in our previous study (15), no adenocarcinoma was not detected in this study in either group. These results suggest that the mice on the isorhamnetin diet recovered from colitis faster than the mice on the control diet and further suggest a chemopreventive effect of isorhamnetin on colorectal tumorigenesis.
Figure 2. Isorhamnetin inhibited AOM/DSS-induced inflammation.

A, MRI showed decreased inflammation in isorhamnetin-fed mice. On day 20, 25 and 33, 12 mice per group were imaged by MRI. Two T1 weighted post-contrast representative images are shown for each diet and time point. Coronal, sagittal and axial views are shown for the indicated mouse (m). Arrow heads shows Gd-DTPA localized to the epithelia tissue. Arrow shows region of coronal view used to generate the axial view. Ct indicates control diet, IsR indicates isorhamnetin diet.
B, Histopathology showed a decrease in inflammation in isorhamnetin fed mice. Colorectal tissues collected on the indicated days were stained with Hematoxylin and Eosin and evaluated for the degree of inflammation and colonic tissue damage produced after AOM/DSS exposure. The tissues were scored for the a degree of inflammation with criteria of 0, 1, 2 or 3, where 1=focal/locally, 2=multifocal, and 3=diffuse for distribution of leukocytes, for AGH/Sq Metaplasia and distribution of epithelial erosion/ulceration (data not shown); 1=mild, 2=moderate and 3=severe for severity of infiltrated and for severity of necrosis (data not shown); and for depth of inflammation 1=mucosa only, 2=extends to submucosa, and 3=extends to muscularis mucosa or serosa. A score of 0 was assigned for each criterion not represented in the section. Data for epithelial erosion and necrosis are not shown because only a minority of mice showed this feature. The overall score is the accumulation of the six individual scores (maximum total 18).
C, Anti-CD45 (1:50) immunohistochemistry (IHC) showed IsR decreased inflammation. Gut-Associated-lymphoid tissue (GALT) is seen in AOM only day 31 mice (top panel 1) and AOM/DSS IsR day 34 mice (bottom panel 1). In the AOM/DSS mice on control diet CD45-positive cells show infiltration. GALT were not detected in these mice (middle panels).
Isorhamnetin decreases CD45-positive leukocytes infiltration in AOM/DSS treated mice
The DSS-induced inflammation results in infiltration of the colon by leukocytes, which was quantified by expression of the cell surface marker of leukocytes, CD45. The leukocyte districution in the colonic mucosa and submucosal tissue was similar at days 21 and 26, irrespective of diet (data not shown). By day 34, however, dietary isorhamnetin decreased infiltration of CD45-positive cells compared to mice on the control diet (Fig. 2C). The expression of CD45 was predominantly localized to the Gut-Associated-lymphoid tissue (GALT) in mice fed isorhamnetin, similar to what is seen in mice not exposed to DSS. The GALT provides immune protection to the gut (20). These results suggests that isorhamnetin prevents the release of CD45-positive leukocytes. Consistent with the histopathology and MRI data (Fig. 2A and B), the decrease in CD45-positive leukocytes infiltration indicates that the mice on the isorhamnetin diet recover more quickly from the colitis than do mice fed the control diet.
Isorhamnetin reduces cell proliferation in AOM/DSS treated mice
In order to assess the effects of isorhamnetin diet on cell proliferation, Ki-67 expression was evaluated by immunohistochemistry on day 34 in the colon tissue of AOM/DSS treated mice. Mice fed the isorhamnetin diet had a 16% crypt proliferation fraction compared to mice on the control diet with a crypt proliferation fraction of 63% (p<0.001) (Fig. 3). Furthermore, Ki-67 expression in the isorhamnetin treated mice was predominantly in the basal portion of the colon crypt that contains multipotent stem cells (21), whereas in mice fed the control diet, Ki67 expression showed aberrant cell proliferation throughout the crypts. Similar results were seen at day 59 (Fig. 3), indicating that the mice on the control diet never completely recovered from the DSS-induced proliferation. The decrease in proliferation suggests that the mice on the isorhamnetin diet recover more quickly from the inflammation-induced cell proliferation response in the colon.
Figure 3. Isorhamnetin inhibits cell proliferation.

A, IHC of representative photomicrographs of colorectal tissues stained anti-Ki-67 (1:100) collected from AOM only (Day 31) and AOM/DSS (Day 34 and 59) treated mice. Mice were fed either control diet (left panels) or an isorhamnetin enriched diet (right panels). B, Microscopically quantification of Ki-67 from days 34 and 59 expressed as percentage of Ki-67-positive cells to total number of cells in the crypt (crypt proliferation fraction). The values are means +/− standard errors after stereological analysis of 10 complete crypts per animal (N=6).
Isorhamnetin inhibits anchorage-independent growth of human colorectal carcinoma HT29 cells
In order to access the effects of isorhamnetin in a human relevant model, human colorectal carcinoma cell lines were treated with varying concentrations of isorhamnetin. Treatment of HT29 cells showed no effect on cell viability up to 40 μM isorhamnetin and very little affect up to 100 μM. HCT116 colorectal cell viability was more sensitive to isorhamnetin than viability of HT29 cells (Supplementary Fig. 2). At 40 μM isorhamnetin significantly inhibited anchorage independent growth of HT29 cells by 62% (p=0.001) compared to solvent control (Supplementary Fig. 2). These results suggest that isorhamnetin at 40 μM can inhibit tumorigenesis without significantly affecting cell viability.
Isorhamnetin inhibits ERK, Akt and Src activity
Previous studies have shown that isorhamnetin inhibits MEK and PI3K in SCC skin cancer (14). Here we show a similar dose-dependent inhibition of phosphorylation of Akt, a PI3K target, and of ERK 1/2, a MEK target in HT29 CRC cells (Fig. 4A). Interestingly, we did not see inhibition of S6kinase phosphorylation, a target of Akt. The tyrosine phosphatase CD45 dephosphorylates Src tyrosine 529 leading to autophosphorylation at tyrosine 417 and activation of Src. Here we show a dose-dependent inhibition of Src Y417 phosphorylation by isorhamnetin (Fig. 4A). Conversely, there was a dose dependent increase in Src Y529 phosphorylation. Src activation was inhibited by isorhamnetin in a dose dependent manner with a 34% inhibition (increase in Y529 phosphorylation) at 40 μM.
Figure 4. Isorhamnetin inhibition of ERK, Akt, and Src activity is CSK dependent.

A, Dose dependent inhibition of ERK, AKT and Src activity. Western blot analysis of whole cell extracts from HT29 cells treated with indicated dose of IsR for 24 hours. p indicates phospho-specific antibody targeting Akt phosphorylated at S473 (pAkt), p70S6K (pp70S6K), ERK1/2 (pERK1/2), activated Src phosphorylated at Y417 (pSrc417), inactivated Src phosphorylated at Y529 (pSrc529)
B, Dose dependent activation of CSK mRNA and protein expression. Upper panel represents real-time PCR analysis of cDNA from CSK mRNA transcript in response to IsR pre-treatment for 24 hours. The data is expressed as fold changes relative to GAPDH control. The values are the mean +/− S.E.M of three independent experiments. IsR treatment at 20 and 40 μM resulted in significantly higher CSK transcript levels (*p<0.05, **p<0.01). Lower panel shows Western blot against CSK with a 2.6 fold increase at 40 μM as determined by densitometry.
C, Isorhamnetin inhibition reversed by siRNA knock down of C-Terminal Src Kinase. Western blot analysis of HT29 cells transfected with control siRNA (siCt) or CSK siRNA (siCSK) and 24 hrs later treated with the indicated concentration of IsR. α indicates antibody.
Isorhamnetin induces C-Terminal Src Kinase expression
Because tyrosine 529 on Src is phosphorylated by C-Terminal Src kinase (CSK), we examined the effect of isorhamnetin on CSK. Isorhamnetin induced a dose dependent increase in CSK mRNA (2.2 fold change at 40 μM) and protein (2.6 fold change at 40 μM) (Fig. 4B). Isorhamnetin at 40 μM also induced CSK expression in the breast cancer cell line MCF7 (data not shown). These results indicate that isorhamnetin activates gene expression of C-Terminal Src Kinase leading to inhibition of Src activity.
Isorhamnetin inhibition of ERK, Akt and Src activity is CSK dependent
To determine if the isorhamnetin inhibition of Src is dependent on CSK expression, we knocked down CSK expression with siRNA and assessed Src activity. Transfection with the control siRNA (siCt) did not affect the dose dependent increase of CSK protein in the isorhamnetin treated cells, while knock-down of the CSK gene blocked the isorhamnetin induced increase in CSK protein (Fig. 4C). Knock-down of CSK expression reversed the inhibition of Src activation by isorhamnetin. Transfection with control siRNA had no effect on the isorhamnetin dependent decrease in Src Y417 phosphorylation. Conversely, transfection of HT29 cells with siRNA against CSK reversed the isorhamnetin induced decrease in Y417 phosphorylation on Src. In fact, siCSK knock down resulted is a slight increase in Y417 phosphorylation (Fig. 4C), demonstrating that inhibitory affects of isorhamnetin on Src is dependent on CSK in HT29 cells.
Transfection of HT29 cells with siCSK also reversed the isorhamnetin inhibition of Akt and ERK activation seen in cells transfected with the control siRNA (Fig. 4C), indicating that activation of the MAPK and PI3K pathways is in part regulated by Src or a Src family kinase in HT29 cells. Src can activate both the extracellular signal-regulated kinase (ERK) and the phosphatidylinositol 3-kinase (PI3K) pathways (22–24). These observation show that in HT29 CRC cells isorhamnetin induces C-Terminal Src Kinase expression leading to inactivation of Src by phosphorylation at Y527 and decrease in ERK1/2 and AKT activity.
Isorhamnetin inhibits Src dependent β-catenin nuclear translocation
The Wnt/β-catenin pathway is activated in most CRC. β-catenin can complex with APC and GSK3 in the cytoplasm. Perturbation of this complex releases a stabilized β-catenin. β-catenin can also be tethered to the plasma membrane by binding with α–catenin to E-cadherin (10). We found a dose-dependent reduction in nuclear β-catenin with a corresponding accumulation of cytoplasmic β-catenin in HT29 cells treated with isorhamnetin (Fig. 5A). Interestingly, isorhamnetin did not change GSK3 activity or E-Cadherin levels (Fig. 5C). Alternatively, isorhamnetin inhibited tyrosine phosphorylation at Y654 on β-catenin (Fig. 5A, nuclear). Tyrosine 654 is phosphorylated by Src which results in release of β-catenin from its complex with E-Cadherin allowing nuclear localization (10). The corresponding increase in cytoplasmic β-catenin did not show an increase in phosphorylation at Y654, consistent with a decrease in Src regulated nuclear translocation of β-catenin. To assess whether nuclear translocation of β-catenin was Src dependent, we transfected HT29 cells with CSK siRNA. Knock-down of CSK expression reversed the inhibitory effects of isorhamnetin treatment on Y654 phosphorylation and nuclear translocation of β-catenin (Fig. 5B). These results indicate that nuclear localization of β-catenin in HT29 CRC cells is a consequence of Src phosphorylation of Y654 on β-catenin and that isorhamnetin induced expression CSK leads to inhibition Src phosphorylation of Y654 on β-catenin, blocking its release from E-cadherin and its nuclear localization.
Figure 5. Isorhamnetin inhibition of nuclear localization of β-catenin is C-Terminal Src Kinase dependent.

A-B, Western blot analysis of nuclear (left panels) and cytoplasmic extracts (right panels) probed for β-catenin and β-catenin phosphorylated at Y654. A, IsR induced a dose dependent decrease in Y654 phosphorylated nuclear β-catenin. B, siRNA knock down of CSK expression reversed isorhamnetin inhibition of Y654 phosphorylation, β-catenin nuclear translocation, and cytoplasmic accumulation. pβ-Cat654 indicate β-catenin phosphorylated by Src at Y654. p84 is displayed as nuclear loading control and β-actin for cytoplasmic loading control. siCt indicates cells transfected with control siRNA, siCSK indicates CSK siRNA transfected cells. C, IsR treatment did not change GSK3 activity (pGSK) or E-Cadherin protein level.
Isorhamnetin induces CSK expression and inhibits Src activation in vivo
To determine if the effects of isorhamnetin on Src activation and β-catenin nuclear localization seen in HT29 cells can be translated to the mouse colon, we measured CSK levels, Src activity and β-catenin levels by IHC in AOM/DSS treated mice. Similar to the HT29 cells, mice consuming isorhamnetin showed higher levels of C-Terminal Src Kinase in both the DSS treated and untreated mice compared to the mice fed the control diet (Fig. 6A). CSK expression was higher in the lamina propria surrounding the crypts compared to the epithelial tissue. CSK was also detected, although at lower levels, in normal human colon tissue. Furthermore, activated Src (phosphorylated at Y417) was higher in the crypts of AOM/DSS treated mice fed the control diet at both day 34 and day 59 (Fig. 6B). Conversely, inactivated Src (phosphorylated at Y529) was higher in crypts of the isorhamnetin fed mice (Fig. 6B). These results indicate that isorhamnetin induces expression of CSK and inhibits Src activation in vivo in the colons of AOM/DSS treated mice.
Figure 6. Isorhamnetin induces C-Terminal Src Kinase expression, inhibits Src phosphorylation at Y417, Src phosphorylation of β-catenin at Y654, and β-catenin nuclear localization in vivo.

A, IHC analysis of CSK levels showed expression was elevated in lamina propria surrounding the crypts and epithelial tissue of mice fed isorhamnetin diet after AOM (Day 31, top panels), and after AOM/DSS (Days 34 and 59, middle panels). Images are representative photomicrographs of colonic sections. Normal colons (no AOM, DSS or IsR treatment) from mouse (bottom left) and human colon tissue (bottom right).
B, DSS-induced phosphorylation of Src Y417 was reduced in isorhamnetin fed mice. Representative photomicrographs depicting activated Src in crypts of AOM/DSS treated mice as measured using IHC analysis of pSrc Y417 (brown staining). Blue counterstained of nuclei indicated activated Src is not nuclear. IsR supplementation reduced the amount of Src activity as measured at days 34 and 59 (left middle panels). C-terminal phosphorylated Src (pScr) was elevated in IsR fed mice at day 34 (bottom panels).
C, Nuclear localization and phosphorylation of β-catenin is reduced in isorhamnetin fed mice. IHC analysis of β–catenin and Y654 phosphorylated β–catenin. β-catenin and pβ-catenin are co-localizing with nuclear counterstained, indicating phosphorylation and nuclear localization of β-catenin in AOM/DSS treated animals (middle panels). In IsR fed mice β-catenin staining was predominantly cytoplasmic and with limited p-β-catenin (bottom panels). Representative serial sections of colonic tissue taken from mice treated with AOM only (day 31) or AOM/DSS (day 34).
Isorhamnetin inhibits β-catenin nuclear localization in vivo
Nuclear β-catenin levels were increased in AOM/DSS treated mice on the control diet, consistent with activation of Src (Fig. 6C middle panels). By day 34 AOM/DSS treated mice on the control diet showed a diffuse staining for β-catenin throughout the cells, indicating both cytoplasmic and nuclear accumulation of β-catenin. Furthermore, IHC staining with the Y654 phospho-specific β-catenin antibody indicated a high level of Src induced phosphorylation. In contrast, in the isorhamnetin fed mice β-catenin remained predominantly in the cytoplasm (Figure 6C bottom panels), with lower amounts of Y654 phosphorylated β-catenin and decreased nuclear localization. These results suggests that Src regulated phosphorylation of β-catenin and its nuclear translocation can be inhibited with an isorhamnetin supplemented diet.
Discussion
In a mouse model of CRC we evaluated the effects of dietary isorhamnetin for CRC prevention. In this model, FVB mice treated with AOM and DSS develop tumors that will progress to adenoma and adenocarcinomas (15, 19) (Table 1). Dietary isorhamnetin reduced inflammation, neutrophil infiltration, cell proliferation, tumor burden, and mortality associated with the AOM/DSS treatment. Src activation and β-catenin nuclear localization induced by AOM/DSS were also reduced in the isorhamnetin-fed mice and in HT29 colon cancer cells treated with isorhamnetin. Isorhamnetin induced the expression of C-terminal Src kinase (CSK), a negative regulator of Src. In HT29 cells isorhamnetin-induced inhibition of Src activity and nuclear localization of β-catenin was dependent on CSK expression (Fig. 4). Isorhamnetin did not affect the expression E-cadherin, activation of GSK3, or activation of S6Kinase. These results suggest the anti-inflammatory and anti-cancer activities of isorhamnetin are linked to inhibition of oncogenic Src activity, which can phosphorylate β-catenin at Y654 leading to its dissociation from the membrane and its nuclear localization.
β-catenin signaling in Colorectal Cancer
The Wnt/β-catenin pathway is activated in most CRC (25–26). This pathway is also activated in the AOM/DSS mouse model (19, 27). β-catenin can complex with α-catenin and E-cadherin at the cytoplasmic membrane providing adherin junction communication (10). Activated Src can phosphorylate β-catenin at Y654 releasing it and α-catenin from the E-cadherin complex. Dissociation of β-catenin from the cadherin complex will cause dysregulation of tyrosine kinase signaling, will affect cell-cell communication, will affect β-catenin-regulated gene expression and can lead to transformation and survival of β-catenin-driven cancer (10).
Activated c-Src in Colorectal Cancer is an indicator of poor prognosis
Src family tyrosine kinases (SFK) are non-receptor tyrosine kinases that are recruited to the membrane by integrin or receptor tyrosine kinase induced phosphorylation of focal adhesion kinase (FAK). Recruitment of SFK to the membrane provides a molecular switch important for regulating proliferation, differentiation, cell adhesion and cell mobility (28). Activated Src in primary CRC is an indicator of poor prognosis (7) and elevated Src activity can be detected in the majority of human colon cancer (29). Inhibition of Src or SFK can enhance cell to cell adhesion and can suppress migration and invasion in vitro and metastasis in vivo, suggesting an anti-invasive role for Src inhibitors (10). Inhibitors of Src are currently being tested in clinical trials (10).
C-Terminal Src Kinase negatively regulates Src
The C-terminal Src Kinase (CSK) is a non-receptor tyrosine kinase that serves as a negative regulator of Src and SFK. CSK phosphorylates the C-terminal regulatory site of SFK resulting in a conformation change and inactivation of the kinase activity (30–33). A reduction in CSK mRNA, protein, and kinase activity in colorectal carcinoma has been shown to be correlated with an increase in Src activity, suggesting that a loss in C-Terminal Src Kinase may influence transformation of colorectal carcinoma (8). Rengifo-Cam et al have shown that CSK regulates signaling from integrin-SFK-mediated cell adhesion, which can influence the metastasis of cancer cells (34). Recruitment to the membrane by scaffolding proteins such as CSK binding protein (Cbp) is required for inactivation of Src by CSK and is crucial for preventing tumorigenesis (31, 35). The fact that none of the other flavonols tested, including mycetin, quercetin and rutin, induced expression of CSK in HT29 cells (results not shown) and were not effective for inhibiting carcinogenesis in vivo is consistent with our conclusion that the chemoprotective effects of isorhamnetin in colon cancer are linked to its anti-inflammatory activities and its inhibition of oncogenic Src activity and consequential loss of nuclear β-catenin, activities that are dependent on CSK expression. While CSK expression can be regulated translationally (36), very little is known about how C-Terminal Src Kinase is regulated transcriptionally. Further investigation is needed to learn how CSK expression is lost in CRC and how isorhamnetin restores CSK expression.
Dietary Isorhamnetin has chemoprotective properties
Isorhamnetin, quercetin, kaempferol, and myricetin are flavonols that are present in a wide variety of fruits and vegetables and have anti-cancer activity (37). Computational and binding assays have shown that isorhamnetin can bind directly to MEK1 and to PI3-K and quercetin can bind to RSK2 (14, 38). Lee et al (39) reported that kaempferol can bind to Src, and Jung et al (40) showed that myricetin can bind to the SFK Fyn. Our findings showed that isorhamnetin can inhibit Src activity, but that this inhibition is dependent on CSK expression, suggesting that unlike the structurally similar flavonols, kaempferol and myricetin, isorhamnetin does not bind directly to Src. The mechanism of how isorhamnetin is up regulating C-Terminal Src Kinase expression is currently under investigation. Our results and those of others (8, 34–35) show the importance of CSK as a negative regulator of SFK and as a tumor suppressor, suggesting that preventing the loss of or restoring the expression of C-Terminal Src Kinase would be beneficial for preventing tumorigenesis, tumor progression and tumor metastasis.
Supplementary Material
Acknowledgments
We would like to thank Craig Driver of the Laboratory Animal Sciences Program of SAIC Frederick Inc., Darlene Green and Tammy Beachley of the Pathology/Histotechnology Laboratory of SIAC Frederick Inc. and Thomas G. McCloud from the Natural Products Support Group of Applied Developmental Research Program of SAIC Frederick Inc. This study was funded by the Office of Complementary and Alternative Medicine, Office of Dietary Supplements, the Division of Cancer Prevention and the Intramural Research Program, National Cancer Institute, NIH, DHHS, Bethesda, MD
Footnotes
Conflict of Interests: The authors of this manuscript have no conflict of interests to disclose.
References
- 1.American CS. Cancer Facts & Figures 2011. Atlanta, GA: American Cancer Society; 2011. [Google Scholar]
- 2.Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park) 2002;16:217–26. 29. discussion 30–2. [PubMed] [Google Scholar]
- 3.Shenoy AK, Fisher RC, Butterworth EA, Pi L, Chang LJ, Appelman HD, et al. Transition from colitis to cancer: high Wnt activity sustains the tumor-initiating potential of colon cancer stem cell precursors. Cancer research. 2012;72:5091–100. doi: 10.1158/0008-5472.CAN-12-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–35. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gyde S, Prior P, Dew MJ, Saunders V, Waterhouse JA, Allan RN. Mortality in ulcerative colitis. Gastroenterology. 1982;83:36–43. [PubMed] [Google Scholar]
- 6.Lieu C, Kopetz S. The SRC family of protein tyrosine kinases: a new and promising target for colorectal cancer therapy. Clin Colorectal Cancer. 2010;9:89–94. doi: 10.3816/CCC.2010.n.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aligayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA, Gallick GE. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer. 2002;94:344–51. doi: 10.1002/cncr.10221. [DOI] [PubMed] [Google Scholar]
- 8.Cam WR, Masaki T, Shiratori Y, Kato N, Ikenoue T, Okamoto M, et al. Reduced C-terminal Src kinase activity is correlated inversely with pp60(c-src) activity in colorectal carcinoma. Cancer. 2001;92:61–70. doi: 10.1002/1097-0142(20010701)92:1<61::aid-cncr1292>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 9.Hermiston ML, Zikherman J, Zhu JW. CD45, CD148, and Lyp/Pep: critical phosphatases regulating Src family kinase signaling networks in immune cells. Immunological reviews. 2009;228:288–311. doi: 10.1111/j.1600-065X.2008.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wadhawan A, Smith C, Nicholson RI, Barrett-Lee P, Hiscox S. Src-mediated regulation of homotypic cell adhesion: implications for cancer progression and opportunities for therapeutic intervention. Cancer Treat Rev. 2011;37:234–41. doi: 10.1016/j.ctrv.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Gill S, Sinicrope FA. Colorectal cancer prevention: is an ounce of prevention worth a pound of cure? Semin Oncol. 2005;32:24–34. doi: 10.1053/j.seminoncol.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 12.American CS. Cancer Facts & Figures 2010. Atlanta, GA: American Cancer Society; 2010. [Google Scholar]
- 13.Bobe G, Sansbury LB, Albert PS, Cross AJ, Kahle L, Ashby J, et al. Dietary flavonoids and colorectal adenoma recurrence in the Polyp Prevention Trial. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive. Oncology. 2008;17:1344–53. doi: 10.1158/1055-9965.EPI-07-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JE, Lee DE, Lee KW, Son JE, Seo SK, Li J, et al. Isorhamnetin suppresses skin cancer through direct inhibition of MEK1 and PI3-K. Cancer Prev Res. 2011;4:582–91. doi: 10.1158/1940-6207.CAPR-11-0032. [DOI] [PubMed] [Google Scholar]
- 15.Young MR, Ileva LV, Bernardo M, Riffle LA, Jones YL, Kim YS, et al. Monitoring of tumor promotion and progression in a mouse model of inflammation-induced colon cancer with magnetic resonance colonography. Neoplasia. 2009;11:237–46. doi: 10.1593/neo.81326. 1p following 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitsunaga M, Kosaka N, Choyke PL, Young MR, Dextras CR, Saud SM, et al. Fluorescence endoscopic detection of murine colitis-associated colon cancer by topically applied enzymatically rapid-activatable probe. Gut. 2012 doi: 10.1136/gutjnl-2011-301795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bobe G, Barrett KG, Mentor-Marcel RA, Saffiotti U, Young MR, Colburn NH, et al. Dietary cooked navy beans and their fractions attenuate colon carcinogenesis in azoxymethane-induced ob/ob mice. Nutrition and cancer. 2008;60:373–81. doi: 10.1080/01635580701775142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reeves PG, Nielsen FH, Fahey GC., Jr AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. The Journal of nutrition. 1993;123:1939–51. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965–73. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salminen S, Bouley C, Boutron-Ruault MC, Cummings JH, Franck A, Gibson GR, et al. Functional food science and gastrointestinal physiology and function. The British journal of nutrition. 1998;80 (Suppl 1):S147–S71. doi: 10.1079/bjn19980108. [DOI] [PubMed] [Google Scholar]
- 21.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 22.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–42. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 23.Karni R, Gus Y, Dor Y, Meyuhas O, Levitzki A. Active Src elevates the expression of beta-catenin by enhancement of cap-dependent translation. Molecular and cellular biology. 2005;25:5031–9. doi: 10.1128/MCB.25.12.5031-5039.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Penuel E, Martin GS. Transformation by v-Src: Ras-MAPK and PI3K-mTOR mediate parallel pathways. Mol Biol Cell. 1999;10:1693–703. doi: 10.1091/mbc.10.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Kim YS, Milner JA. Dietary modulation of colon cancer risk. The Journal of nutrition. 2007;137:2576S–9S. doi: 10.1093/jn/137.11.2576S. [DOI] [PubMed] [Google Scholar]
- 27.Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–36. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 29.Talamonti MS, Roh MS, Curley SA, Gallick GE. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. The Journal of clinical investigation. 1993;91:53–60. doi: 10.1172/JCI116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 31.Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012;8:1385–97. doi: 10.7150/ijbs.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. CSK: a protein-tyrosine kinase involved in regulation of src family kinases. The Journal of biological chemistry. 1991;266:24249–52. [PubMed] [Google Scholar]
- 33.Sabe H, Knudsen B, Okada M, Nada S, Nakagawa H, Hanafusa H. Molecular cloning and expression of chicken C-terminal Src kinase: lack of stable association with c-Src protein. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:2190–4. doi: 10.1073/pnas.89.6.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rengifo-Cam W, Konishi A, Morishita N, Matsuoka H, Yamori T, Nada S, et al. Csk defines the ability of integrin-mediated cell adhesion and migration in human colon cancer cells: implication for a potential role in cancer metastasis. Oncogene. 2004;23:289–97. doi: 10.1038/sj.onc.1207041. [DOI] [PubMed] [Google Scholar]
- 35.Oneyama C, Hikita T, Enya K, Dobenecker MW, Saito K, Nada S, et al. The lipid raft-anchored adaptor protein Cbp controls the oncogenic potential of c-Src. Mol Cell. 2008;30:426–36. doi: 10.1016/j.molcel.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Liang F, Luo Y, Dong Y, Walls CD, Liang J, Jiang HY, et al. Translational control of C-terminal Src kinase (Csk) expression by PRL3 phosphatase. The Journal of biological chemistry. 2008;283:10339–46. doi: 10.1074/jbc.M708285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Birt DF, Hendrich S, Wang W. Dietary agents in cancer prevention: flavonoids and isoflavonoids. Pharmacol Ther. 2001;90:157–77. doi: 10.1016/s0163-7258(01)00137-1. [DOI] [PubMed] [Google Scholar]
- 38.Chen H, Yao K, Nadas J, Bode AM, Malakhova M, Oi N, et al. Prediction of molecular targets of cancer preventing flavonoid compounds using computational methods. PloS one. 2012;7:e38261. doi: 10.1371/journal.pone.0038261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee KM, Lee KW, Jung SK, Lee EJ, Heo YS, Bode AM, et al. Kaempferol inhibits UVB-induced COX-2 expression by suppressing Src kinase activity. Biochem Pharmacol. 2010;80:2042–9. doi: 10.1016/j.bcp.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jung SK, Lee KW, Byun S, Kang NJ, Lim SH, Heo YS, et al. Myricetin suppresses UVB-induced skin cancer by targeting Fyn. Cancer research. 2008;68:6021–9. doi: 10.1158/0008-5472.CAN-08-0899. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.