

Summary
In response to unfavorable environmental conditions such as starvation, crowding and elevated temperature, Caenorhabditis elegans larvae enter an alternative developmental stage known as dauer [1], which exhibit dramatic remodeling of stress resistance and metabolism [2–3]. The genetic dissection of the molecular mechanisms of the C. elegans dauer developmental decision has defined evolutionarily conserved signaling pathways of organismal neuroendocrine physiology [2–4]. Here, we have identified a mechanism by which a dominant mutation in a neuronal insulin gene, daf-28(sa191) [5–7], causes constitutive entry into dauer diapause. We demonstrate that expression of the mutant DAF-28 insulin peptide results in endoplasmic reticulum (ER) stress in the ASI pair of chemosensory neurons. The neuronal ER stress does not compromise cellular survival, but activates PEK-1, the C. elegans ortholog of the mammalian eIF2α kinase PERK, which in turn phosphorylates Ser49 of eIF2α, specifically in the ASI neuron pair, to promote entry into dauer diapause. Our data establish a novel role for ER stress and the Unfolded Protein Response in promoting entry into dauer diapause, and suggest that in addition to cell autonomous activities in the maintenance of ER homeostasis, the Unfolded Protein Response may act in a cell non-autonomous manner to promote organismal adaptation to stress during larval development.
Results
The daf-28(sa191) Mutation Causes ER Stress in the ASI Chemosensory Neuron Pair
The daf-28(sa191) mutation results in constitutive entry into dauer diapause, partially independent of previously identified insulin and TGFβ signaling pathways ([6] and Table S1) but the mechanism is not well understood [5–7]. Molecular characterization of the daf-28(sa191) mutant revealed that the sa191 mutation results in an arginine-to-cysteine substitution at the predicted proteolytic cleavage site of the DAF-28 insulin peptide [7]. Genetic studies established that the daf-28(sa191) allele is semi-dominant and results in altered insulin/DAF-28 activity [5, 7]. Our characterization of two putative null alleles of daf-28, daf-28(tm2308) [8] and daf-28(gk411072), alongside daf-28 (sa191) corroborated these data (Figure 1A and Table S2).
Figure 1. The daf-28 (sa191) Mutation Causes ER Stress in the ASI Neuron Pair.
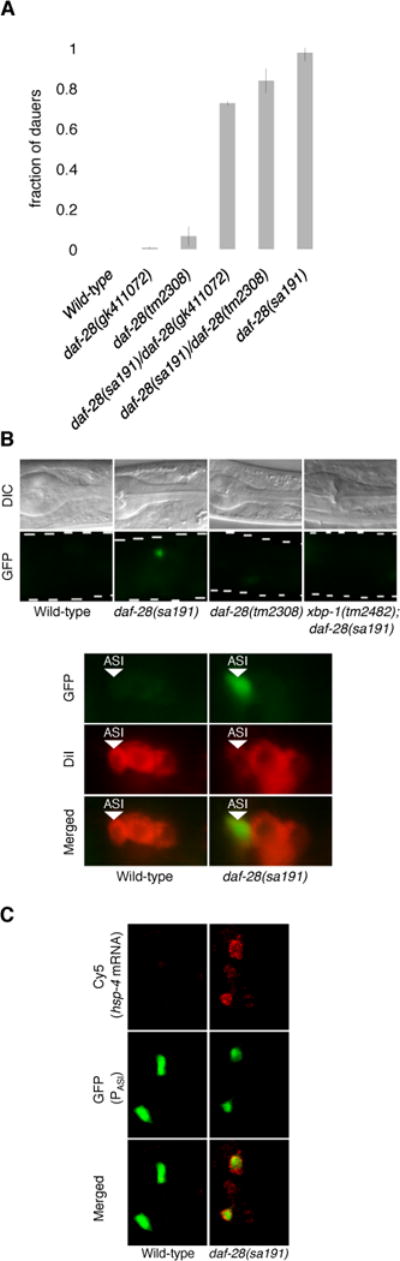
(A) Fractions of the indicated daf-28 mutants that enter the dauer stage at 25ºC. The tm2308 mutation harbors a 158-base pair deletion that results in premature truncation of the DAF-28 peptide, while the gk411702 mutation carries an early nonsense substitution mutation. Plotted is mean ± SD. The number of trials and animals scored is documented in Table S2.
(B) Fluorescence microscopy of the animals with indicated genotypes carrying the hsp-4p::GFP (zcIs4) transgene (GFP). Nomarski images (DIC) are provided for orientation. The amphid chemosensory neurons are identified by dye filling (DiI). The images shown are representative of three or more independent experiments.
(C) Maximum Z projection of smFISH stacked images of pre-dauer larvae with indicated genotypes. The ASI neurons are identified by GFP expression driven by the cell-specific daf-7 promoter [18, 19]. See also Figures S1 and S2.
In considering the gain-of-function nature of the sa191 allele, we hypothesized that the sa191 mutation might cause the production of a toxic insulin peptide that could disrupt protein folding homeostasis in the ER. Such a mechanism would be reminiscent of the Akita mouse model of insulin-dependent diabetes, in which a dominant mutation in an insulin gene causes ER stress in pancreatic beta cells [9]. Expression of the daf-28 gene was previously shown to be restricted to the ASI and ASJ chemosensory neurons [7]. To determine whether the sa191 mutant DAF-28 insulin peptide disrupts ER homeostasis, we utilized the fluorescent reporter transgene in which GFP is under the control of the promoter of hsp-4, a C. elegans ortholog of the ER chaperone BiP. Expression of hsp-4p::GFP(zcIs4) is induced by ER stress in diverse tissues [10]. We observed induction of GFP expression specifically in the ASI neuron pair in the daf-28(sa191) animals, but not in the ASI neuron pairs of wild-type or daf-28 loss-of-function mutant animals (Figure 1B). No hsp-4p::GFP expression was observed in the ASJ neuron pair of daf-28(sa191) animals.
Single molecule fluorescent in situ hybridization (smFISH) [11] confirmed upregulation of endogenous hsp-4 mRNA in the ASI neuron pair in the daf-28(sa191) mutant (Figure 1C). The highly sensitive smFISH method also revealed expression of hsp-4 mRNA in cells other than the ASI neuron pair in the daf-28(sa191) mutant (Figures 1C, S1A and S1B), but we observed that these cells were not the ASJ neuron pair (Figure S1A). We confirmed that the non-ASI, non-ASJ neuron expression of hsp-4 mRNA in the daf-28(sa191) mutant overlapped with additional cells expressing daf-28 as determined by smFISH analysis of daf-28 mRNA expression (Figures S1A and S1B).
We also observed hsp-4p::GFP expression in the ASI neurons in the daf-28(sa191)/+ heterozygote, consistent with the dominant nature of the sa191 allele (Figure S1C). ER stress induces hsp-4 expression in an IRE-1-XBP-1-dependent manner [10]. GFP expression in the ASI neuron pair of the daf-28(sa191) mutant was dependent on XBP-1, demonstrating that the observed upregulation of hsp-4 expression was a consequence of ER stress and UPR activation (Figure 1B). Further evidence that daf-28(sa191) mutant insulin expression is sufficient to trigger ER stress and UPR activation in a cell autonomous manner was obtained from ectopic overexpression of daf-28 in the intestine. We observed that intestinal expression of the sa191 mutant DAF-28 peptide, but not wild-type DAF-28, resulted in the induction of hsp-4p::GFP expression (Figure S1D). The mitochondrial UPR and the cytosolic heat shock response, as well as genes mediating oxidative stress response and autophagy, were not activated in the ASI neurons of the daf-28(sa191) mutant (data not shown), establishing that expression of the mutant DAF-28 insulin peptide specifically causes disruption of ER homeostasis in the ASI neuron pair.
The ASI Neuron Pair Remains Intact in the daf-28 (sa191) Mutant
The ASI neuron pair has been shown to play an important role in the dauer decision [12], and thus we considered whether neuronal ER stress promotes the dauer decision by compromising the survival of the ASI neuron pair. Three lines of evidence argue against such a mechanism. First, while laser ablation of the ASI neurons, in combination with a few other chemosensory neurons, results in constitutive entry into dauer diapause, dauer entry caused by chemosensory neuron ablation is dependent on DAF-3/SMAD [12], whereas the constitutive dauer entry phenotype of the daf-28 (sa191) mutant is independent of DAF-3 ([6] and Table S3). Second, the ASI neurons of the sa191 animals appeared morphologically intact (Figures 1B and S2C) and expressed the daf-28p::GFP transcriptional reporter (Figure S2A). Third, the ASI neuron pair of the daf-28(sa191) mutant are functional for the secretion of the transgenic translational fusion DAF-28::GFP (Figure S2B). These data strongly suggest that neuronal ER stress in the daf-28(sa191) mutant promotes entry into dauer diapause without compromising the survival of the ASI neuron pair.
Activation of the PERK/PEK-1 Branch of the UPR in the ASI Neuron Pair Promotes Entry into Dauer Diapause
Having determined that neuronal ER stress does not cause cell death in the daf-28(sa191) mutant, we sought to define the mechanism by which the ASI-specific ER stress promotes dauer entry. We hypothesized that the dauer developmental decision might be promoted by activation of the UPR, which in metazoans is comprised of three principal branches, mediated by IRE-1-XBP-1, PERK/PEK-1 and ATF-6 [13–15]. We found that loss-of-function mutations in the genes encoding the IRE-1, XBP-1 and ATF-6 did not affect the constitutive dauer entry phenotype of the daf-28(sa191) mutant, indicating that dauer entry is not triggered by the activation of IRE-1-XBP-1 and ATF-6 in response to ER stress (Figure 2A). Of note, at a lower temperature where the daf-28(sa191) mutation is less penetrant, mutations in xbp-1 and atf-6 that disrupt ER homoeostasis enhanced dauer formation in the daf-28(sa191) background (Figure S1E), consistent with our hypothesis that the daf-28(sa191) mutation causes dysregulation of ER homeostasis to promote the dauer decision.
Figure 2. Activation of PERK/PEK-1 in the ASI Neuron Pair of the daf-28(sa191) Mutant Promotes the Dauer Developmental Decision.
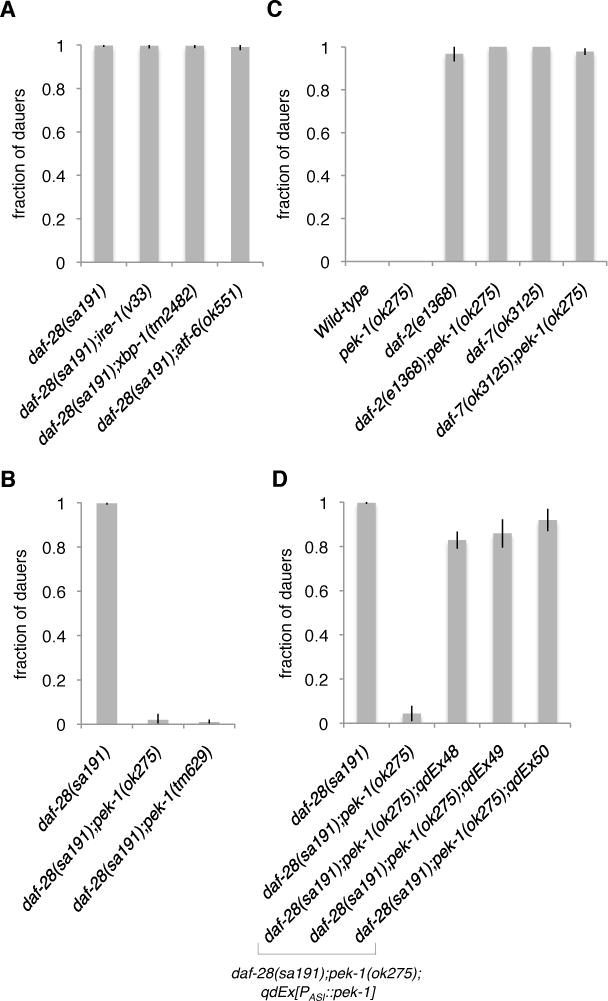
(A) Fractions of the daf-28(sa191) mutants with indicated ire-1, xbp-1, and atf-6 deficiencies that enter the dauer stage at 25ºC.
(B) Fractions of the daf-28(sa191) mutants with indicated pek-1 deficiencies that form dauer at 25ºC.
(C) Fractions of the indicated mutants in the pek-1-deficient background that form dauer at 25ºC.
(D) Fractions of the animals with indicated genotypes that enter the dauer stage at 25ºC. The daf-28(sa191);pek-1(ok275) strain in this figure represents non-transgenic controls from all three lines. Results from the cell-specific gpa-4 promoter are shown in this figure (referred to as PASI), and are confirmed with another ASI-specific promoter, str-3p (Figure S3B). The ASI neurons appeared intact in the animals where the cell-specific pek-1 transgene was expressed (Figure S2C).
Plotted is mean ± SD. The number of trials and animals scored is documented in Table S2. See also Figure S3.
In contrast, loss-of-function mutations in pek-1, which encodes the conserved ER stress-responsive eIF2α kinase PERK/PEK-1 [16], suppressed the constitutive dauer entry phenotype of daf-28(sa191) animals (Figure 2B), suggesting that the constitutive dauer entry phenotype of the daf-28(sa191) mutant is dependent on PEK-1. Mutation of gcn-2, encoding an ER stress-independent eIF2α kinase, had no effect on dauer entry in the daf-28(sa191) mutant (Figure S3A). Mutation of pek-1 had no effect on dauer formation in the wild-type animals or in dauer-constitutive mutants carrying mutations in genes encoding components of insulin and TGFβ signaling pathways (Figure 2C), demonstrating that PEK-1 activity is required for dauer entry specifically in response to neuronal ER stress in the daf-28(sa191) mutant, and that insulin and TGFβ signaling mechanisms for dauer entry are not dependent on PEK-1. Because we observed ER stress specifically in the ASI neuron pair of daf-28(sa191) animals (Figure 1), we anticipated that the corresponding activation of PEK-1 in the ASI neuron pair would be sufficient to promote entry into dauer diapause. Consistent with this expectation, ASI-specific expression of pek-1 was able to restore the constitutive dauer entry phenotype of the daf-28(sa191);pek-1(ok275) mutant, demonstrating that PEK-1 activation by ER stress in the ASI neuron pair is sufficient to promote entry into dauer diapause (Figures 2D and S3B).
The Dauer Developmental Decision Is Dependent on the Phosphorylation State of Ser49 of eIF2α in the ASI Neuron Pair
To further define the mechanism by which ASI-specific UPR activation promotes the dauer developmental decision, we asked whether ASI-specific eIF2α phosphorylation downstream of PEK-1 activation is required for the daf-28(sa191) mutant to constitutively enter dauer diapause. Ser49 is the conserved phosphorylation site of C. elegans eIF2α [17]. We overexpressed an unphosphorylatable version of eIF2α(S49A) specifically in the ASI neurons of the daf-28(sa191) mutant and observed partial suppression of the constitutive dauer entry phenotype (Figure 3A). We then asked whether ASI-specific eIF2α phosphorylation could functionally substitute for PEK-1 activation to promote dauer entry in response to neuronal ER stress. We introduced the ASI-specific phosphomimetic eIF2α(S49D) transgene into the daf-28(sa191);pek-1(ok275) mutant and found that this transgene substantially restored the constitutive dauer entry phenotype of the daf-28(sa191) mutant that was suppressed by pek-1(ok275) (Figure 3B). The ASI neurons appeared intact in the animals where the cell-specific eIF2α(S49D) transgene was expressed in the daf-28(sa191);pek-1(ok275) background (Figure S2C). Importantly, overexpression of the wild-type version of eIF2α in the ASI neuron pair did not influence the dauer developmental decision in response to neuronal ER stress (Table S3). To further establish that phosphorylation of Ser49 of eIF2α in the ASI neuron pair is the key event downstream of ER stress induced by daf-28(sa191) to promote dauer entry, we introduced the ASI-specific phosphomimetic eIF2α(S49D) transgene into the daf-28(tm2308) null mutant and observed that this transgene was sufficient to confer the constitutive dauer entry phenotype (Figure S3C). These data suggest that PEK-1 promotes dauer diapause through the phosphorylation of Ser49 of eIF2α in the ASI neuron pair.
Figure 3. Phosphorylation of Ser49 of eIF2αby PEK-1 in the ASI Neuron Pair Promotes Entry into Dauer Diapause.
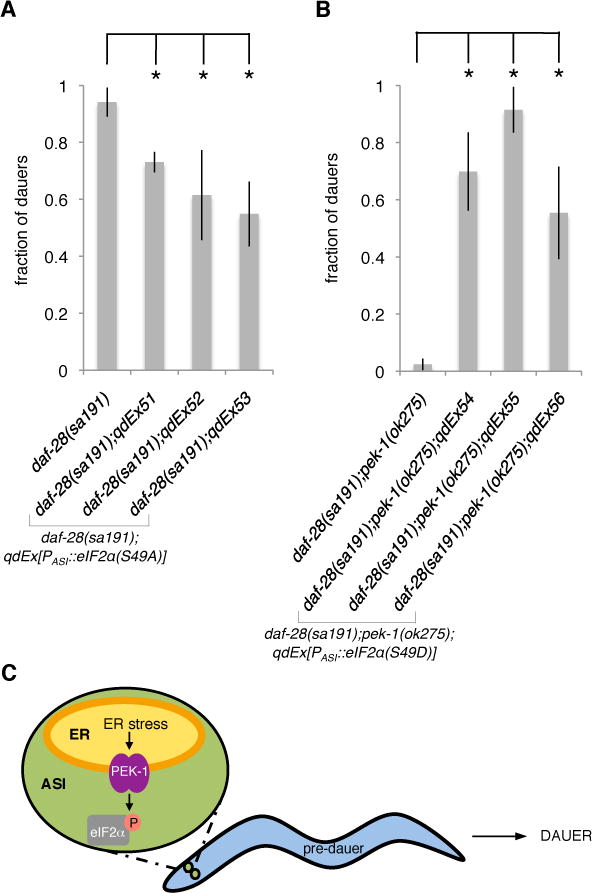
(A) Fractions of the animals with indicated genotypes that enter the dauer stage at 25ºC. The daf-28(sa191) strain in this figure represents non-transgenic controls from all three lines.
(B) Fractions of the animals with indicated genotypes that enter the dauer stage at 25ºC. The daf-28(sa191);pek-1(ok275) strain in this figure represents non-transgenic controls from all three lines. *P < 0.01 was determined by paired, two-tailed Student’s t-test (non-transgenic fraction vs. transgenic fraction for each line). Results from the ASI-specific gpa-4 promoter are shown in this figure (referred to as PASI).
Plotted is mean ± SD. The number of trials and animals scored is documented in Table S2. See also Figure S3.
(C) Schematic for the mechanism by which ER stress in the ASI neuron pair promotes entry into dauer diapause.
Discussion
The genetic study of the dauer diapause in C. elegans has served as an experimental paradigm for understanding how environmental cues influence organismal physiology through conserved neuroendocrine signaling mechanisms [1–4]. Our data demonstrate that ER stress in the ASI neuron pair, arising from the sa191 mutant DAF-28 insulin peptide, triggers activation of the conserved eIF2α kinase PEK-1 to promote entry into dauer diapause (Figure 3C). These data establish a previously uncharacterized mechanism promoting entry into dauer diapause in C. elegans, and moreover, suggest that PEK-1 phosphorylation of eIF2α not only functions to maintain ER homeostasis in the ASI neuron pair, but can have cell non-autonomous effects on larval development and organismal physiology.
Mutations in insulin genes have been shown to result in disruption in proinsulin processing, leading to ER stress and activation of the UPR [9, 18]. In the Akita mouse model [9], dominant mutations in an insulin gene impair proinsulin folding and result in chronic UPR activation that leads to dysfunctional pancreatic beta cells. Such effects of ER stress on pancreatic beta cells are cell autonomous, causing compromised cellular function and subsequent cell death in response to ER stress [14]. Because the ASI neuron pair plays a central neuroendocrine role in the regulation of C. elegans physiology, mediating food sensing, dauer formation and longevity [7, 12, 19–23], we initially considered that death and/or dysfunction of the stressed ASI neurons might underlie the observed daf-28(sa191) phenotype. However, our genetic analysis (Table S3 and Figure S2) suggests that the daf-28(sa191) mutation promotes dauer entry through a distinct mechanism from that which has been observed from ASI ablation. Moreover, the suppression of the daf-28(sa191) constitutive dauer entry phenotype by the pek-1 mutation (Figure 2) strongly suggests that the activation of PEK-1 and phosphorylation of eIF2α, not the toxic consequences of neuronal ER stress on cell survival, regulates the dauer entry decision. Our data suggest that neuronal ER stress in the daf-28(sa191) mutant activates UPR signaling and specifically PEK-1 to phosphorylate Ser49 of eIF2α in the ASI pair of neurons, which has organism-wide effects on larval development without causing death of the ASI neuron pair.
The sa191 mutant DAF-28 peptide triggers ER stress in the ASI neurons, but not in the ASJ neurons (Figures 1, S1A and S1B). The lack of detectable UPR activation in the ASJ neuron pair (Figures S1A and S1B) may be a consequence of lower daf-28 expression levels compared with expression in the ASI neuron pair, or differences in threshold of the ASI and ASJ neuron pairs in response to perturbation in ER homeostasis. The ASI neurons secrete a broad array of neuropeptides [7, 19–21] and thus may be more susceptible to disruptions that affect their secretory functions. We speculate that just as we have observed that infection with pathogenic bacteria and activation of innate immunity in the intestine activates the UPR [24], similarly adverse environmental conditions may trigger neuroendocrine responses in the ASI neuron pair that may induce ER stress and UPR activation. Alternatively, or in addition, by analogy to the induction of ER stress by metabolic dysregulation in mammals [25], fluctuations in nutrients and metabolites may disrupt ER homeostasis of ASI sensory neurons that regulate C. elegans organismal physiology [7, 12, 19–23].
The activation of the mitochondrial UPR, ER UPR and the heat shock response in specific tissues of C. elegans has been shown to alter stress response pathways of distal cells through the secretion of postulated “mitokines,” “secreted ER stress signal (SERSS)” and through a transcription factor FoxA-dependent transcellular chaperone signaling mechanism respectively [26–28]. As the ASI neuron pair does not directly innervate the downstream tissues that are remodeled during the dauer diapause, we speculate that the disruption of ER homeostasis in a pair of neurons may promote entry into dauer diapause through the secretion of neuroendocrine signals that are produced in the ASI neuron pair in response to eIF2α phosphorylation by PEK-1. Previous studies have established that DAF-16/FoxO, downstream of insulin signaling, is required for the constitutive dauer entry of the daf-28(sa191) mutant ([6] and Table S1). The partial suppression of the daf-28(sa191) dauer-constitutive phenotype by daf-16 may in part be due to the reduction of insulin signaling caused by loss of functional DAF-28, which is consistent with the observation that the daf-28(tm2308) null mutation confers a weak constitutive entry into dauer phenotype that is suppressed by daf-16 mutation [8]. PEK-1 phosphorylation of eIF2α in the ASI neuron pair may also activate DAF-16-dependent pathways, for example, through the altered translation of insulin peptides that mediate DAF-16 activity and the dauer developmental decision.
Roles for the mammalian UPR in plasma cell differentiation [29] and the recent report of olfactory receptor neuron fate commitment [30] have underscored the function of the UPR in not only maintaining cellular ER homeostasis, but in triggering developmental cell fate programs. Furthermore, ER stress in liver cells has been shown to influence diverse physiological outputs such as glucose metabolism [31] and iron homeostasis [32]. Our data suggests that the UPR not only functions to restore and maintain cellular ER homeostasis in the two ASI neurons, but that UPR activation in the ASI neuron pair can promote an organismal stress adaptation, namely entry into dauer diapause, during larval development. Evolutionary conservation of the mechanisms described in our studies would implicate a pivotal role for ER homeostasis and UPR signaling in how cellular stress and translational status are perceived and communicated in the physiology and development of more complex animals.
Experimental Procedures
Caenorhabditis elegans Strains
C. elegans strains were maintained as previously described [33]. The following strains were used in Figures 1, 2 and 3 (additional strains used in Figures S1, S2 and S3 and Tables S1, S2 and S3 are listed in Supplemental Experimental Procedures): N2 Bristol wild-type strain, JT191 daf-28(sa191), ZD699 daf-28(tm2308), ZD940 daf-28(gk411072), SJ4005 zcIs4[hsp-4p::GFP], ZD702 daf-28(sa191);zcIs4[hsp-4p::GFP], ZD759 daf-28(tm2308);zcIs4[hsp-4p::GFP], ZD801 xbp-1(tm2482);daf-28(sa191);zcIs4[hsp-4p::GFP], FK181 ksIs2[daf-7p::GFP], ZD1064 daf-28(sa191);ksIs2[daf-7p::GFP], ZD704 xbp-1(tm2482);daf-28(sa191), ZD937 ire-1(v33);daf-28(sa191), ZD846 daf-28(sa191);atf-6(ok551), ZD823 daf-28(sa191);pek-1(ok275), ZD824 daf-28(sa191);pek-1(tm629), MC366 pek-1(ok275), DR1572 daf-2(e1368), ZD915 daf-2(e1368);pek-1(ok275), ZD715 daf-7(ok3125), ZD916 daf-7(ok3125);pek-1(ok275), ZD842-844 daf-28(sa191);pek-1(ok27 5);qdEx48-50[gpa-4p::pek-1::unc-54 3′UTR], ZD928-929 and 932 daf-28(sa191);qdEx51-53[gpa-4p::eIF2α(S49A)::unc-54 3′UTR] and ZD951-953 daf-28(sa191);pek-1(ok275);qdEx54-56[gpa-4p::eIF2α(S49D)::unc-54 3′UTR].
Constructs and Generation of Transgenic Lines
Transgenic animals, carrying extrachromosomal arrays, were generated using standard microinjection methods [34]. Unless otherwise noted, injection mixture includes an indicated PCR fusion construct (injected at 15 ng/μl) and the ofm-1p::GFP co-injection marker (pQZ22, injected at 50 ng/μl), kindly provided by J. Alcedo. PCR fusion was performed as previously described [35]. The final heterologous constructs and their precursors were pooled from at least eight independent PCRs to minimize misincorporation rates, using high-fidelity DNA polymerase. The expected sizes of the constructs were confirmed by agarose gel electrophoresis.
The promoter of the gpa-4 gene was used as a cell-specific promoter to express transgenes. A second, independent ASI-specific promoter, str-3p, was also used to corroborate our results. Previous studies have validated the specificity of both promoters (see Figure S3 and Supplemental References)
Dauer Assay
Six to eight gravid animals were picked to individual well-seeded 6-cm NGM plates, allowed to lay eggs at the assay temperature for three to six hours, and removed. Live E. Coli strain OP50 was used as a food source. For the assays conducted at 25ºC, dauer and L4 larvae were scored at 48 hours after the egg-lay midpoint, as at this time point all the animals have passed the pre-dauer stages and, for the dauer-constitutive mutants that exit dauer, the dauer larvae have not resumed reproductive development. Dauers were discriminated from non-dauers based on radial shrinkage of the body, the absence of pharyngeal pumping, and an overall dark appearance [1]. Resistance to 1% detergent can be used to definitively confirm the dauer identity. To minimize variation in environmental conditions that could influence dauer formation, the same position in the incubator was used in all the experiments for temperature consistency, and the population density on each plate was controlled by the number of gravid adults laying eggs and the duration of egg-lay.
Microscopy
Animals were mounted with 10mM sodium azide onto slides with a 2% agarose pad. The slides were viewed using an AxioImager Z1 fluorescence microscope (Zeiss) primarily with a x40/1.3 (oil) objective. The fluorescence signals were recorded by a CCD camera (AxioCam), using constant exposure time without saturation. The images were captured and processed using the AxioVision image processor software. Lipophilic dye DiI was used to identify amphid neurons.
Supplementary Material
Highlights.
Expression of mutant insulin induces ER stress in two sensory neurons of C. elegans
Neuronal ER stress triggers PERK phosphorylation of eIF2α to promote dauer diapause
Unfolded Protein Response in two neurons promotes organismal stress adaptation
Acknowledgments
We thank J. Alcedo, M. Crowder, G. Ruvkun, S. Takagi and S. Tuck for providing strains and reagents. We thank the Caenorhabditis Genetics Center (CGC), which is supported by the NIH—National Center for Research Resources, and S. Mitani and the National BioResource Project (NBRP) for providing strains used in this study. This work was supported by NIH Grant GM084477 (to D.H.K.) and an Ellison New Scholar in Aging Award (to D.H.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information includes three figures, three tables, Supplemental Experimental Procedures and Supplemental References.
References
- 1.Cassada RC, Russell RL. The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev Biol. 1975;46:326–342. doi: 10.1016/0012-1606(75)90109-8. [DOI] [PubMed] [Google Scholar]
- 2.Hu PJ. Dauer. WormBook. 2007:1–19. doi: 10.1895/wormbook.1.144.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fielenbach N, Antebi A. C. elegans dauer formation and the molecular basis of plasticity. Genes Dev. 2008;22:2149–2165. doi: 10.1101/gad.1701508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riddle DL, Swanson MM, Albert PS. Interacting genes in nematode dauer larva formation. Nature. 1981;290:668–671. doi: 10.1038/290668a0. [DOI] [PubMed] [Google Scholar]
- 5.Malone EA, Thomas JH. A screen for nonconditional dauer-constitutive mutations in Caenorhabditis elegans. Genetics. 1994;136:879–886. doi: 10.1093/genetics/136.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malone EA, Inoue T, Thomas JH. Genetic analysis of the roles of daf-28 and age-1 in regulating Caenorhabditis elegans dauer formation. Genetics. 1996;143:1193–1205. doi: 10.1093/genetics/143.3.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W, Kennedy SG, Ruvkun G. daf-28 encodes a C. elegans insulin superfamily member that is regulated by environmental cues and acts in the DAF-2 signaling pathway. Genes Dev. 2003;17:844–858. doi: 10.1101/gad.1066503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornils A, Gloeck M, Chen Z, Zhang Y, Alcedo J. Specific insulin-like peptides encode sensory information to regulate distinct developmental processes. Development. 2011;138:1183–1193. doi: 10.1242/dev.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 11.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bargmann CI, Horvitz HR. Control of larval development by chemosensory neurons in Caenorhabditis elegans. Science. 1991;251:1243–1246. doi: 10.1126/science.2006412. [DOI] [PubMed] [Google Scholar]
- 13.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 14.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- 16.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 17.Nukazuka A, Fujisawa H, Inada T, Oda Y, Takagi S. Semaphorin controls epidermal morphogenesis by stimulating mRNA translation via eIF2alpha in Caenorhabditis elegans. Genes Dev. 2008;22:1025–1036. doi: 10.1101/gad.1644008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, Arvan P. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010;21:652–659. doi: 10.1016/j.tem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren P, Lim CS, Johnsen R, Albert PS, Pilgrim D, Riddle DL. Control of C. elegans larval development by neuronal expression of a TGF-beta homolog. Science. 1996;274:1389–1391. doi: 10.1126/science.274.5291.1389. [DOI] [PubMed] [Google Scholar]
- 20.Schackwitz WS, Inoue T, Thomas JH. Chemosensory neurons function in parallel to mediate a pheromone response in C. elegans. Neuron. 1996;17:719–728. doi: 10.1016/s0896-6273(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 21.Pierce SB, Costa M, Wisotzkey R, Devadhar S, Homburger SA, Buchman AR, Ferguson KC, Heller J, Platt DM, Pasquinelli AA, et al. Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 2001;15:672–686. doi: 10.1101/gad.867301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alcedo J, Kenyon C. Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron. 2004;41:45–55. doi: 10.1016/s0896-6273(03)00816-x. [DOI] [PubMed] [Google Scholar]
- 23.Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- 24.Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463:1092–1095. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Oosten-Hawle P, Porter RS, Morimoto RI. Regulation of organismal proteostasis by transcellular chaperone signaling. Cell. 2013;153:1366–1378. doi: 10.1016/j.cell.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 30.Dalton RP, Lyons DB, Lomvardas S. Co-opting the unfolded protein response to elicit olfactory receptor feedback. Cell. 2013;155:321–332. doi: 10.1016/j.cell.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. doi: 10.1038/nature08111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, Pietrangelo A. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325:877–880. doi: 10.1126/science.1176639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hobert O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques. 2002;32:728–730. doi: 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.