

Abstract
Paget’s disease of bone (PDB) is characterized by abnormal osteoclasts with unique characteristics that include: increased sensitivity of osteoclast progenitors to 1,25(OH)2D3, RANKL and TNF-α, increased osteoclast numbers, increased expression of IL-6 and several transcription factors. We recently reported that measles virus nucleocapsid protein (MVNP) plays a key role in the development of these abnormal osteoclasts. MVNP can induce the pagetic osteoclast phenotype in vitro and in vivo in TRAP-MVNP transgenic mice. However, the molecular mechanisms by which MVNP generates pagetic osteoclasts have not been determined. TANK-binding kinase 1 (TBK1) and IκB kinase-ɛ (IKKɛ) are IKK family members which complex with MVNP and activate both IRF3 and NF-κB pathways. MVNP increases the amount of TBK1 protein in bone marrow monocytes (BMM). Interestingly, we found that RANKL increased TBK1 and IKKɛ early in osteoclast differentiation, suggesting a possible role in normal osteoclastogenesis. However, only TBK1 is further increased in osteoclasts formed by TRAP-MVNP BMM due to increased TBK1 protein stability. TBK1 over-expression induced IL6 promoter reporter activity, and elevated endogenous IL6 mRNA and p65 NF-κB, TAF12 and ATF7 proteins in several cell lines. Over-expression of TBK1 was insufficient to induce pagetic osteoclasts from WT BMM, but synergized with MVNP to increase pagetic osteoclast formation from TRAP-MVNP BMM. BX795 inhibition of TBK1 impaired MVNP-induced IL-6 expression in both NIH3T3 cells and BMM, and shRNA knockdown of Tbk1 in NIH3T3 cells impaired IL-6 secretion induced by MVNP and decreased TAF12 and ATF7, factors involved in 1,25(OH)2D3 hypersensitivity of pagetic osteoclasts. Similarly, Tbk1 knockdown in BMM from TRAP-MVNP and WT mice specifically impaired development of the MVNP-induced osteoclast pagetic phenotype. These results demonstrate that TBK1 plays a critical role in mediating the effects of MVNP on osteoclast differentiation, and on the expression of IL-6, a key contributor to the pagetic osteoclast phenotype.
Keywords: TBK1, MVNP, IL-6, osteoclasts, Paget’s disease
INTRODUCTION
Paget’s disease of bone is characterized by abnormal osteoclasts that express a “pagetic phenotype” that distinguishes them from normal osteoclasts, which includes increased sensitivity of osteoclast progenitors to 1,25(OH)2D3, RANKL, and TNF-α, increased rate of formation, numbers of osteoclasts and nuclei/osteoclast, enhanced bone resorption capacity, and increased expression of the transcription regulator TAF12 and the cytokine IL-6 (1–3). Both environmental factors, such as measles virus, and genetic alterations have been implicated in the pathogenesis of Paget’s disease of bone (4, 5). The only gene in which protein-coding mutations are causally linked to Paget’s disease of bone is sequestosome 1, which encodes p62, a scaffold protein that is involved in signaling by several cytokines, including RANKL. The most common mutation is p62P392L, an autosomal dominant mutation with variable penetrance (6). However, mutant p62 alone does not appear to be capable of inducing Paget’s disease of bone because we found that mice with the murine equivalent of p62P392L (p62P394L) knocked-in or over-expressing p62P392L have increased osteoclast numbers, but the osteoclasts do not have a pagetic phenotype, nor do the mice develop the bone lesions characteristic of Paget’s disease (1, 7). Further, transfection of p62P392L cDNA into normal osteoclast precursors does not result in formation of pagetic-like osteoclasts in vitro (8). Previously, we reported that 70–80% of Paget’s disease patients expressed measles virus nucleocapsid protein (MVNP) (1, 9) and transduction of normal osteoclast precursors with MVNP cDNA results in development of osteoclasts that express the characteristics of pagetic osteoclasts (10). In addition, we also demonstrated that MVNP can induce the pagetic osteoclast phenotype in vitro and in vivo in TRAP-MVNP transgenic mice, in which the TRAP promoter drives MVNP expression in osteoclast lineage cells (11). Most importantly, we recently showed that antisense knockdown of MVNP expression in MVNP-positive osteoclasts from Paget’s patients harboring a p62 mutation resulted in loss of the pagetic phenotype (1, 9). Further, when mice carrying the mutant p62 knock-in were bred to TRAP-MVNP mice, the p62/MVNP mice developed greater numbers of pagetic osteoclasts and dramatic pagetic bone lesions that were strikingly similar to those seen in Paget’s disease (1, 9). However, the molecular mechanisms responsible for the capacity for MVNP to induce osteoclasts that express a pagetic phenotype are not well understood.
MVNP activates the antiviral response pathway by interacting with a complex containing the IκB kinase (IKK)-related homologous kinases, TANK-binding kinase 1 (TBK1) and IKKɛ (gene name IKBKE), and the transcription factor interferon regulatory factor 3 (IRF3) (12, 13). MVNP binding results in activation of TBK1 and IKKɛ, which in turn phosphorylate the IRF3 C-terminus, thereby activating IRF3 and resulting in nuclear translocation and transcriptional activation of its target genes, such as interferon β (IFNβ) (12, 13). Gene expression profiling studies indicated that IFNβ and associated molecules are up-regulated in pagetic peripheral blood monocytes (14). TBK1 and IKKɛ can also activate NF-κB both indirectly and directly. They interact with the TRAF-binding protein TANK (also called I-TRAF) complexed with TRAF2, and function upstream of IKKα and IKKβ to activate NF-κB indirectly (15, 16). TBK1 and IKKɛ also can directly phosphorylate p65 NF-κB at Ser468 and Ser536 (17–21), which allows IκB-independent nuclear translocation and potentiates transactivation of transcription (22).
We previously reported that MVNP increases p65 NF-κB, phospho-Ser536-p65 NF-κB, nuclear NF-κB and the activation of NF-κB-responsive reporters, including the IL6 gene (10, 23). In addition, the high levels of IL-6 observed in Paget’s disease of bone (2, 24) are induced in osteoclasts by MVNP but not by mutant p62 (1, 7, 8). We have shown that loss of IL-6 expression in TRAP-MVNP mice abrogated the formation of pagetic osteoclasts and bone lesions in vivo (1, 7, 8)). Therefore the mechanism by which MVNP elevates IL-6 is an important part of the generation of Paget’s disease. We hypothesized that MVNP activation of TBK1 and/or IKKɛ contributes to the formation of pagetic osteoclasts either by upstream regulation of IKK-dependent NF-κB activation or by increasing the IKK-independent activity of p65 NF-κB via direct phosphorylation, resulting in increased IL-6 expression. Therefore, we investigated whether TBK1 and/or IKKɛ mediates the effects of MVNP in pagetic osteoclasts, with a particular focus on MVNP up-regulation of IL-6 expression.
MATERIAL AND METHODS
Reagents
Cell culture media, penicillin and streptomycin (pen/strep) were from GIBCO-BRL (Grand Island, NY). Fetal calf serum (FCS), protease inhibitor cocktail and cycloheximide (CHX) were from Sigma-Aldrich (St. Louis, MO). Recombinant murine macrophage colony-stimulating factor (M-CSF), recombinant murine RANKL, recombinant murine IL-6, and quantikine mouse IL-6 ELISA kits were purchased from R&D Systems (Minneapolis, MN). Antibodies were from the following vendors: anti-TBK1 (#3013), anti-phospho-Ser172-TBK1 (#5483), anti-p65 NF-κB (#3034), anti-phospho-Ser536-p65 NF-κB (#3033) and anti-GFP (#2956) from Cell Signaling Technology (Beverly, MA); anti-IKKɛ (HPA015788) and mouse monoclonal antibody against β-actin (A5316) from Sigma-Aldrich; anti-TAF12 (12353-1-AP) was from Protein Tech (Chicago, IL); and anti-ATF7 (ab65064) and anti-MVNP antibody [3E1] (ab9397) were from Abcam (Cambridge, MA). HRP-conjugated IgG secondary antibodies were from GE Healthcare Life Sciences (Little Chalfont, UK). ECL reagents were from GE Healthcare Life Sciences (Buckinghamshire, UK). Polyvinyl difluoride (PVDF) membranes were from Bio-Rad Laboratories (Hercules, CA). The TBK1 inhibitor BX795 was from Invivogen (San Diego, CA).
Mice
The Institutional Animal Care and Use Committees at the Virginia Commonwealth University, University of Pittsburgh School of Medicine, and VA Pittsburgh Healthcare System approved all animal studies. Transgenic mice expressing MVNP under the control of the mouse TRAP promoter (TRAP-MVNP mice) were previously described (11).
Osteoclast formation from mouse bone marrow monocytes (BMM)
Nonadherent BMM were isolated from the tibias of 4–6 month old mice as previously described (25) and cultured at 2 × 105 per well in 96-well plates in 0.1 mL of α-modified Eagle medium (α-MEM) with 10% FCS and M-CSF (50 ng/mL) for 3 days at 37°C in 5% CO2. Osteoclast differentiation was induced by culturing in α-MEM with 10% FCS, M-CSF (50 ng/mL), and RANKL (50 ng/mL), with half of the media changed every other day. After 4 days of osteoclastogenic culture, the cells were fixed with 36.5% formaldehyde for 10 min. Osteoclast-like cells were stained for TRAP expression using a leukocyte acid phosphatase kit (Sigma). The TRAP-positive multinucleated cells with ≥3 nuclei were scored. All osteoclast formation experiments were scored from three independent wells.
Cell lines and culture conditions
NIH3T3 cell lines stably transduced with MVNP or empty vector (EV) were previously described (26). RAW264.7 cell lines stably transduced with MVNP or EV were made using the same vectors and methods described for the NIH3T3 cell lines (26). Mouse MC3T3-E1 subclone-4 (MC4) pre-osteoblasts were described previously (27) and maintained in ascorbic acid-free α-MEM–10% FCS-1% pen/strep (proliferation media). All the other cell lines, including HEK293, were grown in Dulbecco’s modified Eagle medium (DMEM) containing 10% FCS, 1% pen/strep, and 2 mM L-glutamine.
Plasmids and luciferase reporter assay
Constructs expressing wild-type and dominant negative (K38A) human IKKɛ and TBK1 as well as shRNA against human TBK1 were generously received from Dr. Tom Maniatis, Harvard University (28). The pcDNA3.1-MVNP (11) and FLAG-MVNP (23) expression constructs were previously described. Transfection of HEK293, EV/MVNP-NIH3T3, and EV/MVNP-RAW264.7 was done using Lipofectamine 2000 (Invitrogen). The −1200 IL-6 promoter driven pGL2 reporter (29) was a gift from Dr. Jian Zhang, University of Pittsburgh. All transfections included pRL-CMV as an internal control. Cell lysates were harvested with the Passive Lysis Buffer (Promega), and the luciferase activity of the IL-6 promoter reporter was determined by the dual luciferase assay (Promega) using a GloMax luminometer (Promega).
Lentivirus production and transduction
Mouse Tbk1 shRNA (GCTGGGTGAGATTTCAGACAT) and scrambled shRNA (CCTAAGGTTAAGTCGCCCTCG) lentiviruses (made from vector pLKO.1-puro) were generated by the UPCI lentivirus core facility. For TBK1 over-expression experiments, we inserted the human TBK1 cDNA into the FUGW lentiviral construct (which contains a GFP cDNA) kindly provided by Dr. Guoshang Xu, University of Pittsburgh (30), such that a TBK1-GFP fusion protein was encoded. To produce the lentivirus, supernatants were harvested from 293T cells (6.0 × 106/per 10-cm culture dish) transfected with FUGW or FUGW-TBK1 construct (10 µg), and VSV-G (2.5 µg), RSV-REV (3.5 µg), and PRRE (6.5 µg) constructs in antibiotic-free DMEM via lipofectamine2000 reagent for 48 h..
NIH3T3 cells were infected with cell-free lentiviral supernatants (106 pfu/mL) in the presence of polybrene (8 µg/mL) as previously described (31). To transduce mouse primary nonadherent BMM, the lentivirus was first concentrated with lenti-X™ concentrator (Clontech) according to the manufacturer’s instructions. The concentrated lentivirus transduction of BMM was aided by ViroMag R/L based on Magnetofection™ technology (Oz Biosciences, France) carried out according to the manufacturer’s instructions (32). For lentiviral transduction, the nonadherent cells (2 × 105/well) were mixed per well in 100 µL αMEM containing 10% FCS, M-CSF (10 ng/mL), ViroMag R/L beads (2 µL), and 10 µL concentrated lentiviral transduction particles (5 × 107/mL), and incubated for 15 min at RT before plating in 96-well culture plates. The plated cell-virus particle-magnet mixtures were incubated on a magnet plate for 60 min, and then the cells incubated with or without RANKL at 37°C with 5% CO2 for 4 days.
Enzyme-linked immunosorbent assay (ELISA)
Conditioned media from NIH3T3 cell lines was harvested 38 days after lentivirus infection and conditioned media from osteoclast cultures was collected 4 days after the addition of RANKL. The concentration of IL-6 present was determined using a Quantikine mouse IL-6 ELISA kit (R&D) according to the manufacturer's instructions.
Protein extraction and Western blot analysis
Collected cells were centrifuged at 500×g for 5 min, washed with cold phosphate-buffered saline, and lysed in cell lysis buffer (20 mM Tris HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 50 mM NaF, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, 1 mM ethylenediaminetetraacetic acid, 1 mM β-glycerophosphate, 2.5 mM sodium pyrophosphate, 1 mM orthovanadate, 1× protease inhibitor cocktail, 1 mM phenylmethylsulfonyl fluoride). After centrifugation at 14,000×g for 15 min at 4°C, equal amounts (10 µg) boiled protein samples were run on a 10% SDS polyacrylamide gel, transferred to PVDF membranes, probed with primary and the appropriate HRP-conjugated secondary antibodies. Signals were detected using ECL reagents and film. Band densitometric analysis was performed using Image J software (NIH).
RNA extraction and quantitative reverse transcription polymerase chain reaction (qPCR)
Total RNA was isolated from cell pellets using Trizol® reagent according to the manufacturer’s instructions. Reverse transcription was conducted using a kit (#A3500) from Promega (Madison, WI). An aliquot of each cDNA product was used for qPCR analysis to measure the relative mRNA levels using the iQ™ SYBR Green Supermix and iCycler iQ PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Samples were normalized to 18S RNA expression. The gene specific primers used are listed in Table 1.
Table 1.
Primer sequences for real-time RT-PCR (qPCR)
| MOUSE GENE NAME |
SENSE | ANTISENSE |
|---|---|---|
| IL6 | 5’-GAGTCCTTCAGAGAGATACAG-3’ | 5’-TGGTCTTGGTCCTTAGCC-3’ |
| Tnfrsf11a | 5’-GTGGAAATAAGGAGTCCTCAG-3’ | 5’-CACCGTCTTCTGGAACCATC-3’ |
| Ctsk | 5’-AATACGTGCAGCAGAACGGAGGC-3’ | 5’-CTCGTTCCCCACAGGAATCTCTCGTAC-3’ |
| Tbk1 | 5’-ACTGGTGATCTCTATGCTGTCA-3’ | 5’-TTCTGGAAGTCCATACGCATTG-3’ |
| 18S | 5’-CGCTTCCTTACCTGGTTGAT-3’ | 5’-GAGCGACCAAAGGAACCAA-3’ |
|
HUMAN GENE NAME |
||
| TBK1 | 5’-GCAGTTTGTTTCTCTGTATGGC-3’ | 5’-AATGTTACCCCAATGCTCCA-3’ |
Statistical analysis
All the experiments were repeated at least two independent times. Data are presented as the means ± standard deviation. Statistical significance was evaluated by Student’s t-test with ρ<0.05 and ρ<0.01 considered to be significant and highly significant, respectively.
RESULTS
MVNP increases total and activated TBK1, in part, by stabilizing TBK1 protein
Since MVNP can activate TBK1 and IKKɛ, we hypothesized that MVNP activation of one or both of these proteins plays a major role in transducing the signals that generate the pagetic phenotype in osteoclasts. We first investigated whether MVNP influenced TBK1 protein levels in RAW264.7 cells (pre-osteoclasts) and NIH3T3 cells. MVNP stably transduced RAW264.7 cells (MVNP-RAW264.7) were generated as previously published for the MVNP-NIH3T3 cells (10) and confirmed by qPCR for MVNP mRNA expression (Supplemental Figure S1). Western blot analysis indicated that both cell types stably expressing MVNP had increased levels of TBK1 protein as compared to cells carrying the empty-vector (EV) (Figure 1A, left panel). In addition, transient transfection of HEK293 cells with a MVNP expression plasmid dose-dependently increased total TBK1 levels (Figure 1A, right panel). Importantly, we found BMM isolated from transgenic TRAP-MVNP (TG-MVNP) mice, which selectively express MVNP in osteoclasts and their precursors, had increased TBK1 but not IKKɛ protein levels when compared to BMM from WT mice (Figure 1B day 0). Interestingly, RANKL treatment increased both TBK1 and IKKɛ protein levels in WT and TRAP-MVNP mice, with similar kinetics, both peaking at day 1, but again only TBK1 was increased to a greater extent in MVNP-expressing cells (Figure 1B). A more detailed time course analysis revealed that RANKL treatment of BMM increased the TBK1 protein level very rapidly in MVNP-containing BMM, with a 2-fold increase within 5 min and a 4-fold increase at 60 min after RANKL addition, while in WT BMM the TBK1 protein level response to RANKL was slower and more modest (Figure 1C). Of note, the serum starvation (2% FCS) and lack of M-CSF for 16 hr prior to addition of RANKL in this experimental protocol resulted in a smaller MVNP effect on TBK1 protein levels at time 0 compared to that observed in Figure 1B. Together, these data indicate that MVNP increases TBK1 protein levels and potentiates the capacity of RANKL to further increase TBK1 protein.
Figure 1. TBK1 expression is increased in TRAP-MVNP mouse osteoclast precursors, MVNP-RAW264.7 cells, and MVNP-NIH3T3 cells.

(A) Left panel, Cell lysates of RAW264.7 and NIH3T3 cells stably transduced with empty vector (EV) or MVNP expression constructs were immunoblotted for TBK1 and β-actin. Right panel, Cell lysates from HEK293 cells (12-well plates) transfected with MVNP expression construct (0–0.8 µg) for 36 h were immunoblotted for TBK1, MVNP, and β-actin. (B) BMM obtained from TRAP-MVNP (TG-MVNP) and wild-type (WT) mice and depleted of adherent cells were expanded for 3 d in M-CSF (50 ng/mL) and then cultured for osteoclast formation in the presence of RANKL (50 ng/mL) plus M-CSF (50 ng/mL) for 0, 1, 3, and 6 d. Cell lysates were immunoblotted for IKKε, TBK1, and β-actin. (C, D) Cell lysates were harvested and analyzed by Western blot for TBK1 and β-actin (C) After 16 h serum starvation, TG-MVNP and WT BMM obtained as in 1B were treated with RANKL (RL) for 0–60 min before harvest. (D) Left panel, EV- and MVNP-NIH3T3 cells treated with 100 µg/mL cyclohexamide (CHX) for the indicated times. Right panel, Following 24 h RANKL treatment, WT and TG-MVNP BMM were treated with CHX for the indicated times. Data are expressed as the mean ± SD (n=3). * (ρ<0.05) or ** (ρ<0.01), significantly different between samples as indicated.
We found that MVNP did not affect the levels of Tbk1 mRNA in NIH3T3 cells (Supplemental Figure S2), so we examined if MVNP affected TBK1 protein stability. Treatment of EV-NIH3T3 and MVNP-NIH3T3 cells with the protein synthesis inhibitor cyclohexamide (CHX) revealed that MVNP prolonged the half-life of TBK1 protein from 2.5 to 3.5 hours (Figure 1D, left panel). We then examined the stability of TBK1 protein in BMM from WT and MVNP mice. BMM cultured in the presence of RANKL for 24 hrs were treated with CHX for the indicated times. TBK1 protein half-life was about 4 hours in early WT osteoclasts and was significantly prolonged in MVNP-expressing cells (Figure 1D, right panel).
To confirm that there was more activated TBK1 in MVNP-containing cells, we analyzed the levels of phosphorylated-Ser172-TBK1 (P-TBK1) before and during TNF-α treatment of stably transduced MVNP-NIH3T3 and EV-NIH3T3 cells (Supplemental Figure S3). Before TNF-α addition, both the phospho-TBK1/β-actin (Supplemental Figure S3, left graph) and the total TBK1//β-actin (Supplemental Figure S3, middle graph) levels were higher in cells expressing MVNP, indicating that the total amount of steady-state activated TBK1 was higher in the MVNP containing cells. The phospho-TBK1/total TBK1 ratio (Supplemental Figure S3, right graph) was increased rapidly by TNF-α in a time-dependent manner in both EV- and MVNP-NIH3T3 cells. Although the phospho-TBK1/total TBK1 ratio was similar in both cell types, total TBK1 and phospho-TBK1 were much higher in MVNP-NIH3T3 cells. These results indicate that the total amount of activated TBK1 was greater in the MVNP-NIH3T3 cells than in EV-NIH3T3 cells.
TBK1 over-expression stimulated IL-6 promoter activity and increased the endogenous expression of IL-6, p65 NF-κB, ATF7 and TAF12
We and others have shown that MVNP can increase IL-6 expression when introduced into cells (23, 33). We recently reported that MVNP modulation of IL6 mRNA levels is via altered transcription rather than mRNA stabilization (23). Therefore, we determined if IL6 promoter regulation by MVNP could be mediated by TBK1 activity. We co-transfected expression constructs for TBK1, IKKɛ, and MVNP (or empty vector) with an IL6 promoter-luciferase reporter into HEK293 and NIH3T3 cells and assessed the relative activity of the transfected IL6 promoter. We found that the co-transfected IL6 promoter was activated by over-expressed TBK1, IKKɛ or MVNP in both HEK293 and NIH3T3 cells (Figure 2A). We then examined by qPCR analysis if transient transfection of TBK1, IKKɛ, and MVNP expression constructs stimulated the endogenous IL6 gene in NIH3T3 and RAW264.7 cells. We observed that, similar to the effects of MVNP expression, TBK1 and IKKɛ over-expression activated the endogenous IL6 gene in both cell types (Figure 2B).
Figure 2. TBK1 over-expression stimulated IL6 promoter-driven reporter activity and increased the endogenous expression of IL6, p65 NF-κB, and ATF7.

Cell lines HEK293, NIH3T3, and RAW264.7 were transfected with pCDNA3.1 (EV) or expression constructs for IKKɛ, TBK1, and MVNP as indicated. (A) Regulation of co-transfected IL6 reporter expression was assessed by luciferase assays. (B) Endogenous IL6 mRNA expression levels were analyzed by qPCR. (C) Cell lysates were immunoblotted for total (p65) and phospho-Ser536-p65 NF-κB (P-p65), ATF7, TAF12 and β-actin. Upper panel, Lysates of RAW264.7 cells stably transduced with EV or MVNP expression constructs. Lower panel, RAW264.7 cells transiently transfected with EV, IKKɛ, TBK1, and MVNP constructs. (D) HEK293 cells in 12-well plates were transfected with TBK1 expression constructs (0, 0.25, 0.5, 1 µg) or MVNP expression constructs (0, 0.2, 0.4, 0.8 µg) for 36 hrs, and cell lysates were immunoblotted for TBK1, total and P-p65, ATF7, TAF12, MVNP, and β-actin.
We previously reported that part of the pagetic phenotype induced by MVNP includes increases in the protein levels of the transcription factors TAF12, ATF7, and p65 NF-κB as well as increased phosphorylation of Ser536-p65 NF-κB (1, 23, 26, 34). Comparison of stably transfected EV- and MVNP-RAW264.7 cells demonstrated that MVNP increased all 3 transcription factors and the amount of phospho-Ser536-p65 NF-κB (Figure 2C, top panel). Consistent with the effects of transiently and stably expressed MVNP, both transient TBK1 and IKKɛ over-expression also increased phospho-Ser536-p65 NF-κB and total p65 NF-κB protein expression in RAW264.7 cells. However, only TBK1 over-expression mimicked the effect of MVNP in elevating ATF7 and TAF12 (Fig 2C, bottom panel). We then compared the effects of transient expression of MVNP and TBK1 in HEK293 cells on p65 NF-κB, TAF12, and ATF7 proteins. Similar to its effects in osteoclast precursors, MVNP increased all three of these transcription factors as well as phospho-Ser536-p65 NF-κB (Figure 2D). Unlike in RAW264.7 cells, TBK1 over-expression by itself in HEK293 cells dose-dependently increased p65 NF-κB (total and phospho-) and ATF7, but did not affect TAF12 protein levels (Figure 2D). In summary, TBK1 over-expression increased IL6 reporter expression, endogenous IL6 mRNA levels, and phospho-Ser536-p65 NF-κB, total p65 NF-κB, ATF7, and TAF12 protein levels. These results indicate that TBK1 is a likely candidate for mediating many of the downstream effects of MVNP in the cell.
Over-expression of TBK1 in WT mouse BMM is not sufficient by itself to induce pagetic osteoclasts, but synergizes with MVNP
To discern if TBK1 over-expression in BMM is sufficient to induce a pagetic osteoclast phenotype, we infected BMM from both WT and TG-MVNP mice with a lentivirus expressing a hTBK1-GFP fusion protein. The GFP tag allowed us to visualize the virus infection efficiency and hTBK1 expression. After viral transduction, expression of hTBK1-GFP was detectable by fluorescence microscopy and hTBK1-GFP mRNA expression was detectable by RT-PCR (Figure 3A). However, we could not detect the hTBK1-GFP protein by Western blot of lysates from the BMM with either anti-TBK1 or anti-GFP, although Western blotting with both of these antibodies detected the hTBK1-GFP protein in lysates from HEK293 and MC4 cells infected with this lentivirus (Supplemental Figure S4A). Cotransfection of the lentiviral plasmid encoding the hTBK1-GFP protein activated an IL6-promoter reporter in HEK293 cells, indicating that the hTBK1-GFP protein was functional (Supplemental Figure S4B). The above results suggest that the transduced BMM are expressing low levels of the exogenous hTBK1-GFP protein and/or that there is more proteolytic degradation of the protein during isolation from BMM resulting in lysates levels that are below the detection limits of Western blotting. TRAP staining after 4 days of RANKL treatment revealed that this low level of exogenous TBK1 protein was not sufficient by itself to increase the number of TRAP+ multinuclear osteoclasts formed. However, it was able to synergize with MVNP to significantly increase osteoclast number (~3-fold) compared with MVNP alone (Figure 3B). Similar results were obtained for the production of mouse IL6 mRNA (Figure 3C). Importantly, the transduction of TBK1 in WT cells increased the mRNA levels of Tnfrsf11a (RANK) by 3-fold and cathepsin K (Ctsk) by 2-fold (Figure 3D). The transduced TBK1 synergized with MVNP to increase all 3 mRNAs. Interestingly, IL6 and Ctsk mRNA were increased ~3-fold over MVNP alone (similar to osteoclast number), while RANK mRNA was increased 21-fold. These results indicate that increasing TBK1 by itself did not recapitulate the effects of MVNP, and the synergy with MVNP suggests that MVNP activation of TBK1 is important.
Figure 3. TBK1 over-expression enhanced MVNP-induced osteoclastogenesis in BMM.

BMM from WT and TRAP-MVNP (TG-MVNP) mice prepared as described in Figure 1 were infected with FUGW-GFP (EV) and FUGW-TBK1-GFP (TBK1) expression lentiviruses. After 4 d RANKL, (A) hTBK1-GFP fusion protein expression was visualized using fluorescence microscopy, and hTBK1-GFP mRNA expression was detected (along with endogenous 18S RNA) by qPCR and UV light visualization of the products separated on a 2% agarose gel. (B) TRAP+ multinucleated cells with ≥3 nuclei were scored as osteoclasts per well. Data are expressed as the mean ± SD (n=3). (C, D) Isolated RNA was analyzed by qPCR for endogenous murine (C) IL6, (D) Tnfrsf11a (RANK) and Ctsk mRNA levels. LV, lentivirus; GT, BMM genotype.
Knockdown of TBK1 with a shRNA lentivirus impaired MVNP’s capacity to increase IL-6 secretion, as well as TAF12 and ATF7 protein expression
To test whether TBK1 is necessary for MVNP’s modulation of IL-6 expression, and other aspects of the pagetic osteoclast phenotype, we first tested the effects of a pharmacological catalytic activity inhibitor of TBK1/IKKɛ, BX795, on IL6 mRNA expression by EV- and MVNP-NIH3T3 cells. BX795 dose-dependently inhibited the MVNP-induced increase in steady-state IL6 mRNA (Figure 4A). We also investigated if decreasing TBK1 amount or activity in HEK293 cells blocked the effects of transfected MVNP by co-transfecting MVNP with a shTBK1 plasmid and a dominant-negative TBK1 (DN-TBK1) expression vector, which is a kinase-inactive TBK1(K38A) mutant (28). These results showed that MVNP increased phospho-p65 NF-κB, total p65 NF-κB, and ATF7 protein levels (Figure 4B). Co-transfection with either DN-TBK1 or shTBK1 blocked these MVNP-induced changes (Figure 4B). Together these results indicate that MVNP requires TBK1 signal transduction for at least some of the observed effects of MVNP expression.
Figure 4. Inhibition or knockdown of TBK1 in NIH3T3 and HEK293 cells impaired MVNP effects.

(A) EV-NIH3T3 and MVNP-NIH3T3 cells were treated with TBK1 inhibitor BX795 (0, 0.1, 1 µM) for 24 hrs and endogenous IL6 RNA expression was analyzed by qPCR. (B) Lysates from HEK293 cells transfected with the following vectors for 36 h: EV, MVNP alone, 1:1 MVNP:TBK1(K38A) (DN-TBK1), or MVNP plus shTBK1 plasmid were immunoblotted for TBK1, total and phosphorylated-p65 NF-κB (P-p65), ATF7, and β-actin. (C) Lysates from MVNP-NIH3T3 cells infected with 2 different shTBK1 lentiviruses were immunoblotted for ATF7, TAF12, TBK1 and β-actin. (D, E) EV- and MVNP-NIH3T3 cells infected with scrambled (SCR) control and shTBK1 lentiviruses were treated with puromycin (2 µg/mL) at 6 d post-infection and thereafter. (D) At 6 d post-infection, cell lysates were immunoblotted for TBK1 protein (lower band is TBK1; upper band is non-specific) and β-actin, and (E) secreted IL-6 levels in cell culture supernatants were detected by ELISA assay at 38 d post-infection. NS, not significant; ** (ρ<0.01), significantly different between samples as indicated.
Infection of MVNP-NIH3T3 cells with two different shTBK1 lentivirus clones decreased the levels of TBK1, ATF7, and TAF12 protein, although shTBK1 lentivirus #2 was more effective (Figure 4C). Therefore, we utilized shTBK1 lentivirus #2 for all our additional studies. EV- and MVNP-NIH3T3 cells were transduced with both scrambled control (SCR) and shTBK1 lentiviruses. Puromycin was added at 6 days post-infection and thereafter. At 38 days post-infection, TBK1 protein expression levels were assessed by Western blotting, which revealed that the MVNP-increased TBK1 protein was significantly decreased by the shTBK1 (Figure 4D-lower band in doublet). ELISA analysis of cell culture supernatants revealed that secreted IL-6 protein levels were equivalent in supernatants from SCR and shTBK1 lentivirus transduced EV-NIH3T3 cells, indicating that TBK1 signaling does not regulate the basal production of IL-6 in these cells (Figure 4E). As expected, there was much higher IL-6 in the supernatants from SCR-lentivirus transduced MVNP-NIH3T3 cells than in shTBK1-lentivirus transduced EV-NIH3T3 cells. Most importantly, TBK1 down-regulation by shTBK1 lentivirus decreased the ability of MVNP to increase IL-6 so dramatically that the MVNP-NIH3T3 cells produced only slightly more than the EV-NIH3T3 cells (Figure 4E).
Knockdown of TBK1 in MVNP-expressing osteoclast progenitors impaired the increase in osteoclast formation induced by RANKL and TNF-α
To determine whether TBK1 is required for MVNP induction of a pagetic osteoclast phenotype, we first treated BMM from WT and TRAP-MVNP mice with the TBK1/IKKɛ inhibitor BX795 and analyzed the effect on IL6 gene expression. Consistent with our results in NIH3T3 cells (Figure 4A), the MVNP-elevated IL6 mRNA expression was dose-dependently inhibited by BX795 (Figure 5A). We then used the shTBK1 and SCR lentiviruses to infect, aided by magnetofection, the nonadherent BMM from WT and TG-MVNP mice. The shTBK1 lentivirus markedly reduced the TBK1 protein in both WT and TG-MVNP cells after 1 day RANKL exposure (Figure 5B). TRAP staining of RANKL-treated BMM showed that MVNP still increased the osteoclast number obtained from BMM infected with the SCR lentivirus when compared to WT. Importantly, TBK1 knockdown blocked the increased osteoclast formation from TG-MVNP BMM (Figure 5C and Supplemental Figure S5A). The SCR lentivirus did not prevent MVNP from increasing the mRNA levels of IL6 (Figure 5D) and the osteoclast marker genes RANK and Ctsk (Figure 5E). The shTBK1 lentivirus in WT osteoclasts did not affect the mRNA levels of IL6 and the osteoclast marker gene RANK, but was slightly inhibitory for expression of Ctsk mRNA. Importantly, the shTBK1 lentivirus strongly decreased the MVNP effects on expression of IL6, RANK, and Ctsk. To confirm these effects are not caused by a variation in transduction efficiency, we did qPCR to assay the mRNA levels of the puromycin resistance gene (Puro) expressed by the lentiviruses used and found similar levels in each of the groups, indicating that the transduction efficiencies in the four cell populations were similar (Supplemental Figure S5B).
Figure 5. Knockdown of TBK1 impaired MVNP-induced increased response of osteoclast progenitors to RANKL.

BMM from WT and TRAP-MVNP (TG-MVNP) mice were prepared the same as in Figure 1. (A) IL6 mRNA levels were assayed by qPCR following BX795 treatment (0, 0.1, 0.5 µM) for 24 h. (B–E) The nonadherent cells were infected with shTBK1 lentivirus and scrambled (SCR) lentivirus. After RANKL treatment for 4 days, (B) endogenous TBK1 protein expression levels were probed by Western blot. (C) Micrograph of TRAP stained osteoclasts (oiginal magnification 100×) and graph of TRAP+ multinucleated cells with 3 or more nuclei scored as osteoclasts per well. (D) IL6 and, (E) RANK and Ctsk endogenous mRNAs were analyzed by qPCR. Data are expressed as the mean ± SD (n=3). NS, not significant; * (ρ<0.05) or ** (ρ <0.01), significantly different between samples as indicated.
A similar experiment with TNF-α revealed that knockdown of TBK1 also impaired the MVNP-induced increased response of osteoclast progenitors to TNF-α, completely blocking the MVNP effects on osteoclast formation, as well as IL6, RANK, and Ctsk mRNA expression (Supplemental Figure S6). These results demonstrate that there is an even tighter requirement for TBK1 function for MVNP to induce pagetic osteoclasts in response to TNF-α treatment than observed with RANKL. Together, these data demonstrate that TBK1 mediates the MVNP-induced increased response of osteoclast progenitors to RANKL and TNF-α.
DISCUSSION
Osteoclasts expressing MVNP display a pagetic phenotype (1, 10, 11). We found that MVNP activates NF-κB, increases IL-6 production, and induces pagetic osteoclast formation through an alternative pathway that involves activation of TBK1. The presence of MVNP increased TBK1 in the four cell types examined (RAW264.7, NIH3T3, HEK293, and primary BMM) and TBK1 activation as reflected in the increased amount of phosphorylated-Ser172-TBK1. Over-expression of TBK1 in the cell lines increased expression of both co-transfected IL6 promoter luciferase reporters and the endogenous IL6 gene, indicating that the large increase in TBK1 promoted auto-activation of TBK1 that resulted in activation of the IL6 gene. Increased expression of IL-6 is not only a marker of a pagetic osteoclast, but also is required for MVNP expressing mice to form pagetic osteoclasts and bone lesions and is not induced by the p62P392L mutant (1, 2, 10, 11). Inhibition of TBK1 by BX795 or knockdown of TBK1 by shTBK1 lentivirus blocked the capacity of MVNP to up-regulate IL6 gene expression in both NIH3T3 cells and primary BMM, as well as blocked the MVNP-induced increase in osteoclast numbers after RANKL or TNF-α stimulation. Interestingly, the presence of IKKɛ in the TBK1 knockdown cells could not compensate for the loss of TBK1, suggesting that TBK1 and IKKɛ are not functionally redundant in their capacity to transduce the MVNP signal to regulate IL6 expression and osteoclast formation. However, these data do not indicate that IKKɛ has no role in osteoclasts. RANKL rapidly upregulated both TBK1 and IKKɛ protein levels in BMM. Interestingly, we did not observe an effect of TBK1 knockdown on the osteoclast differentiation of BMM from WT mice. This may indicate that IKKɛ can compensate for loss of TBK1 during normal osteoclastogenesis. Together these data demonstrate that MVNP-activation of TBK1 is required for upregulation of IL6 expression and formation of pagetic osteoclasts (Figure 6).
Figure 6. MVNP-induced increase in TBK1 protein and activity contributes to the generation of pagetic osteoclasts.
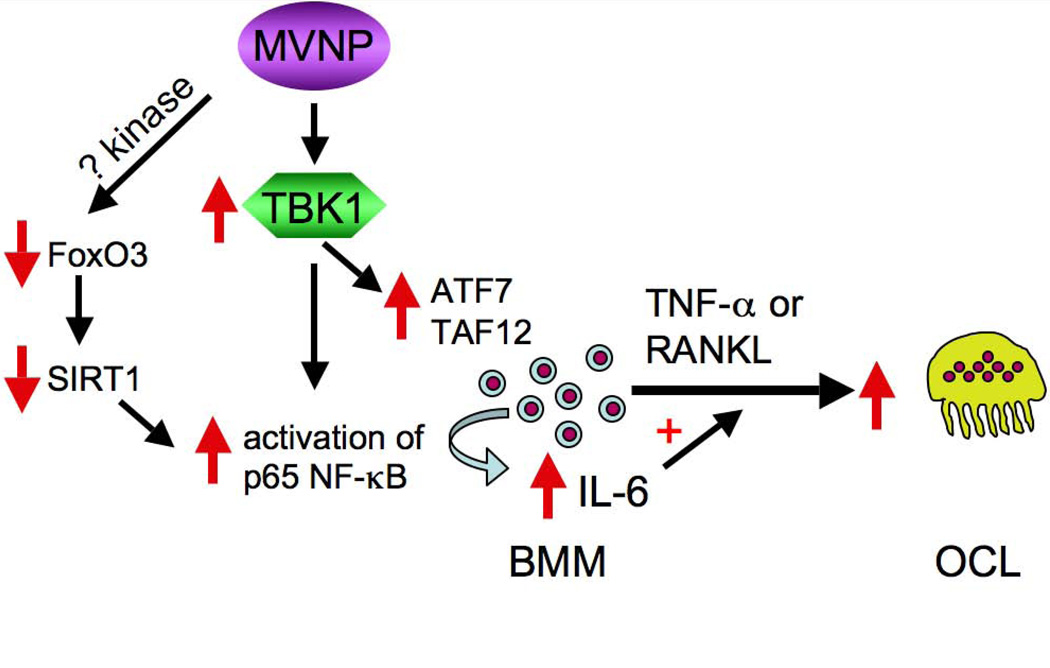
MVNP stabilization of TBK1 protein and stimulation of TBK1 activation in BMM results in increased total and phospho-Ser536-p65 NF-κB, ATF7 and TAF12. These changes induce elevation of IL-6 expression, which acts to increase the number of osteoclasts formed in response to RANKL and TNF-α. ATF7 and TAF12 increases also result in hypersensitivity to 1,25(OH)2D (34). While TBK1 directly phosphorylates Ser536-p65 NF-κB, MVNP decreases Sirt1 via activation of phosphorylation-induced proteolysis of the Sirt1 regulator FoxO3, which also leads to increased NF-κB activity by decreasing its deacetylation, further contributing to increased IL6 expression. The identity of the FoxO3 kinase activated by MVNP is not known. These two pathways stimulated by MVNP act in concert to increase IL-6 production, a key feature and requirement for developing pagetic osteoclasts.
TBK1/IKKɛ have been reported to activate both IRF3/IRF7 (12) and NF-κB (17, 19, 21), and possibly both C/EBPβ and C/EBPδ (35). We focused on NF-κB in this study because we have shown that MVNP increases both total p65 NF-κB and phospho-Ser536-p65 NF-κB, and NF-κB is a crucial regulator of IL6 gene expression (36). We confirmed that TBK1 up-regulates p65 NF-κB in RAW264.7 cells and in HEK293 cells. More significantly, use of a dominant-negative TBK1 or TBK1 knockdown in HEK293 cells blocked the MVNP-induced increase in both total p65 NF-κB and phospho-Ser536-p65 NF-κB, suggesting that MVNP requires TBK1 to enhance NF-κB activity, and thereby increase IL6 transcription.
We have recently published that MVNP can induce phosphorylation of the transcription factor FoxO3, leading to its proteolysis and to a decrease in the expression of its target Sirtuin 1 (Sirt1), an NF-κB deacetylase, thereby increasing NF-κB activity and IL6 expression (10, 23). Among the kinases reported to phosphorylate FoxO3 are AKT and IKKɛ (37–39). Further TBK1 and IKKɛ have been reported to activate AKT (40, 41). Our preliminary data (not shown) revealed that over-expression of TBK1 did not decrease Sirt1 and loss of TBK1 activity did not rescue Sirt1 expression from MVNP repression. These data indicate that MVNP does not act via TBK1 to regulate Sirt1 expression. Therefore, both decreased Sirt1 activity (10, 23) and increased TBK1 activity are required for MVNP to increase IL6 gene expression (Figure 6).
TAF12, a VDR co-activator that plays an important role in the increased osteoclast formation in Paget’s disease by increasing the sensitivity of osteoclast precursors to 1,25-(OH)2D3, is increased by the presence of MVNP in the cell (26, 42, 43). We recently reported that MVNP increases ATF7, which interacts with TAF12, and that ATF7 increases TAF12 expression and contributes to the hypersensitivity of osteoclast precursors to 1,25-(OH)2D3 in Paget’s disease (34). Therefore, we assessed whether TBK1 mediates MVNP regulation of TAF12 and/or ATF7. In both RAW264.7 and HEK293 cells, TBK1 over-expression increased ATF7, but only in RAW264.7 cells did we observe increased TAF12. In the HEK293 cells, a dominant-negative TBK1 or TBK1 knockdown repressed the MVNP-induced increase of ATF7. TBK1 knockdown decreased both ATF7 and TAF12 in MVNP-NIH3T3 cells. These data indicate that MVNP increases ATF7 via TBK1 activity in all three cell types. However, MVNP acts through TBK1 to elevate TAF12 in both RAW264.7 and NIH3T3 cells, but not in HEK293 cells, perhaps reflecting cell-type differences. Since little is known about how TAF12 and ATF7 expression is regulated, we do not know if TBK1 activation of NF-κB or another transcription factor is involved in elevating ATF7 and TAF12 expression.
Protein stability assays showed that MVNP slowed TBK1 degradation. TBK1 stability was reported to be regulated by the presence of K63-linked versus K48-linked polyubiquitination, which is dynamically altered by a number of E3 ligases, such as K63 E3 ligases MIB1 and 2 (44), K48 E3 ligase TRIP (45), and de-ubiquinylation enzyme A20 (with the help of TAX1BP1) (46, 47). Which E3 ligases or DUBs are differentially recruited in the presence of MVNP remains to be elucidated. RANKL more rapidly up-regulated TBK1 protein in the presence of MVNP, with a ~2-fold increase within 5 min, followed by a continual increase through 60 min. The rapidity of the change suggests that RANKL potentiates the MVNP stabilization of TBK1 or increases translation.
Infection of BMM from WT and TRAP-MVNP mice with a lentivirus expressing a hTBK1-GFP fusion protein resulted in potentiation of the MVNP effects on osteoclast formation and IL-6 expression, but did not alter these in WT cells, suggesting that the low level of ectopic hTBK1-GFP protein in these cells was not able to trans-autophosphorylate and auto-activate (48) and required MVNP-induced activation. However, the ectopic hTBK1-GFP levels were capable of increasing RANK and Ctsk mRNA above EV in WT cells after 4 days of RANKL induction. Together these results indicate TBK1 over-expression by itself is not sufficient to induce aberrant osteoclast formation, but requires either activation by MVNP, or a partner that is altered by MVNP.
Our data indicates that MVNP-induced activation of TBK1, and the consequent increased IL-6 production via enhanced NF-κB is important for the generation of pagetic osteoclasts. However, activated TBK1 and IKKɛ have several other functions that may also contribute to the generation of pagetic osteoclasts. TBK1 has been reported to be involved in regulating autophagic maturation and to phosphorylate p62 at Ser403 in the ubiquitin binding domain, thereby enhancing binding and clearance (49). TBK1 also is critical for K-Ras dependent non-small lung cancer cells because it drives basal autophagy via the cargo receptors NDP52 and TAX1BP1, which promotes non-canonical NF-κB signaling (50). This may be another pathway by which MVNP regulation of TBK1 enhances NF-κB activation of the IL6 gene. Both TBK1 and IKKɛ can activate AKT via a PI3K-indendent pathway (40) and a PI3K-dependent pathway (51). The PI3K/AKT signaling pathway plays a critical role in activating osteoclast differentiation and bone resorption in normal and diseased states (52). Increased osteoclast survival may contribute to the pagetic osteoclast phenotype. Of great interest is that TBK1 binds optineurin (OPTN), a NEMO-like IKK family member, that both regulates TBK1 activity and is phosphorylated by TBK1 on Ser177, which enhances LC3 binding affinity and promotes autophagy (53, 54). Genome-wide association studies have identified variants in the OPTN gene as a genetic risk factor for Paget’s disease, although the variations are not in the coding region (55). It remains to be elucidated if MVNP alters the interaction of TBK1 and OPTN.
Our data from both cell lines and mouse BMM demonstrated that TBK1 plays a critical role in mediating the effects of MVNP on the expression of p65 NF-κB, TAF12, ATF7, and IL-6, key contributors to the pagetic osteoclast phenotype. It has been shown that TBK1 and IKKɛ are potential therapeutic targets in rheumatoid arthritis (56, 57). Therapeutics that inhibit TBK1 activity, like the inhibitor BX795, and other more specific inhibitors that are in the pipeline (58, 59) may prevent the development of Paget’s disease of bone.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully thank Dr. Tom Maniatis, for TBK1 and IKKɛ plasmids, Dr. Jian Zhang for the IL6 promoter reporters, Dr. Guoshuang Xu for FUGW, RSV-REV, PRRE, the UPCI Lentivirus Facility for the GFP, SCR and shTBK1 lentiviruses, and the Veterans Administration Pittsburgh Healthcare System, Research and Development for use of the facilities. We thank other members of our laboratories for helpful discussion. Support for the UPCI Lentiviral Facility was provided by the Cancer Center Support Grant from the National Institutes of Health (P30 CA047904). This work was supported by the National Institutes of Health (NIAMS grants AR057310 to D.L.G. and AR057308 to G.D.R.) and Research Funds from the Veteran’s Administration (to G.D.R.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIAMS, NIH, the Department of Veterans Affairs, or the United States Government.
Footnotes
DISCLOSURES
G.D.R. is a consultant to Amgen and develops CME materials for Clinical Care Options. All other authors state that they have no conflicts of interest.
Authors’ roles: Study design: QS, GDR, and DLG. Study conduct: QS, BS, FW, NK, JJW, DLG. Data collection: QS and BS. Developed and provided the transgenic mice: JJW. Created the EV and MVNP-RAW264.7 cell lines: NK. Data analysis: QS and DLG. Data interpretation: QS, GDR, and DLG. Drafting manuscript: QS and DLG. Revising manuscript content: QS, FW, JJW, GDR, and DLG. Approving final version of manuscript: all authors. DLG takes responsibility for the integrity of the data analysis.
REFERENCES
- 1.Kurihara N, Hiruma Y, Yamana K, Michou L, Rousseau C, Morissette J, Galson DL, Teramachi J, Zhou H, Dempster DW, Windle JJ, Brown JP, Roodman GD. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget's disease. Cell Metab. 2011 Jan 5;13(1):23–34. doi: 10.1016/j.cmet.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roodman GD, Kurihara N, Ohsaki Y, Kukita A, Hosking D, Demulder A, Smith JF, Singer FR. Interleukin 6. A potential autocrine/paracrine factor in Paget's disease of bone. J Clin Invest. 1992 Jan;89(1):46–52. doi: 10.1172/JCI115584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrlich LA, Roodman GD. The role of immune cells and inflammatory cytokines in Paget's disease and multiple myeloma. Immunological reviews. 2005 Dec;208:252–266. doi: 10.1111/j.0105-2896.2005.00323.x. [DOI] [PubMed] [Google Scholar]
- 4.Chung PY, Van Hul W. Paget's disease of bone: evidence for complex pathogenetic interactions. Semin Arthritis Rheum. 2012 Apr;41(5):619–641. doi: 10.1016/j.semarthrit.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Roodman GD, Windle JJ. Paget disease of bone. J Clin Invest. 2005 Feb;115(2):200–208. doi: 10.1172/JCI24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002 Jun;70(6):1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurihara N, Hiruma Y, Zhou H, Subler MA, Dempster DW, Singer FR, Reddy SV, Gruber HE, Windle JJ, Roodman GD. Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest. 2007 Jan;117(1):133–142. doi: 10.1172/JCI28267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiruma Y, Kurihara N, Subler MA, Zhou H, Boykin CS, Zhang H, Ishizuka S, Dempster DW, Roodman GD, Windle JJ. A SQSTM1/p62 mutation linked to Paget's disease increases the osteoclastogenic potential of the bone microenvironment. Hum Mol Genet. 2008 Sep 2;17(23):3708–3719. doi: 10.1093/hmg/ddn266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget's disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab. 1995 Jul;80(7):2108–2111. doi: 10.1210/jcem.80.7.7608263. [DOI] [PubMed] [Google Scholar]
- 10.Kurihara N, Reddy SV, Menaa C, Anderson D, Roodman GD. Osteoclasts expressing the measles virus nucleocapsid gene display a pagetic phenotype. J Clin Invest. 2000 Mar;105(5):607–614. doi: 10.1172/JCI8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurihara N, Zhou H, Reddy SV, Garcia Palacios V, Subler MA, Dempster DW, Windle JJ, Roodman GD. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget's disease-like bone lesions in mice. J Bone Miner Res. 2006 Mar;21(3):446–455. doi: 10.1359/JBMR.051108. [DOI] [PubMed] [Google Scholar]
- 12.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003 May 16;300(5622):1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 13.tenOever BR, Servant MJ, Grandvaux N, Lin R, Hiscott J. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J Virol. 2002 Apr;76(8):3659–3669. doi: 10.1128/JVI.76.8.3659-3669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagy ZB, Gergely P, Donath J, Borgulya G, Csanad M, Poor G. Gene expression profiling in Paget's disease of bone: upregulation of interferon signaling pathways in pagetic monocytes and lymphocytes. J Bone Miner Res. 2008 Feb;23(2):253–259. doi: 10.1359/jbmr.071021. [DOI] [PubMed] [Google Scholar]
- 15.Nomura F, Kawai T, Nakanishi K, Akira S. NF-kappaB activation through IKK-i-dependent I-TRAF/TANK phosphorylation. Genes Cells. 2000 Mar;5(3):191–202. doi: 10.1046/j.1365-2443.2000.00315.x. [DOI] [PubMed] [Google Scholar]
- 16.Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. Embo J. 1999 Dec 1;18(23):6694–6704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adli M, Baldwin AS. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-kappaB activity via regulation of Ser-536 p65/RelA phosphorylation. J Biol Chem. 2006 Sep 15;281(37):26976–26984. doi: 10.1074/jbc.M603133200. [DOI] [PubMed] [Google Scholar]
- 18.Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004 Dec 31;279(53):55633–55643. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- 19.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004 May;5(5):392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 20.Mattioli I, Geng H, Sebald A, Hodel M, Bucher C, Kracht M, Schmitz ML. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J Biol Chem. 2006 Mar 10;281(10):6175–6183. doi: 10.1074/jbc.M508045200. [DOI] [PubMed] [Google Scholar]
- 21.Wietek C, Cleaver CS, Ludbrook V, Wilde J, White J, Bell DJ, Lee M, Dickson M, Ray KP, O'Neill LA. IkappaB kinase epsilon interacts with p52 and promotes transactivation via p65. J Biol Chem. 2006 Nov 17;281(46):34973–34981. doi: 10.1074/jbc.M607018200. [DOI] [PubMed] [Google Scholar]
- 22.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007 Jan;8(1):49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 23.Wang FM, Sarmasik A, Hiruma Y, Sun Q, Sammut B, Windle JJ, Roodman GD, Galson DL. Measles virus nucleocapsid protein, a key contributor to Paget's disease, increases IL-6 expression via down-regulation of FoxO3/Sirt1 signaling. Bone. 2013 Dec 20;53(1):269–276. doi: 10.1016/j.bone.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoyland JA, Freemont AJ, Sharpe PT. Interleukin-6, IL-6 receptor, and IL-6 nuclear factor gene expression in Paget's disease. J Bone Miner Res. 1994 Jan;9(1):75–80. doi: 10.1002/jbmr.5650090111. [DOI] [PubMed] [Google Scholar]
- 25.Mundy GR, Roodman GD, Bonewald LF, Oreffo RO, Boyce BF. Assays for bone resorption and bone formation. Methods Enzymol. 1991;198:502–510. doi: 10.1016/0076-6879(91)98049-c. [DOI] [PubMed] [Google Scholar]
- 26.Kurihara N, Reddy SV, Araki N, Ishizuka S, Ozono K, Cornish J, Cundy T, Singer FR, Roodman GD. Role of TAFII-17, a VDR binding protein, in the increased osteoclast formation in Paget's Disease. J Bone Miner Res. 2004 Jul;19(7):1154–1164. doi: 10.1359/JBMR.040312. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Christensen K, Chawla K, Xiao G, Krebsbach PH, Franceschi RT. Isolation and characterization of MC3T3-E1 preosteoblast subclones with distinct in vitro and in vivo differentiation/mineralization potential. J Bone Miner Res. 1999 Jun;14(6):893–903. doi: 10.1359/jbmr.1999.14.6.893. [DOI] [PubMed] [Google Scholar]
- 28.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature immunology. 2003 May;4(5):491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Johnston G, Stebler B, Keller ET. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase. Antioxid Redox Signal. 2001 Jun;3(3):493–504. doi: 10.1089/15230860152409121. [DOI] [PubMed] [Google Scholar]
- 30.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002 Feb 1;295(5556):868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 31.Xu G, Liu K, Anderson J, Patrene K, Lentzsch S, Roodman GD, Ouyang H. Expression of XBP1s in bone marrow stromal cells is critical for myeloma cell growth and osteoclast formation. Blood. 2012 May 3;119(18):4205–4214. doi: 10.1182/blood-2011-05-353300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofmann A, Wenzel D, Becher UM, Freitag DF, Klein AM, Eberbeck D, Schulte M, Zimmermann K, Bergemann C, Gleich B, Roell W, Weyh T, Trahms L, Nickenig G, Fleischmann BK, Pfeifer A. Combined targeting of lentiviral vectors and positioning of transduced cells by magnetic nanoparticles. Proc Natl Acad Sci U S A. 2009 Jan 6;106(1):44–49. doi: 10.1073/pnas.0803746106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helin E, Vainionpaa R, Hyypia T, Julkunen I, Matikainen S. Measles virus activates NF-kappa B and STAT transcription factors and production of IFN-alpha/beta and IL-6 in the human lung epithelial cell line A549. Virology. 2001 Nov 10;290(1):1–10. doi: 10.1006/viro.2001.1174. [DOI] [PubMed] [Google Scholar]
- 34.Teramachi J, Hiruma Y, Ishizuka S, Ishizuka H, Brown JP, Michou L, Cao H, Galson DL, Subler MA, Zhou H, Dempster DW, Windle JJ, Roodman GD, Kurihara N. Role of ATF7-TAF12 interactions in the vitamin D response hypersensitivity of osteoclast precursors in Paget's disease. J Bone Miner Res. 2013 Jun;28(6):1489–1500. doi: 10.1002/jbmr.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kravchenko VV, Mathison JC, Schwamborn K, Mercurio F, Ulevitch RJ. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J Biol Chem. 2003 Jul 18;278(29):26612–26619. doi: 10.1074/jbc.M303001200. [DOI] [PubMed] [Google Scholar]
- 36.Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, Haegeman G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998 Feb 6;273(6):3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- 37.Luron L, Saliba D, Blazek K, Lanfrancotti A, Udalova IA. FOXO3 as a new IKK-epsilon-controlled check-point of regulation of IFN-beta expression. Eur J Immunol. 2012 Apr;42(4):1030–1037. doi: 10.1002/eji.201141969. [DOI] [PubMed] [Google Scholar]
- 38.Yang JY, Hung MC. Deciphering the role of forkhead transcription factors in cancer therapy. Curr Drug Targets. 2011 Aug;12(9):1284–1290. doi: 10.2174/138945011796150299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999 Mar 19;96(6):857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 40.Guo JP, Coppola D, Cheng JQ. IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J Biol Chem. 2011 Oct 28;286(43):37389–37398. doi: 10.1074/jbc.M111.287433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, Brekken R, Wurz R, Tasker A, Polverino T, Tan SL, White MA. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011 Feb 18;41(4):458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurihara N, Ishizuka S, Demulder A, Menaa C, Roodman GD. Paget's disease-a VDR coactivator disease? J Steroid Biochem Mol Biol. 2004 May;89–90(1–5):321–325. doi: 10.1016/j.jsbmb.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 43.Kurihara N, Yamana K, Roodman GD. Expression of TAFII-17 and IL-6 in osteoclast (OCL) precursors is sufficient for formation of pagetic-like OCL. J Bone Miner Res. 2005;20(Sup 1):1207. Abstract. [Google Scholar]
- 44.Wang L, Li S, Dorf ME. NEMO binds ubiquitinated TANK-binding kinase 1 (TBK1) to regulate innate immune responses to RNA viruses. PLoS One. 2012;7(9):e43756. doi: 10.1371/journal.pone.0043756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang M, Wang L, Zhao X, Zhao K, Meng H, Zhao W, Gao C. TRAF-interacting protein (TRIP) negatively regulates IFN-beta production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J Exp Med. 2012 Sep 24;209(10):1703–1711. doi: 10.1084/jem.20120024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saitoh T, Yamamoto M, Miyagishi M, Taira K, Nakanishi M, Fujita T, Akira S, Yamamoto N, Yamaoka S. A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol. 2005 Feb 1;174(3):1507–1512. doi: 10.4049/jimmunol.174.3.1507. [DOI] [PubMed] [Google Scholar]
- 47.Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 2010 May 14;285(20):14999–15009. doi: 10.1074/jbc.M110.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A. 2012 Jun 12;109(24):9378–9383. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012 Aug 24;37(2):223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newman AC, Scholefield CL, Kemp AJ, Newman M, McIver EG, Kamal A, Wilkinson S. TBK1 Kinase Addiction in Lung Cancer Cells Is Mediated via Autophagy of Tax1bp1/Ndp52 and Non-Canonical NF-kappaB Signalling. PLoS One. 2012;7(11):e50672. doi: 10.1371/journal.pone.0050672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, Wang CY, Guan KL. IkappaB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci U S A. 2011 Apr 19;108(16):6474–6479. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cicek M, Vrabel A, Sturchio C, Pederson L, Hawse JR, Subramaniam M, Spelsberg TC, Oursler MJ. TGF-beta inducible early gene 1 regulates osteoclast differentiation and survival by mediating the NFATc1, AKT, and MEK/ERK signaling pathways. PLoS ONE. 2011;6(3):e17522. doi: 10.1371/journal.pone.0017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011 Jul 8;333(6039):228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kachaner D, Genin P, Laplantine E, Weil R. Toward an integrative view of Optineurin functions. Cell Cycle. 2012 Aug 1;11(15):2808–2818. doi: 10.4161/cc.20946. [DOI] [PubMed] [Google Scholar]
- 55.Albagha OM, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R, Dunlop MG, Fraser WD, Hooper MJ, Isaia G, Nicholson GC, del Pino Montes J, Gonzalez-Sarmiento R, di Stefano M, Tenesa A, Walsh JP, Ralston SH. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget's disease of bone. Nat Genet. 2010 Jun;42(6):520–524. doi: 10.1038/ng.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corr M, Boyle DL, Ronacher L, Flores N, Firestein GS. Synergistic benefit in inflammatory arthritis by targeting I kappaB kinase epsilon and interferon beta. Ann Rheum Dis. 2009 Feb;68(2):257–263. doi: 10.1136/ard.2008.095356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Migita K, Nakamura T. TBK1: a potential therapeutic target in RA. Rheumatology. 2012 Apr;51(4):588–589. doi: 10.1093/rheumatology/ker207. [DOI] [PubMed] [Google Scholar]
- 58.Hutti JE, Porter MA, Cheely AW, Cantley LC, Wang X, Kireev D, Baldwin AS, Janzen WP. Development of a high-throughput assay for identifying inhibitors of TBK1 and IKKepsilon. PLoS One. 2012;7(7):e41494. doi: 10.1371/journal.pone.0041494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McIver EG, Bryans J, Birchall K, Chugh J, Drake T, Lewis SJ, Osborne J, Smiljanic-Hurley E, Tsang W, Kamal A, Levy A, Newman M, Taylor D, Arthur JS, Clark K, Cohen P. Synthesis and structure-activity relationships of a novel series of pyrimidines as potent inhibitors of TBK1/IKKepsilon kinases. Bioorg Med Chem Lett. 2012 Dec 1;22(23):7169–7173. doi: 10.1016/j.bmcl.2012.09.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.