

Factor V (FV) is primarily synthesized in the liver and is the precursor of factor Va (FVa), a cofactor that accelerates conversion of prothombin to thrombin by activated factor X [1]. Most FV in blood is in the plasma, but up to 25% is found in platelet α-granules[1]. Although megakaryocytes are capable of synthesizing FV, the majority of platelet FV in humans is of hepatic origin, internalized from plasma via clathrin-dependent endocytosis [2]. FV deficiency may be hereditary or acquired. Inherited severe FV deficiency is a rare autosomal recessive disorder, with an estimated prevalence of 1:1,000,000 individuals due to homozygosity or compound heterozygosity for FV gene mutations [1]. Interestingly, the bleeding phenotype in FV deficient patients is highly variable and difficult to predict based on plasma FV activity levels. Acquired FV deficiency is mainly due to the development of antibody inhibitors. FV inhibitors have been described in patients with previous exposure to bovine thrombin preparations, underlying rheumatologic disease or malignancy, or after treatment with antibiotics [3]. The inhibitors may directly affect FV/Va activity or storage and processing of the protein [3]. We report a case of acquired FV deficiency with low plasma FV activity and no bleeding diathesis that does not appear to be due to a classic FV inhibitor.
A 64 year old Caucasian female was referred to our hematology clinic for evaluation of a markedly prolonged prothrombin time (PT, 36.0 s [normal 9.1-11.6 s]) and activated partial thromboplastin time (APTT, 94.4 s [normal 24.2-35.6 s]), identified during preoperative evaluation for left hip replacement. Despite these impressive abnormalities, she did not have a history of significant bleeding with prior surgical procedures, including right hip replacement, spinal surgery and partial left knee replacement as an adult; and tonsillectomy and appendectomy as a child. Her PT and APTT were normal on multiple occasions in the past, suggesting that the current results reflected an acquired process. The platelet count, thrombin time, fibrinogen level, and d-dimer level were within normal range. The PT and APTT corrected on mixing with normal plasma (0 and 2 hour incubation) indicating a factor deficiency, rather than the presence of an inhibitor. Plasma V activity was markedly reduced (1% of normal), while levels of factors II and X were within normal range. A standard Bethesda assay failed to identify a FV inhibitor, consistent with the results of the mixing study.
The patient’s medical history and clinical examination did not indicate the presence of autoimmune disease. She was up to date with all age appropriate cancer screening. She may have been exposed to bovine thrombin during prior surgical procedures, but we were unable to confirm this. Results of functional studies for lupus anticoagulants, and ELISAs for anticardiolipin and anti-beta-2-glycoprotein antibodies (IgG, IgM) were unremarkable. ELISA and western blots (Figure 1A) demonstrated absence of FV antigen in the patient’s plasma. While the initial studies did not support the presence of an inhibitor that neutralized FV activity, we investigated the possibility that the patient had an antibody that cleared FV from plasma. However, an ELISA designed to detect anti-FV antibodies in the patient plasma yielded negative results.
Figure 1. Factor V and TFPI in Patient Plasma. (A and B) Western blots of plasma for FV.
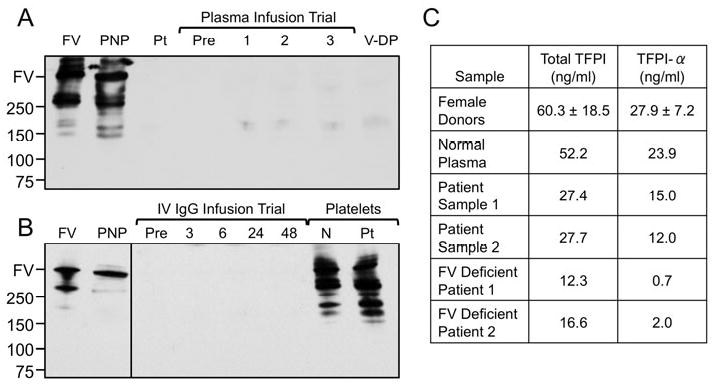
Plasma samples were size-fractionated on 4-20% polyacrylamide SDS-gels, followed by western blotting with polyclonal goat-anti-human FV IgG. (A) Lanes were loaded with purified FV (FV); pooled normal plasma (PNP); patient plasma (Pt); patient plasma before (Pre), and 1, 2, or 3 hours after FFP infusion; and plasma from a patient with known severe FV deficiency (V-DP). (B) Lanes are loaded with purified FV (FV); pooled normal plasma (PNP); patient plasma before (Pre), and 3, 6, 24 and 48 hours after intravenous IgG; normal platelets; and patient platelets. For both panels, positions of molecular mass standards in kilo-Daltons are shown on the left, along with an indicator for the normal position of migration for human FV. (C) TFPI. Total TFPI and TFPIα in normal plasma, patient plasma from two separate draws, and plasma from two different FV deficient patients were measured by ELISA. Results are compared to those for a cohort of normal female donors (mean ± 1 S.D., n = 15). Patient Sample 1 corresponds to the “Pre“ sample in panel A. Patient Sample 2 corresponds to the “Pre” sample in panel B.
Our inability to identify an antibody suggested that the absence of FV antigen in the patient’s plasma represented a true production problem. However, there was no increase in plasma FV activity or antigen after infusion of three units of fresh frozen plasma (FFP) (Figure 1A), indicating that the FV deficiency was due, at least in part, to rapid selective removal from the circulation. Possible causes included an antibody that rapidly clears FV (undetected by our ELISA) without neutralizing its activity or selective binding of FV to tissues. During a 30-day course of high dose corticosteroids, FV levels remained low, with a peak of 7% midway through the course but with a subsequent drop to her baseline value of 1%. While the response to steroids was modest, these data confirm that the patient’s liver is capable of synthesizing FV. We empirically treated the patient with intravenous immunoglobulin (IV IgG), which also had little effect (Figure 1B).
Emori et al. [4] described a Japanese man with primary amyloidosis, acquired FV deficiency and gastrointestinal bleeding. Importantly, infusion of FFP resulted in a modest transient increase in plasma FV level. The transient nature of the response was attributed to FV binding to amyloid fibrils. Such a mechanism has been reported most frequently for acquired factor X deficiency in patients with amyloidosis [5]. We evaluated the patient for amyloidosis by obtaining bone marrow and fat pad biopsies, and by performing serum and urine protein electrophoresis. None of these studies supported a diagnosis of amyloidosis. A relatively mild bleeding phenotype in some patients with severe deficiency of plasma FV has been attributed to normal levels of platelet FV [6]. On western blot (Figure 1B) and ELISA, FV antigen was detectable in the patient’s platelets at a roughly comparable amount to FV in normal control platelets. As FV in human platelets appears to be primarily of plasma origin, the results in Figure 1B are consistent with the premise that our patient’s liver makes FV, and suggest that the absence of bleeding symptoms may be related to a normal platelet FV pool.
Recent evidence suggests that the bleeding tendency in patients with severe congenital FV deficiency may in part be mitigated by concomitant defects in anticoagulant mechanisms. Duckers et al. [7] described low tissue factor pathway inhibitor-alpha (TFPIα) antigen and activity in the plasma of patients with congenital FV deficiency. We also noted reduced TFPIα in plasma from two patients with severe congenital FV deficiency (Figure 1C). TFPI is a Kunitz-type protease inhibitor that binds and inhibits the activities of factor Xa and the factor VIIa/tissue factor complex [8]. A substantial fraction of plasma TFPIα circulates as a complex with FV in plasma, and absence of FV might reduce its stability or enhance its clearance [8]. We measured total TFPI and TFPIα in our patient’s plasma (Figure 1C) at two time points corresponding to the pre-plasma infusion (Sample 1) and pre-IVIG infusion (Sample 2) in Figures 1A and 1B, respectively. Total TFPI and TFPIα were below the normal range established with a gender-matched cohort, but were not as low as those observed in plasma from congenital FV deficient patients. These results are consistent with the reported effects of reduced plasma FV on TFPI, and indicate that acquired FV deficiency in this patient is unlikely to be due to an antibody to TFPI that clears the TFPI-FV complex.
Because of increasing discomfort, the patient underwent left hip replacement without plasma FV replacement, and received standard post-operative anticoagulation therapy with enoxaparin. She did not experience excessive bleeding. Interestingly, two weeks postoperatively, she was readmitted for a periprosthetic fracture that required surgical revision. She tolerated the procedure without bleeding complications. While we were not able to define the mechanism behind her FV deficiency, the clinical data support a process in which plasma FV (either the patient’s endogenous FV or FV in FFP infusions) is rapidly cleared from the circulation. The deficiency could be related to rapid immune-mediated clearance of FV, or to FV binding to vascular or extravascular tissues. It is unlikely that the findings are related to an abnormality in the patients FV molecules, as FV administered with FFP was also cleared rapidly. The normal PT/APTT results in the past, the relatively normal level of FV in platelets, and the transient elevation in FV during steroid therapy indicate that the patient’s liver makes FV. These observations, along with the normal levels of other coagulation factors, argue against a FV synthesis defect.
This patient’s case highlights the discrepancy between standard coagulation assay results and clinical presentation that can occur in severe FV deficiency. Despite the lack of circulating plasma FV, the patient did not experience abnormal bleeding when challenged with a procedure that can be associated with significant blood loss. This is consistent with the observation that some patients with congenital FV deficiency with normal FV levels in their platelets have a relatively mild phenotype [9]. The low TFPI level caused by the FV deficiency may also have contributed to the absence of a bleeding diathesis in this patient, similar to what has been described in congenital FV deficient patients.
Acknowledgments
I.R. Sosa is supported by NIH T32 GM007569-36 training grant.
Footnotes
Authorship
I.R.S reviewed the medical record and literature, contributed to data analysis, and wrote and revised the manuscript. P.E. performed and interpreted assays of plasma TFPI. A.E.M and A.T.N contributed to experimental design and preparation of the manuscript. D.G. designed and performed experiments, analyzed data and revised manuscript. All authors have approved the final submitted version.
Disclosures
Dr. Gailani receives consultant’s fees for projects unrelated to this manuscript from pharmaceutical companies: Merck & Co., Bayer Pharma AG and Novartis Pharmaceuticals Corporation. Dr. Mast receives research funding from Novo Nordisk for projects unrelated to this manuscript. The other authors have no interests which might be perceived as posing a conflict or bias.
References
- 1.Lippi G, Favaloro EJ, Montagnana M, Manzato F, Guidi GC, Franchini M. Inherited and acquired factor V deficiency. Blood Coagul Fibrinolysis. 2011;22:160–166. doi: 10.1097/MBC.0b013e3283424883. [DOI] [PubMed] [Google Scholar]
- 2.Duckers C, Simioni P, Rosing J, Castoldi E. Advances in understanding the bleeding diathesis in factor V deficiency. Br J Haematol. 2009;146:17–26. doi: 10.1111/j.1365-2141.2009.07708.x. [DOI] [PubMed] [Google Scholar]
- 3.Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis. 2011;31:449–457. doi: 10.1007/s11239-010-0529-6. [DOI] [PubMed] [Google Scholar]
- 4.Emori Y, Sakugawa M, Niiya K, Kiguchi T, Kojima K, Takenaka K, Shinagawa K, Ishimaru F, Ikeda K, Tanimoto M, Yamasaki R, Ohara N, Harada M. Life-threatening bleeding and acquired factor V deficiency associated with primary systemic amyloidosis. Blood Coagul Fibrinolysis. 2002;13:555–559. doi: 10.1097/00001721-200209000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Greipp PR, Kyle RA, Bowie EJ. Factor-X deficiency in amyloidosis: a critical review. Am J Hematol. 1981;11:443–450. doi: 10.1002/ajh.2830110414. [DOI] [PubMed] [Google Scholar]
- 6.Duckers C, Simioni P, Spiezia L, Radu C, Dabrilli P, Gavasso S, Rosing J, Castoldi E. Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood. 2010;115:879–886. doi: 10.1182/blood-2009-08-237719. [DOI] [PubMed] [Google Scholar]
- 7.Duckers C, Simioni P, Spiezia L, Radu C, Gavasso S, Rosing J, Castoldi E. Low plasma levels of tissue factor pathway inhibitor in patients with congenital factor V deficiency. Blood. 2008;112:3615–3623. doi: 10.1182/blood-2008-06-162453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broze GJ, Jr, Girard TJ. Tissue factor pathway inhibitor: structure-function. Front Biosci. 2012;17:262–280. doi: 10.2741/3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomassen MC, Castoldi E, Tans G, Magdeleyns EJ, Delaunoit C, Debusscher L, Van Assche KJ, Rosing J. Endogenous factor V synthesis in megakaryocytes contributes negligibly to the platelet factor V pool. Haematologica. 2003;88:1150–1156. [PubMed] [Google Scholar]