

Abstract
BACKGROUND
Less than 30% of the cases of Brugada syndrome (BrS) have an identified genetic cause. Of the known BrS-susceptibility genes, loss-of-function mutations in SCN5A or CACNA1C and their auxiliary subunits are most common. On the basis of the recent demonstration that fibroblast growth factor (FGF) homologous factors (FHFs; FGF11–FGF14) regulate cardiac Na+ and Ca2+ channel currents, we hypothesized that FHFs are candidate BrS loci.
OBJECTIVE
The goal of this study was to test whether FGF12 is a candidate BrS locus.
METHODS
We used quantitative polymerase chain reaction to identify the major FHF expressed in the human ventricle and then queried a phenotype-positive, genotype-negative BrS biorepository for FHF mutations associated with BrS. We queried the effects of an identified mutant with biochemical analyses combined with electrophysiological assessment in a novel rat ventricular cardiomyocyte system in which we swapped the endogenous FHF with the identified mutant on multiple ionic currents in their native milieu and on the cardiac action potential.
RESULTS
We identified FGF12 as the major FHF expressed in the human ventricle. In 102 individuals in the biorepository, we identified a single missense mutation in FGF12-B (Q7R-FGF12). The mutant reduced binding to the NaV1.5 C terminus, but not to junctophilin-2, which mediates Ca2+ channel regulation. In rats, adult cardiac myocytes Q7R-FGF12, but not wild-type FGF12, reduced Na+ channel current density and availability without affecting Ca2+ channel function. Furthermore, the mutant, but not wild-type FGF12, reduced action potential amplitude, which is consistent with a mutant-induced loss of Na+ channel function.
CONCLUSIONS
These multilevel investigations strongly suggest that Q7R-FGF12 is a disease-associated BrS mutation. Moreover, these data suggest for the first time that FHF effects on Na+ and Ca2+ channels are separable. Most significantly, this study establishes a new method to analyze effects of human arrhythmogenic mutations on cardiac ionic currents.
Keywords: Brugada syndrome, Na+ channels, Ca2+ channels, Electrophysiology
Introduction
Brugada syndrome (BrS) is a potentially life-threatening inherited cardiac arrhythmia characterized by ST-segment elevation in the right precordial leads of the electrocardiogram (ECG).1 BrS is a channelopathy: the most commonly mutated locus is SCN5A that encodes the pore-forming subunit of the major cardiac voltage-gated Na+ channel, NaV1.5, responsible for the phase 0 upstroke of the ventricular action potential. SCN5A mutations associated with BrS are loss-of-function, decreasing NaV1.5 channel availability or surface expression.2 Loss-of-function mutations have also been found in the CACNA1C-encoded pore-forming subunit of the cardiac CaV1.2 L-type Ca2+ channel.3,4 In addition, genes associated with BrS include those encoding channel modulators, such as Na+ and Ca2+ channel β subunits and proteins responsible for channel trafficking and targeting within the cardiomyocyte. For many patients with BrS, however, genetic analysis does not identify a cause, suggesting the existence of additional, yet unidentified, BrS loci.
Among candidate genes are those encoding fibroblast growth factor (FGF) homologous factors (FHFs; FGF11–FGF14) that can modulate both Na+ and Ca2+ channels.5–7 FHFs are part of the FGF superfamily, but are not secreted, cannot bind or activate FGF receptors, and do not function as growth factors.8 Instead, FHFs remain intracellular where they perform multiple tasks. Their best-characterized intracellular binding partner is the C-terminal domain (CTD) of Na+ channels.9 Knockdown of the predominant FHF expressed in murine ventricular myocytes, FGF13, decreased Na+ channel current density and channel availability as well as reduced conduction velocity in a myocyte monolayer.6 In addition, FGF13 knockdown reduces CaV1.2 current density and prevents proper subcellular targeting of CaV1.2 channels.5
Because FHFs modulate 2 cardiac currents that, when perturbed, can lead to BrS, we hypothesized that loss-of-function mutations in FHFs would be associated with BrS. To test this hypothesis, we identified the major FHF expressed in the human ventricle (FGF12-B) and applied a candidate gene approach to patients with phenotype-positive but heretofore genotype-negative BrS. In 102 unrelated patients with BrS, we found a single, rare missense mutation in FGF12-B (Q7R-FGF12). To test the physiological effects of Q7R-FGF12, we developed a system to query the effects of the Q7R-FGF12 or wild-type (WT) FGF12 in an adult rat cardiomyocyte by replacing the endogenous FGF13 with the human variants. With this novel approach, we show that Q7R-FGF12 mutation leads to a Na+ channel loss-of-function phenotype consistent with BrS, thereby suggesting that FGF12 may be a BrS locus.
Methods
Study population
The study population consisted of 102 unrelated patients with BrS who were referred either to the Molecular Cardiology Laboratory, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy, or to the Windland Smith Rice Sudden Death Genomics Laboratory at Mayo Clinic, Rochester, MN, for laboratory-based genetic testing (Table 1). All patients with BrS included in this study remained genotype negative after comprehensive genotyping for mutations in the 14 known BrS-susceptibility genes listed in the Online Supplemental Methods. This study was approved by both the Mayo Foundation Institutional Review Board and the Medical Ethical Committee of Fondazione IRCCS Policlinico San Matteo. Informed consent was obtained for all patients.
Table 1.
Demographic characteristics of genotype-negative patient cohort with BrS
| Patient Demographic | Cohort |
|---|---|
| No. of probands | 102 |
| Age at diagnosis (y) | 45 ±13 |
| Range (y) | 9–81 |
| Man | 84 (82%) |
| Woman | 18 (18%) |
| Mean corrected QT interval (ms) | 407 ± 27 |
| Mean PQ interval (ms) | 169 ± 27 |
| Symptomatic patients (%) | 12 (12%) |
| Family history of cardiac events/unexplained sudden death | 27 (26%) |
| Type 1 ST-segment elevation at baseline | 46 (45%) |
| Type 1 ST-segment elevation with sodium blockade | 50 (49%) |
| Type 1 ST-segment elevation with fever | 6 (6%) |
Data are presented as mean ± standard error of the mean and as n (%).
Mutational analysis and control population
Comprehensive open reading frame/splice site mutational analysis of all amino acid coding exons and intron borders of FGF12 was performed by using polymerase chain reaction, denaturing high-performance liquid chromatography, and DNA sequencing, as described previously.10
The criteria for considering any FGF12 variant as a putative pathogenic mutation are outlined in the Online Supplemental Methods.
Subcloning and adenovirus production
Human FGF12-B (accession no. NM_004113.5) in pIRES2-AcGFP11 was mutated by using QuikChange II Site-Directed Mutagenesis (Agilent Technologies) to form Q7R-FGF12 and then both were subcloned into the pAdRFP adenovirus shuttle vector. The adenoviruses expressing FGF13 short hairpin RNA with GFP has been described previously.6 WT-FGF12 and Q7R viruses were generated similarly by using the AdEasy system (Agilent). The adenoviral plasmid was packaged in HEK293 cells. The recombinant virus was isolated by multiple freeze/thaw cycles, which was further amplified and then purified and concentrated by using Vivapure AdenoPACK 20 (Sartorius Stedim Biotech). The viral titer was determined by using optical density. All constructs were confirmed by sequencing.
HEK293T cell transfection, electrophysiology, and co-immunoprecipitation
Transfection, NaV1.5 Na+ current recording with FGF12-B, and immunoprecipitation techniques have been described previously in HEK293T cells.11 The construct encoding WT human junctophilin-2 (JPH2) was provided generously by Xander Wehrens (Baylor College of Medicine, Houston, TX).
Isothermal titration calorimetry
Isothermal titration calorimetry of the NaV1.5 CTD with FHFs has been described previously.12
Cardiomyocyte isolation
Animals were handled according to National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. The study was approved by Duke University Animal Care and Welfare Committee. Cardiomyocytes were isolated from 6- to 8-week-old Sprague-Dawley rats and cultured as described previously.5
Cardiomyocyte electrophysiology
Ca2+ currents and Na+ currents were recorded by using the whole-cell voltage clamp technique in cardiomyocytes after 36–48 hours, as described previously.5,6 Cardiac action potentials were recorded in current clamp as described previously.5 Input resistance was not different between the groups, and junction potential was calculated to be 5.6 mV but not corrected.
Immunocytochemistry
Immunocytochemistry was performed as described previously.5
Statistical analysis
Results are presented as mean ± standard error of the mean; the statistical significance of differences between groups was assessed by using 1-way analysis of variance and was set at P < .05.
Results
FGF12, the most highly expressed FHF in the human ventricle, is a BrS candidate locus
We have shown that in adult ventricular cardiomyocytes, FHFs regulate NaV1.5 trafficking and function as well as CaV1.2 targeting to T tubules, likely through a JPH2-mediated process.5,6 Given this important role of FHFs in the heart and the understanding that alterations in the cardiac Na+ and L-type Ca2+ channel function underlie BrS pathogenesis, we hypothesized that loss-of-function mutations in FHFs could perturb NaV1.5 and/or CaV1.2 function and lead to BrS. To determine the complement of FHFs expressed in the human ventricle, we extracted mRNA from nonfailing human ventricular tissue and performed quantitative polymerase chain reaction (qPCR). Because each of the 4 FHFs undergoes alternative splicing of their first exon, which confers unique Na+ channel modulatory properties,13 we also designed primers for specific FHF splice variants (see the Online Supplemental Table for primer sequences). As shown in Figure 1A, qPCR demonstrated that FGF12-B is the most highly expressed FHF splice variant. There was a negligible amount of the alternatively spliced FGF12-A. FGF13-Y and/or FGF13-VY (the qPCR strategy cannot distinguish between these 2 variants) mRNAs were also detected at approximately 40% of the amount of FGF12-B.
Figure 1.
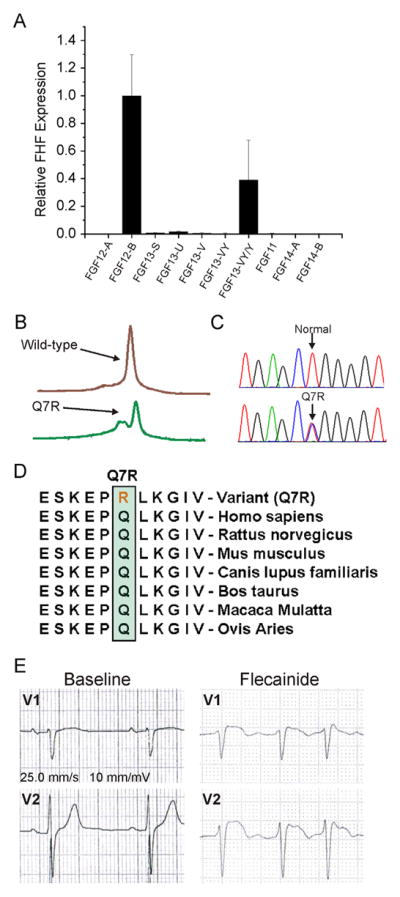
FGF12 is a candidate locus for BrS. A: FGF12-B is the most highly expressed FHF in the human ventricle by using the quantitative polymerase chain reaction. B: Denaturing high-performance liquid chromatography profiles. C: DNA sequence chromatograms showing the nucleotide change at position resulting in a glutamine (Q) to arginine (R) substitution at position 7 (Q7R) versus normal. D: Sequence conservation across species for Q7R in FGF12B. E: Leads V1 and V2 of an electrocardiogram from the proband during a diagnostic flecainide challenge. BrS = Brugada syndrome; FGF = flbroblast growth factor; FHF = fibroblast growth factor homologous factor; WT = wild type.
We therefore performed comprehensive mutational analysis of the major FGF12 splice variant (FGF12-B) in 102 unrelated patients with BrS in whom genetic testing had not previously detected a known BrS locus. The clinical demographic characteristics of the study cohort are summarized in Table 1. In a single patient, 1 rare missense mutation (Q7R) resulting in a substitution of glutamine (Q) at amino acid residue 7 by arginine (R) was identified (Figures 1B and 1C). This mutation involved a residue that is highly conserved across species (Figure 1D) and was absent in more than 1000 ethnically matched, internal control individuals and all publically available exome databases (>12,000 individuals).
The Q7R-FGF12 index case was a 61-year-old man who exhibited abnormalities on the ECG suggestive of BrS during flecainide therapy for atrial fibrillation. He subsequently underwent a flecainide challenge that induced a diagnostic type 1 Brugada ECG (Figure 1E) during his electrophysiology study and atrial fibrillation ablation. His personal history revealed an episode of supraventricular tachycardia during surgery for a hiatal hernia; however, he had a negative family history of sudden cardiac death.
The Q7R mutant in FGF12-B decreases binding to the NaV1.5 CTD
The Q7R mutant was of particular interest because it directly abuts the NaV1.5 interaction surface and leads to a change in charge (neutral to positive) near an important conserved electrostatic interaction site, K9 (Figure 2A), which we previously showed forms a critical salt bridge with E1890 in NaV1.5. Disruption of this salt bridge markedly reduced the affinity of FGF12 for NaV1.5 and affected the regulation of NaV1.5 by FGF12.14 We therefore suspected that Q7R might adversely affect the interaction with NaV1.5 and the consequent regulation of NaV1.5 function. We therefore generated purified recombinant WT-FGF12 or Q7R-FGF12 and tested affinity for a purified recombinant NaV1.5 CTD by isothermal titration calorimetry. As shown in Figure 2B, the mutant showed a 2-fold reduction in binding affinity for the NaV1.5 CTD compared to WT-FGF12.
Figure 2.

Q7R-FGF12 decreases affinity for the NaV1.5 CTD. A: Surface rendering of the interface between the NaV1.5 CTD (green) and the FHF core domain (yellow). Q7 (red) is in the same plane as the salt bridge between the NaV1.5 CTD and the FHF core domain, which is made of E1890 in the NaV1.5 CTD and K9 in FGF13 (blue). B: Isothermal titration calorimetry data demonstrate a 2-fold decrease (P < .05) in binding affinity for Q7R-FGF12 versus WT from 3 independent experiments. C: Representative co-immunoprecipitation and Western blot of FGF12-B WT or Q7R with JPH2 from at least 3 independent experiments. CTD = C-terminal domain; FGF = fibroblast growth factor; FHF = fibroblast growth factor homologous factor; HA = XXXX; IP = XXXX; JPH2 = junctophilin-2; WT = wild type.
Because FHFs also regulate CaV1.2 in cardiomyocytes, likely through JPH2,5 we tested whether the Q7R mutant affected the interaction with JPH2. We expressed His6-tagged WT-FGF12 or Q7R-FGF12 with JPH2 and performed co-immunoprecipitations. We observed no difference in the co-immunoprecipitation of JPH2 for WT-FGF12 and Q7R-FGF12 (Figure 2C). These data show that Q7R-FGF12 specifically affects the interaction with NaV1.5 and suggest that the functional perturbation of cardiac ion currents would likely be limited to NaV1.5 rather than to CaV1.2.
A system to study FHF modulation of ion channels in their native milieu
Although a common method to assess functional effects of mutant channels or auxiliary subunits is expression in a heterologous cell system, we discovered (data not shown) that the coexpression of FHFs and NaV1.5 or CaV1.2 in HEK293T cells did not accurately recapitulate the effects of FHFs on Na+ or Ca2+ currents in adult ventricular cardio-myocytes.5,6 We therefore designed a system to explore the physiological effects of FHFs in adult rat ventricular cardiomyocytes. Our aim was to “replace” the endogenous FGF13 with human WT-FGF12 or Q7R-FGF12 and query the resulting effect on Ca2+ and Na+ currents. We began by using our established adenovirally mediated short hairpin RNA knockdown of FGF13 in cultured adult rat ventricular cardiomyocytes (marked by GFP), in which we achieve a greater than 50% reduction of the FGF13 protein within 2 days in culture,6 and coinfected with a separate adenovirus expressing either WT-FGF12 or Q7R-FGF12 (marked by mRFP). We confirmed effective knockdown of the endogenous rat FGF13 and proper subcellular localization of expressed FGF12 by immunocytochemistry with antibodies to FGF13 or to the Hisx6 tag on the expressed FGF12 C termini (Figure 3).
Figure 3.

A system to study the effects of FGF12 on ventricular cardiomyocyte physiology. Control cell stained for FGF13 in cyan shows endogenous FGF13 distribution in the cardiomyocyte at the intercalated disc, T tubules, and nucleus; the magnified inset (2×) emphasizes distribution at the intercalated disc and in a striated pattern. Knockdown with FGF13 using a virus that expresses GFP and shRNA reduces the reactivity of the FGF13 antibody, even with the overexpression of WT-FGF12, indicating no cross-reactivity. By using a virus expressing FGF13 shRNA without GFP and overexpressing WT-FGF12 (indicated by RFP), there is a decreased immunoreactivity for FGF13 (green) and a signal using the His6 antibody that correlates with endogenous FGF13 expression. A similar pattern of His6 immunoreactivity is observed for Q7R-FGF12. Insets are magnified 2× to emphasize pattern of distribution. Scale bar is 50 μm for large images and 12.5 μm for inset. FGF = fibroblast growth factor; GFP = XXXX; Hisx6 = XXXX; KD = knocked down; RFP = XXXX; shRNA = short hairpin.
Q7R-FGF12 differentially affects Ca2+ and Na+ currents in a ventricular cardiomyocyte
Since the Q7R mutation did not appear to affect the interaction of FGF12 with JPH2 as shown in Figure 2, we expected that the mutant would have minimal effects on Ca2+ current density. We tested this hypothesis by using the replacement strategy. Consistent with our previous data,5 in adult ventricular myocytes in which FGF13 was knocked down (FGF13 KD), Ca2+ current density was reduced and CaV1.2 was mislocalized compared to uninfected control cardiomyocytes (Figures 4A–4C). The adenoviral coexpression of WT-FGF12 rescued the effect of FGF13 KD. Ca2+ channel current density reverted to control levels, and the colocalization of CaV1.2 with RyR2 was restored. The coexpression of Q7R-FGF12 also restored Ca2+ currents and CaV1.2 localization. Although current density was approximately 30% higher than that in control cells or rescue with WT-FGF12, this difference was not significantly different from WT-FGF12 (Figure 4; P = .09). Not only did these results demonstrate the utility of the adenoviral “replacement” approach, but they also showed that WT-FGF12 completely recapitulated the effects of endogenous FGF13 in rat adult ventricular myocytes. Moreover, these results established that Q7R-FGF12 did not adversely affect Ca2+ currents or CaV1.2 localization.
Figure 4.

Both WT and Q7R-FGF12 rescue Ca2+ current density and localization from FGF13 KD. A: Representative Ca2+ current traces from the 4 groups. B: I–V curve depicting the rescue of CaV1.2 current density with WT and Q7R-FGF12. Peak current densities at 0 mV are −13.01 ± 0.88 pA/pF (n = 9) for control, −8.09 ± 1.30 pA/pF (n = 13) for FGF13 KD, −12.86 ± 1.50 pA/pF (n = 13) for FGF13 KD + WT-FGF12, and −17.05 ± 1.46 (n = 13) for FGF13 KD + Q7R-FGF12. C: Immunostaining for CaV1.2 (green) and ryanodine receptor (red) showing that CaV1.2 is mislocalized with FGF13 KD and the localization is rescued with WT and Q7R-FGF12. *P < .05 versus control. FGF = fibroblast growth factor; KD = knocked down; WT = wild type.
We next recorded voltage-gated Na+ currents under the same conditions. Consistent with our previous results, FGF13 KD reduced Na+ current density compared to control and decreased channel availability, as indicated by a −4 mV shift in the V1/2 of steady-state inactivation (Figures 5A–5C). As with Ca2+ currents, the coexpression of WT-FGF12 compensated for FGF13 KD and restored the V1/2 of steady-state inactivation compared to FGF13 KD (Table 2 and Figure 5E). Indeed, the coexpression of FGF12 increased Na+ current density beyond the level observed in control cells. To explain these data, we considered several possibilities: FGF12 is an even more potent FHF for Na+ currents than is FGF13; FGF12 has additional effects on Na+ currents compared to FGF13; or NaV1.5 is not saturated by endogenous FGF13 so that FGF12 overexpression modulates additional NaV1.5 channels that were not regulated by a limiting pool of endogenous FGF13. To distinguish among these, we overexpressed WT-FGF12 without knocking down FGF13 and measured the effects on Na+ current density. The overexpression of WT-FGF12 did not significantly increase Na+ channel current density under these conditions (see the Online Supplemental Figure), indicating that FGF12 does not exert additional effects on NaV1.5 beyond FGF13 and that the effects of FGF13 on NaV1.5 are saturated in control cells. Moreover, the lack of an effect of FGF12 in the presence of FGF13 is consistent with our previous data, showing that FGF13 has a higher affinity for the NaV1.5 CTD than does FGF12.11 Thus, we conclude that in the absence of FGF13, FGF12 is more potent than FGF13 for increasing NaV1.5 at the sarcolemma.
Figure 5.

Q7R-FGF12 cannot rescue reduced NaV1.5 current density and availability from FGF13 KD while WT can. A: Representative Na+ current traces for the 4 groups tested. B and C: I–V curve and summarized peak current data. D: Activation curve. E: Steady-state inactivation curve. *P < .05 versus control; **P < .05 versus WT-FGF12B; †P < .01 versus WT-FGF12B. FGF = fibroblast growth factor; KD = knocked down; WT = wild type.
Table 2.
Summary of INa electrophysiology data from cardiac myocyte recordings
| Type of cell | INa peak at −55 mV (pA/pF) | V1/2 of activation (mV) | k of activation (pA/mV) | V1/2 of inactivation (mV) | k of inactivation (pA/mV) |
|---|---|---|---|---|---|
| Control | −69.5 ± 9.0 (14) | −62.6 ± 1.4 (12) | 8.8 ± 0.4 (12) | −99.7 ± 1.4 (15) | 6.2 ± 0.3 (15) |
| FGF13 KD | −34.5 ± 5.1 (9)* | −57.1 ± 1.7 (9) | 4.6 ± 0.5 (9) | −103.6 ± 1.3 (10)* | 6.6 ± 0.3 (10) |
| FGF13 KD + WT-FGF12-B | −100.3 ± 18.7 (9)* | −60.8 ± 1.8 (9) | 3.7 ± 0.5 (9) | −97.2 ±1.9 (6) | 5.8 ± 0.4 (6) |
| FGF13 KD + Q7R-FGF12-B | −38.2 ± 4.3 (9)* | −61.2 ± 1.5 (8) | 4.7 ± 0.3 (8) | −104.5 ± 1.5 (12)* | 6.4 ± 0.3 (12) |
Number of cells is indicated in parentheses. FGF = fibroblast growth factor; INa = Na+ current; KD = knocked down; WT = wild type.
P < .05 vs NaV1.5 only.
In contrast to the ability of WT-FGF12 to restore the effects of FGF13 KD, the coexpression of Q7R-FGF12 failed to rescue current density or the hyperpolarizing shift in steady-state inactivation (Table 2 and Figure 5E). On the basis of the detection of the Hisx6-tagged Q7R-FGF12 by immunocytochemistry (Figure 3) and the success of Q7R-FGF12 (like WT-FGF12) in restoring Ca2+ currents (Figure 4), we could immediately eliminate the possibility that Q7R-FGF12 failed to restore Na+ currents because it did not express well in cardiomyocytes.
Because a spinocerebellar ataxia mutation in the related FGF14 exerts a dominant negative effect on Na+ currents in hippocampal neurons,15 we considered the possibility that the Q7R-FGF12–mediated reduction in Na+ current density resulted from a dominant negative effect on the remaining pool of endogenous FGF13 still present after knockdown rather than because of a reduction in the amount of FGF12 that interacted with NaV1.5. We therefore tested whether Q7R-FGF12 was capable of exerting a dominant negative effect by overexpressing Q7R-FGF12 without knocking down FGF13. Under these conditions, the effects of Q7R-FGF12 were negligible (see the Online Supplemental Figure), ruling out a dominant negative effect.
Q7R-FGF12 leads to changes in action potential morphology consistent with BrS
Finally, we assessed the effects of WT-FGF12 and Q7R-FGF12 on the ventricular action potential. As we reported previously,5 FGF13 KD decreased the action potential peak amplitude and half-width, which is consistent with observed reductions in Na+ and Ca2+ channels. Rescue with WT-FGF12 restored both peak amplitude and half-width to control levels. In contrast, rescue with Q7R-FGF12 failed to restore the peak amplitude, which is consistent with its lack of efficacy in modulating NaV1.5 currents, and increased the half-width (Figures 6A–6C); we hypothesized these results from the potentiating effect of Q7R-FGF12 on CaV1.2 Ca2+ current density (see Discussion).
Figure 6.

Q7R-FGF12 recapitulates the BrS action potential. A: Representative evoked action potentials from cardiomyocytes of the 4 groups. B and C: Summarized data for action potential peak amplitude and half-width, respectively. *P < .05 versus control; **P < .01 versus control; †P < .01 versus FGF13 KD + WT-FGF12. BrS = Brugada syndrome; CON = control; FGF = fibroblast growth factor; KD = knocked down; WT = wild type.
Discussion
Inherited cardiac arrhythmias such as BrS can result from mutations in ion-channel pore-forming subunits or their modulator proteins, providing consequent functional changes in ionic currents. Although still only accounting for less than 25% of BrS, the most commonly affected current in BrS is the voltage-gated Na+ channel and most identified mutations are loss-of-function in SCN5A or in genes that encode regulators of NaV1.5 currents, leading to a reduction in Na+ current density.5 Less commonly, loss-of-function mutations in CaV1.2 and its β subunit have been linked to BrS. In this context, we suspected that loss-of-function mutations in FHFs, which we showed can modulate the cardiac voltage-gated Na+ and Ca2+ channels,5,6 were likely BrS candidate loci. After identifying the FGF12-B splice variant as the most abundantly expressed FHF in the human ventricle, we performed comprehensive mutational analysis of patients in a BrS repository and discovered a Q7R mutation in FGF12-B that leads to a Na+ channel loss-of-function phenotype consistent with the BrS diagnosis by using a novel adult ventricular cardiomyocyte system.
By demonstrating that this rare FGF12 variant (identified in a patient with BrS) has a reduced affinity for the NaV1.5 CTD, cannot potentiate Na+ currents like WT-FGF12, and adversely affects the ventricular action potential, our data suggest that FGF12 is a new candidate BrS locus. Specifically, our data suggest that the haploinsufficiency of FGF12 in the identified heterozygous patient leads to a reduction in Na+ current. Our model system, in which FGF13 knockdown leads to an approximately 50% reduction of the endogenous FGF13 protein, provides an accurate recapitulation of the patient’s heterozygous condition.
We also demonstrated that Q7R-FGF12 is capable of modulating CaV1.2 Ca2+ currents. Thus, this mutation appears to contribute to BrS in this patient by loss-of-function effects on Na+ channel currents without affecting Ca2+ currents. An important corollary of these results is the first demonstration of a separation between FHF-dependent effects on Na+ channels from Ca2+ channels. We hypothesize that other FGF12 mutants may exert effects through the dysregulation of Ca2+ channels without perturbing Na+ channels. Because the Q7R mutation is on the NaV1.5 CTD interface and affects the affinity of FGF12 for the CTD, we further hypothesize that the modulatory actions of FHFs on CaV1.2 channels result from a different FHF domain. The means by which FHFs regulate CaV1.2 are not clear, although our previous data suggest that FHFs may exert their effect through JPH2. The interaction site between FHFs and JPH2 has not yet been mapped, but the Q7R mutation in FGF12 did not affect co-immunoprecipitation with JPH2 (Figure 2), thereby suggesting that the JPH2 interaction site is distinct from the NaV1.5 CTD interaction site. As such, this BrS mutation will be a useful tool for further dissecting the distinct FHF pools within a cardiomyocyte.
The increase in action potential half-width in the context of the Q7R-FGF12 (Figure 6) was unexpected. The reasons are not clear, but we speculate that the potentiating effect of the mutant FGF12 on cardiac Ca2+ current density (Figure 4) may contribute. That hypothesis would be consistent with recent data, suggesting that increased Ca2+ channel current density due to a gain-of-function mutant in CACNA1C can cause long QT syndrome, which would manifest as a longer ventricular action potential duration at the cellular level.16 Finally, as the results herein and our recent data demonstrate that FHFs are modulators of multiple ion channels,6,10 it is possible that FGF12 could affect additional ion channels or 2 properties of 1 ion channel, not necessarily in the same direction, which is reminiscent of overlap syndromes that result from mutations in SCN5A.17
Critical to our ability to define the pathogenesis of this FGF12 mutant was the development of an adult cardiomyocyte-based system for investigation. While heterologous expression systems can show changes in intrinsic channel properties and its regulators isolated from the complex environment of a cardiomyocyte, these systems cannot accurately recapitulate the ion “channelsomes” and are not suited to query combinations of unanticipated cardiomyocyte-specific factors or the particular anatomy of a cardiomyocyte (eg, T tubules). Our preliminary data in HEK cells failed to yield mechanistic insights into how FHFs modulate either Na+ or Ca2+ channels (not shown) and thus were not suitable for exploring the roles of this mutant. Indeed, differences in results from a heterologous expression system and from native cell types have been previously reported for the FHF regulation of Na+ channel currents13,15 and likely derive from a requirement for aspects of the native channel environment for complete expression of the FHF-regulated phenotype. While induced pluripotent stem cells offer a similar strategy for testing endogenous effects of arrhythmogenic mutations,18 the resulting cardiomyocytes are often immature, precluding definitive assessment of the adult phenotype, and induced pluripotent stem cell technology does not currently offer a high-throughput method for the analysis of multiple mutants. Thus, the knockdown and replacement strategy demonstrated here may be of utility in dissecting the roles of other arrhythmia mutations in channels or their regulators.
Conclusions and limitations
The data obtained by using a native cardiomyocyte system suggest that FGF12 is a BrS locus and that the Q7R mutant affects Na+ channel trafficking and kinetics with minimal effects on Ca2+ channel function. Because the clinical data are derived from a single patient from whom family data are not available, we do not have cosegregation of the phenotype and genotype that is necessary to establish FGF12 as a definitive BrS locus. Buttressed by biochemical, cellular, and biophysical investigations, our data nevertheless provide strong motivation for candidate screening of FGF12 in genotype-negative cases of BrS, in which further identification of mutations would help establish FGF12 as a BrS locus. Our results in cardiomyocytes also establish that the effects of FHFs on Na+ channels and Ca2+ channels are clearly separable. Since BrS and other inherited arrhythmias can result from changes in Na+ channel or Ca2+ channel function, we hypothesize that other mutations in FGF12 may underlie genotype-negative cases of inherited arrhythmias.
Supplementary Material
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) R01 HL71165 and R01 HL112918 (to Dr Pitt), the Gertrude Elion Mentored Medical Student Award and NHLBI F30 HL112540-01 (to Dr Hennessey), and the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program (to Dr Ackerman). Dr Ackerman is a consultant/advisory board member for Boston Scientific, Medtronic, and St Jude Medical. Intellectual property derived from Dr Ackerman’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals, now Transgenomic).
ABBREVIATIONS
- BrS
Brugada syndrome
- CTD
C-terminal domain
- ECG
electrocardiogram
- FGF
fibroblast growth factor
- FHF
fibroblast growth factor homologous factor
- GFP
green fluorescent protein
- JPH2
junctophilin-2
- KD
knocked down
- Q
glutamine
- qPCR
quantitative polymerase chain reaction
- R
arginine
- WT
wild type
Appendix
Supplementary data
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.hrthm.2013.09.064.
References
- 1.Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. doi: 10.1161/01.CIR.0000152479.54298.51. [DOI] [PubMed] [Google Scholar]
- 2.Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2010;460:223–237. doi: 10.1007/s00424-009-0761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7:1872–1882. doi: 10.1016/j.hrthm.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hennessey JA, Wei EQ, Pitt GS. Fibroblast growth factor homologous factors modulate cardiac calcium channels. Circ Res. 2013;113:381–388. doi: 10.1161/CIRCRESAHA.113.301215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C, Hennessey JA, Kirkton RD, et al. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res. 2011;109:775–782. doi: 10.1161/CIRCRESAHA.111.247957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan H, Pablo JL, Pitt GS. FGF14 regulates presynaptic Ca(2+) channels and synaptic transmission. Cell Rep. 2013;4:66–75. doi: 10.1016/j.celrep.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsen SK, Garbi M, Zampieri N, et al. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003;278:34226–34236. doi: 10.1074/jbc.M303183200. [DOI] [PubMed] [Google Scholar]
- 9.Liu C, Dib-Hajj SD, Waxman SG. Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1. 9a (NaN) J Biol Chem. 2001;276:18925–18933. doi: 10.1074/jbc.M101606200. [DOI] [PubMed] [Google Scholar]
- 10.Ackerman MJ, Tester DJ, Jones G, Will MK, Burrow CR, Curran M. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc. 2003;78:1479–1487. doi: 10.4065/78.12.1479. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, Wang C, Hoch EG, Pitt GS. Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J Biol Chem. 2011;286:24253–24263. doi: 10.1074/jbc.M111.245803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Chung BC, Hennessey JA, Lee SY, Pitt GS. Structural basis for Ca2+-dependent regulation of NaV channels. In press. [Google Scholar]
- 13.Lou JY, Laezza F, Gerber BR, et al. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol. 2005;569:179–193. doi: 10.1113/jphysiol.2005.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Chung BC, Yan H, Lee SY, Pitt GS. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure. 2012;20:1167–1176. doi: 10.1016/j.str.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laezza F, Gerber BR, Lou JY, et al. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci. 2007;27:12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boczek NJ, Best JM, Tester DJ, et al. Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ Cardiovasc Genet. 2013;6:279–289. doi: 10.1161/CIRCGENETICS.113.000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.