

ABSTRACT
Small RNAs play a critical role in host-pathogen interaction. Indeed, small RNA-mediated silencing or RNA interference (RNAi) is one of the earliest forms of antiviral immunity. Although it represents the main defense system against viruses in many organisms, the antiviral role of RNAi has not been clearly proven in higher vertebrates. However, it is well established that their response to viral infection relies on the recognition of viral RNAs by host pattern recognition receptors (PRRs) to trigger activation of the interferon pathway. In the present work, we report the existence of a novel small noncoding RNA population produced in mammalian cells upon RNA virus infection. Using Sindbis virus (SINV) as a prototypic arbovirus model, we profiled the small RNA population of infected cells in both human and African green monkey cell lines. Here, we provide evidence for the presence of discrete small RNAs of viral origin that are not associated with the RNA-induced silencing complex (RISC), that are highly expressed and detected by Northern blot analysis, and that accumulate as 21- to 28-nucleotide (nt) species during infection. We report that the cellular antiviral endoribonuclease RNase L cleaves the viral genome, producing in turn the small RNAs. Surprisingly, we uncovered the presence of a modification on the 3′-end nucleotide of SINV-derived viral small RNAs (SvsRNAs) that might be at the origin of their stability. Altogether, our findings show that stable modified small viral RNAs could represent a novel way to modulate host-virus interaction upon SINV infection.
IMPORTANCE
In a continuous arms race, viruses have to deal with host antiviral responses in order to successfully establish an infection. In mammalian cells, the host defense mechanism relies on the recognition of viral RNAs, resulting in the activation of type I interferons (IFNs). In turn, the expression of many interferon-stimulated genes (ISGs) is induced to inhibit viral replication. Here we report that the cytoplasmic, interferon-induced, cellular endoribonuclease RNase L is involved in the accumulation of a novel small RNA population of viral origin. These small RNAs are produced upon SINV infection of mammalian cells and are stabilized by a 3′-end modification. Altogether, our findings indicate that in our system RNA silencing is not active against Sindbis virus (SINV) and might open the way to a better understanding of the antiviral response mediated by a novel class of small RNAs.
INTRODUCTION
In order to successfully establish an infection, viruses engage in a continuous arms race with their hosts, the outcome of which might ultimately result in either viral persistence, clearance of the viral infection, or death of the infected cells. The arsenal of antiviral defenses and viral counter defense mechanisms is vast and very diverse among the different phyla of life. Plants, insects, nematodes, and fungi mostly (if not exclusively) rely upon an innate immune response, whereas mammals have evolved an adaptive response in addition to this first line of defense. In the former organisms, the antiviral component of the innate immune response is mainly based on a highly conserved process commonly referred to as RNA silencing, or RNA interference (RNAi). In this phenomenon, long double-stranded RNAs of viral origin are cleaved by the RNase Dicer into small interfering RNAs (siRNAs), which are then assembled into effector complexes that contain a member of the Argonaute (Ago) family; this results in targeted degradation of viral messenger or genomic RNAs (1).
In mammals, another type of innate immunity has evolved to control viruses (2). The initial recognition of nonself viral nucleic acids by extra- and intracellular sensors triggers the activation of type I interferons (IFNs) (3). This leads in turn to the upregulation of numerous IFN-stimulated genes (ISGs) (4), which either directly trigger a signaling cascade directed against the viral infection or mobilize other cells of the immune system. ISGs encode proteins involved in apoptosis induction, protein synthesis blocking, or in the regulation of mRNA editing or RNA degradation (5). One key factor involved in IFN type I-mediated host defense is RNase L, a latent cytoplasmic endoribonuclease that is activated by 2′,5′-oligoadenylates in response to double-stranded RNA (dsRNA) sensing and that cleaves both viral and cellular RNAs (6).
Although extensive deep sequencing analyses have been performed to identify siRNAs in mammalian cells infected with several RNA viruses, there is so far no good evidence for a possible antiviral role of RNAi in mammalian somatic cells (7). Nevertheless, another class of small noncoding RNAs, microRNAs (miRNAs), has been shown to play an important role during viral infection in mammals. miRNAs are evolutionarily conserved small RNAs derived from large primary transcripts, which are sequentially processed by the respective nuclear and cytoplasmic RNase III enzymes, Drosha and Dicer, to generate mature single-stranded ~21- or 22-nucleotide (nt) RNAs (8, 9). Similar to siRNAs, they are incorporated into an effector Ago-containing RNA-induced silencing complex (RISC), whereby they mediate posttranscriptional regulation of target mRNAs via partially complementary sites (10). On one hand, miRNAs of cellular origin can directly or indirectly regulate viral infections (11, 12). On the other hand, some viruses have evolved the capacity to encode their own miRNAs, which represent an ideal tool to stealthily modulate the cellular environment. To date, viral miRNAs have been almost exclusively identified in the genomes of DNA viruses, mostly herpesviruses (13–17), with the notable exception of bovine leukemia virus, a retrovirus with an RNA genome (18). It has been commonly assumed that cytoplasmic RNA viruses cannot encode miRNAs, not only because they do not have access to the nuclear biogenesis machinery but also because their genomic integrity would be destabilized by miRNA processing (19). Nevertheless, it has recently been demonstrated that the insertion of a cellular miRNA precursor into the 3′-end nontranslated region (NTR) of Sindbis virus (SINV) genomic RNA leads to its cytoplasmic processing without affecting the viral replication (20, 21).
The arthropod-borne SINV is a small, enveloped, positive, single-stranded RNA virus and is the prototype for the alphavirus genus. Alphaviruses represent a group of widely distributed human and animal pathogens, which pose a serious public health threat (22). Some of them induce febrile and arthritogenic diseases, while others can cause highly debilitating diseases, such as encephalitis. The SINV genomic RNA is capped and polyadenylated and is infectious as naked RNA. Upon entry into the cytoplasm by endocytosis, the host translational machinery recognizes the genomic RNA, and four nonstructural proteins are produced (23). Their expression is sufficient for the establishment of replication complexes and the synthesis of a complementary antigenomic negative RNA strand. The antigenome is necessary for the replication of the viral genome and the production of a subgenomic RNA that ensures the translation of the structural proteins during the late phase of infection (24, 25).
To date, SINV has not been shown to give rise to any small RNA species in mammalian cells. As such, we decided to investigate the small RNA profile of SINV-infected human and African green monkey cell lines. Using small RNA cloning and deep sequencing techniques, we provide evidence that the SINV genome is a source of small RNAs in infected mammalian cells. Some of these small RNAs accumulate to levels detected by Northern blot analysis. We set out to identify the factors involved in their biogenesis and show that they do not depend on the RNA silencing machinery, but rather, that they are downstream products of the cytoplasmic IFN-induced endoribonuclease RNase L. Finally, we also present experimental evidence that these viral small RNAs are modified at their 3′ extremity. Altogether, our results indicate that the SINV-derived small RNAs could represent key markers of the host defense mechanism.
RESULTS
SINV-derived small RNAs accumulate in infected mammalian cells.
In order to investigate the presence and nature of virus-derived small RNAs in mammalian cells, we used a small RNA cloning and deep sequencing approach. We generated and analyzed small RNA libraries from both HEK293 and Vero cells infected with SINV or not infected with SINV.
Total RNA was isolated from uninfected cells or cells infected with SINV at a multiplicity of infection (MOI) of 0.01 plaque forming units (PFU) per cell 16 h postinfection (hpi). Using these conditions, we could detect cells with a high viral load without observing any apparent cell death as assessed by propidium iodide staining (see Fig. S1 in the supplemental material). We also confirmed that until 24 hpi, cells were still actively proliferating (data not shown). High-throughput sequencing yielded 201,793 and 1,193,893 reads that mapped to the SINV genome in infected HEK293 and Vero cells, respectively (Fig. 1A); these two numbers of reads represented a percentage of 1.66 and 7.99%, respectively, of total mapped small RNAs in HEK293 and Vero cells. The greater fraction of viral reads in Vero cells is consistent with the fact that these cells are known to be more permissive to viral infections due to a defect in interferon production (26). The distribution of the reads on the viral genomic (positive) and antigenomic (negative) strands is indicated in Fig. 1A. Interestingly, the vast majority of the reads (98 to 99%) originate from the genomic strand of the virus. Nonetheless, we identified limited sequencing reads that were derived from the negative strand, albeit at extremely low levels in both libraries (Fig. 1A). Their genomic distribution showed a peak at the 3′ end of the antigenome (see Fig. S2A in the supplemental material). This specific region corresponds to the viral promoter, necessary for the replication of the viral genome and is known to fold into a conserved 44-nt stem-loop structure (25). The length distribution of the antigenomic small RNA reads peaked at a size of 22 nt (Fig. S2B), which corresponds to the 5′ arm of the stem-loop structure (Fig. S2C). Although we could validate the presence of the 44-nt-long RNA by Northern blot analysis, we were unable to detect the accumulation of the small 22-nt RNA as a discrete species (Fig. S2D).
FIG 1 .

Identification of SINV-derived small RNAs. (A) Virus-specific small RNA reads were recovered from each library (HEK293 cells infected with SINV [HEK293-SINV] and Vero-SINV) and were mapped to both positive (+) and negative (−) strands of the viral genome. (B) Size distribution of SINV-derived small RNA populations. The proportion of viral sRNAs that were derived from the genomic sense strand in each size class is shown as a percentage of the total viral small RNA reads for this strand. (C) Distribution of viral reads mapping to the SINV genomic strand. The abundance of small RNAs was calculated and plotted as the sum of normalized reads (per 105 viral reads) in each single-nucleotide sliding window along the viral genome. The small black arrows indicate the peaks chosen for Northern blot validation (see Fig. S1 in the supplemental material). A schematic diagram represents the organization of SINV genome. The two open reading frames (ORF), which encode the nonstructural (ns) and structural proteins, are shown. The nontranslated region (NTR) at the 3′ end of the virus is shown as a small black bar. (D) Northern blot validation of SINV candidates. Total RNA was isolated at the indicated time points from HEK293 cells infected with SINV at an MOI of 0.01. Noninfected cells (−) were used as a negative control. U6 snRNA was used as a loading control. (E) Quantification of the SINV genome during infection by reverse transcription-quantitative PCR (RT-qPCR). The relative quantification is normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels and is represented on a logarithmic scale as a mean of three replicates. NI, not infected.
The size distribution of the main population of viral reads mapping on the positive strand of the virus was broad with no defined peak at a specific length (Fig. 1B). However, a closer inspection revealed that the overall size and genomic distribution of the viral reads were highly similar in both HEK293 and Vero cells (Fig. 1C). Moreover, a greater number of reads were found between nucleotides 7000 and 11700 of the viral genome, corresponding to the abundantly expressed subgenomic RNA.
We selected 14 candidates among the most abundant peaks obtained in both cell types for validation by Northern blot analysis (see Fig. S3A in the supplemental material). We could detect several SINV-derived viral small RNAs (SvsRNAs) of 20 to 30 nt in length that were derived from all along the viral genome (Fig. S3B). Among them, SvsRNA-1, -2, and -3 were detected in both HEK293 and Vero cells (Fig. S3C). The presence of abundant larger bands accumulating between 40 and 100 nt was also evidenced. Of note, the expression of both larger and smaller viral RNAs increased proportionally to the accumulation of the viral genome during a time course of infection (Fig. 1D and E). The identification of SINV-derived small RNAs prompted us to analyze more closely their origin and biochemical properties.
The production of SvsRNAs does not rely on the miRNA biogenesis machinery.
The identification of small RNAs from RNA viruses could be seen either as a result of a productive mechanism for the pathogen (as for virus-encoded miRNAs) or as the consequence of a host response to control the infection by degrading viral RNAs. SINV expresses four enzymes that play key functions during the viral replicative cycle. However, none of these enzymes is known to possess an endoribonuclease activity (22) that could enable the processing of small RNAs from a viral RNA precursor. Therefore, cellular enzymes have to be involved in the biogenesis of SINV sRNAs. In order to identify them, we decided to knock down cytoplasmic enzymes with a known RNase activity. We first focused on proteins important for the production of small RNAs, such as miRNAs. Hence, we transfected siRNAs against Drosha, Dicer, or Ago2 followed by an infection with SINV and an analysis of viral small RNA accumulation. Although Drosha is known to be mostly nuclear, we also took it into consideration because it was recently shown to relocalize to the cytoplasm upon SINV infection (21). Although we were able to downregulate very efficiently Drosha, Dicer, and Ago2 (see Fig. S4A to C in the supplemental material), the knockdown of these factors neither affected the viral load nor the accumulation of SvsRNA-1, -2 and -3 (Fig. S4D to G). We also examined whether we could detect an siRNA signature among all viral reads by looking for small RNAs that could perfectly base pair on 19 or 20 nt and present a 2-nt overhang in 3′. A very limited number of such duplexes could be found, but they were not more represented than larger duplexes presenting with 3′ or 5′ overhangs (data not shown). It has been reported that small RNAs, which are not generated by Dicer or Drosha cleavage, could nevertheless be assembled into Argonaute proteins (e.g., tRNA fragments [27]). We therefore immunoprecipitated Argonaute proteins followed by Northern blot analysis to see whether SvsRNAs could be detected within the RISC. However, although we could detect the accumulation of the cellular miRNA miR-16, using both an antibody specific for Ago2 (Fig. S4H, left panel) and an antibody able to recognize all four Ago proteins (Fig. S4H, right panel), we were unable to detect SvsRNAs within immunoprecipitated RISC.
RNase L is involved in the production of SvsRNAs.
We then hypothesized that an endoribonuclease involved in the antiviral response could be implicated in SvsRNA production. RNase L is a ubiquitous, cytoplasmic, interferon-induced endonuclease that plays a key role in the cellular response to viral infections. It cleaves single-stranded RNA regions after the dinucleotides UA/UU, thereby preventing virus accumulation (28). As such, we knocked down RNase L using siRNAs, before infecting the cells with SINV. As expected, RNase L knockdown (KD) resulted in the upregulation of the SINV genome by approximately 2.5-fold (Fig. 2A and B). Interestingly, it also resulted in a dramatic reduction of the viral small RNAs SvsRNA-1, -2, and -3 and their precursors (Fig. 2C).
FIG 2 .
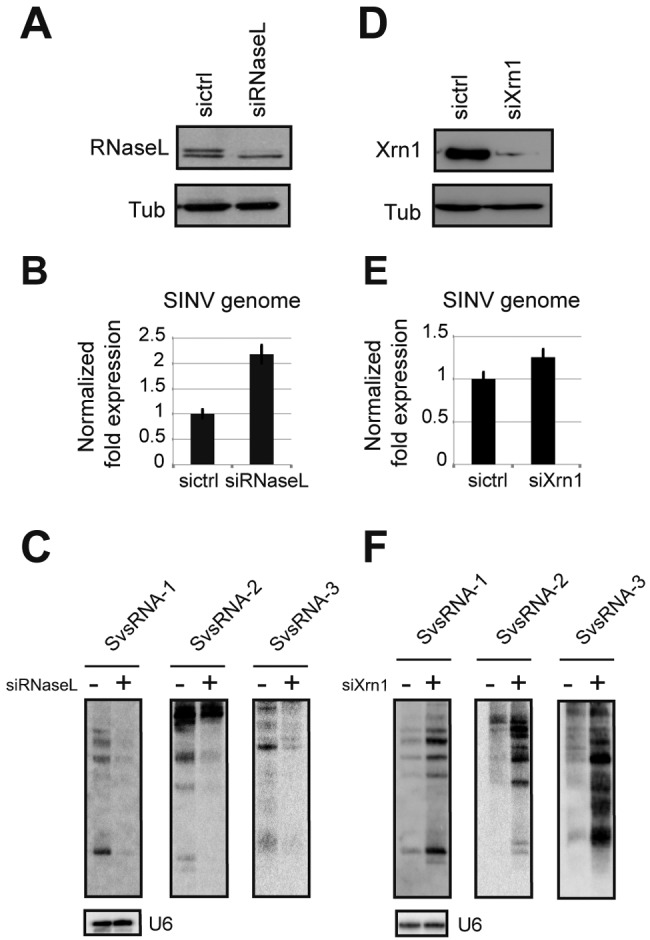
Effects of RNase L and Xrn1 knockdown on SvsRNA accumulation. (A and D) RNase L and Xrn1 knockdown in SINV-infected HEK293 cells was verified by Western blotting. Tubulin (Tub) was used as a normalizer. sictrl, control siRNA; siRNaseL, RNase L-specific siRNA; siXrn1, Xrn1-specific siRNA. (B and E) SINV genomic RNA was quantified by RT-qPCR, before and after RNase L or Xrn1 KD. The relative quantification is normalized to GAPDH levels and is represented on a logarithmic scale as a mean of three replicates. (C and F) Northern blot analysis on total RNA from infected HEK293 cells before and after RNase L or Xrn1 KD. U6 was used as a normalizer.
In order to extend this observation and assess the role of RNase L in SvsRNA generation, we also produced small RNA libraries from HEK293 cells treated with a control or an RNase L-specific siRNA prior to SINV infection. In accordance with the striking effect seen on the three specific SvsRNAs, reduction in RNase L protein level decreased the entire population of clonable viral sequences from 2.75 to 1.19% (see Fig. S5A in the supplemental material). However, RNase L knockdown affected neither the size distribution of viral reads nor their genomic distribution (Fig. S5B and C).
Given the cleavage properties of RNase L (see next section), it was unlikely that the identified small RNAs were direct products of this enzyme. We therefore looked for other factors implicated in RNA degradation that could affect SvsRNA accumulation. We used RNAi to downregulate the expression of the major cytoplasmic 5′-3′ exoribonuclease Xrn1 (Fig. 2D). As expected, the capped viral genome was not affected by downregulation of Xrn1 (Fig. 2E). However, the levels of SvsRNA-1, -2, and -3 and their precursors were stabilized (Fig. 2F). We then looked at the effect of combining knockdown of these factors (see Fig. S6 in the supplemental material). We found that the double knockdown of both RNase L and Dicer did not affect the accumulation of SvsRNA-1 compared to the single RNase L knockdown, which confirms that Dicer does not play a role (Fig. S6A). On the contrary, the accumulation of SvsRNA-1 upon double knockdown of both RNase L and Xrn1 increased compared to the single RNase L knockdown (Fig. S6A). This finding suggests that both RNase L and Xrn1 can act upon viral RNAs and have opposite activities in terms of generation and stabilization of the SvsRNAs. Xrn1 might act upstream of RNase L in degrading, for example, uncapped viral RNAs that would otherwise be a substrate for RNase L; alternatively, it could directly act upon the RNase L products. The former hypothesis is more likely though because Xrn1 has a preference for 5′ phosphorylated RNAs, whereas RNase L leaves a 5′ OH after cleavage. We also performed a high-molecular-weight Northern blot analysis to look at the effects of the various knockdowns on the accumulation of the genomic and subgenomic viral RNAs. The results confirm the quantitative PCR data, in that only the RNase L KD has an impact on the accumulation of both genomic and subgenomic RNAs (Fig. S6B).
We also tested several cytoplasmic 3′-5′ exoribonucleases, including DIS3L, DIS3L2, and Rrp40 (also known as EXOSC3, a core subunit of the exosome); knockdown of these factors had no effect on SvsRNA accumulation (data not shown). This indicated that at least the main cytoplasmic 3′-5′ exoribonucleases do not seem to be implicated in the production of the viral small RNAs.
The 3′ extremity of SvsRNA-1 is modified.
RNase L processing produces a typical signature: the enzyme leaves a 5′ hydroxyl (OH) and a 3′ phosphate (P) after cleavage (6). However, these features are not compatible with our small RNA cloning protocol (29), which strictly relies on the presence of a 5′ P and a 3′ OH on the small RNA extremities. This prompted us to characterize the extremities of SvsRNAs to better understand their biogenesis. We initially decided to restrict our analysis to SvsRNA-1 because it accumulated as a major form of 23 nt. The assay used for the analysis of RNA 5′ ends involved digestion with the Terminator exonuclease, a processive 5′-3′ exonuclease that specifically targets and degrades RNAs with a 5′ monophosphate, but not RNAs with 5′ triphosphates, 5′ OH, or a 5′ CAP (30).
For a control, the Terminator reaction was carried out on both a 5′ OH and a 5′ P oligoribonucleotide with the same sequence as SvsRNA-1. Terminator treatment did not result in the degradation of the 5′ OH synthetic oligoribonucleotide, whereas the 5′ P RNA oligonucleotide was almost completely degraded. Accordingly, SvsRNA-1 is prone to degradation by the Terminator exonuclease, which is consistent with the presence of a monophosphate at the 5′ end, while the accumulation of the precursor bands was less affected by the treatment (Fig. 3A).
FIG 3 .
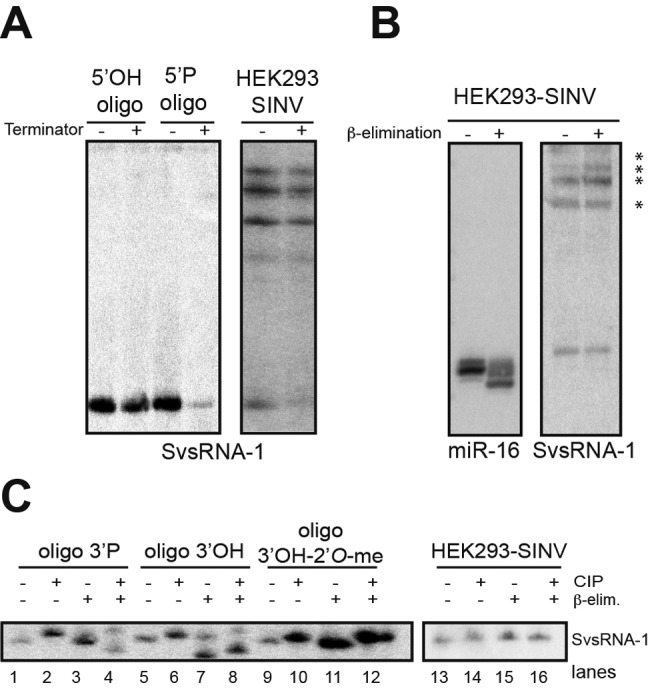
Characterization of 5′ and 3′ extremities of SvsRNA-1. (A) Synthetic oligonucleotides that have 5′ OH or 5′ P ends or total RNA were treated with Terminator exonuclease (+) or not treated with Terminator (−) before being analyzed by Northern blotting. (B) β-elimination on HEK293-SINV total RNA followed by Northern blotting of SvsRNA-1 and miR-16 after migration on 17.5% polyacrylamide gel. Asterisks denote putative precursors of SvsRNA-1. (C) Synthetic SvsRNA-1 molecules that have 3′ P, 3′ OH, or 3′ OH 2′-O-me ends were treated with calf intestinal phosphatase (CIP) (lanes 2, 4, 6, 8, 10, 12, 14, and 16). Dephosphorylation was followed by β-elimination (lanes 4, 8, 12, and 16). oligo, oligonucleotide; me, methyl; elim, elimination.
Next, we performed sodium periodate (NaIO4) treatment followed by β-elimination to specifically examine the 3′ end of SvsRNA-1. The presence of hydroxyl groups in positions 2′ and 3′ on the last ribose is needed for the periodate reaction. β-elimination of the oxidized RNA results in an RNA product one or two nucleotides shorter. On the contrary, the presence of modifications on the ribose of the 3′ terminal residue renders the RNA insensitive to periodate oxidation and impairs the chain scission (31). For a control for the reaction, we measured its effect on the unmodified cellular miRNA miR-16. As expected, the electrophoretic mobility of the miRNA strongly changed after treatment (Fig. 3B). In contrast, SvsRNA-1 mobility was found to be insensitive to periodate oxidation and β-elimination (Fig. 3B). This indicated the presence of a modification on the terminal ribose, most likely in the 2′ position, which is perfectly compatible with our cloning protocol. Interestingly, it also seems that the intermediate RNA products are insensitive to periodate oxidation and are consequently modified (see Fig. S7 in the supplemental material). Moreover, we were also able to assess the existence of a modified 3′ end for other SvsRNAs by Northern blotting (Fig. S8).
In order to gain more insight into the nature of this modification, we treated our samples with calf intestinal phosphatase (CIP) (Fig. 3C, lanes 2, 4, 6, 8, 10, 12, 14, and 16) prior to β-elimination (Fig. 3C, lanes 4, 8, 12, and 16). The double treatment strongly affected the mobility of a control oligoribonucleotide with a 3′ P (Fig. 3C, lane 4), while the mobility of the 3′ OH oligonucleotide was affected independently of the CIP treatment (Fig. 3C, lanes 7 and 8). Of note, the combined reactions changed neither the mobility of a control oligoribonucleotide bearing a CIP-insensitive modification (methyl group in position 2′ of the last ribose) (Fig. 3C, lane 12) nor the mobility of SvsRNA-1 (Fig. 3C, lane 16).
Taken together, these results show that SvsRNA-1 is modified at its 3′ extremity but that the modification is not due to a monophosphate group caused by a direct RNase L cut.
Viral genomic RNA is modified on A8333.
The presence of posttranscriptional modifications, such as 2′-O-methylation, is a characteristic of several cellular RNAs in mammals, such as rRNAs (32), snRNAs (33), and Piwi-interacting RNAs (piRNAs) (34, 35). Recently, it has also been reported for several viruses that their genomic RNA can be 2′-O-methylated at specific positions (36, 37). We hypothesized that the modification on the small RNA could be derived from a modification at the level of the genomic RNA, which is reasonable given the fact that SvsRNA-1 precursor RNAs also appear to be modified (see Fig. S7 in the supplemental material). We thus looked for modifications in the genomic region spanning SvsRNA-1 using the RTL-P (reverse transcription at low deoxyribonucleoside triphosphate concentrations followed by polymerase chain reaction) technique (38): the tendency of the reverse transcriptase to pause immediately before a 2′-O-methylated nucleotide is exploited to establish the efficiency of retrotranscription by semiquantitative PCR, using differential deoxynucleoside triphosphate (dNTP) concentrations.
As shown in Fig. 4A, we designed a reverse transcription primer specific for the viral genome, as well as primers for semiquantitative PCR in the region surrounding SvsRNA-1 (shown in red): one reverse primer (R) and three forward primers, upstream (FU) or downstream of the putative modification (FD1 and FD2). After retrotranscription with low and high dNTP concentrations, we performed multiplex PCR using primers FU/FD1 (in the SvsRNA-1 region) or primers FD1/FD2 (in the control region) and then compared the signal intensity ratios of PCR products amplified with different numbers of PCR cycles (Fig. 4B). Our data indicated that only the FU/FD1 ratio changed depending on the RT conditions (Fig. 4B, top panel), suggesting that at a low dNTP concentration, the reverse transcriptase paused in the SvsRNA-1 region, most probably due to the presence of a modification on the viral genome in the region of interest.
FIG 4 .
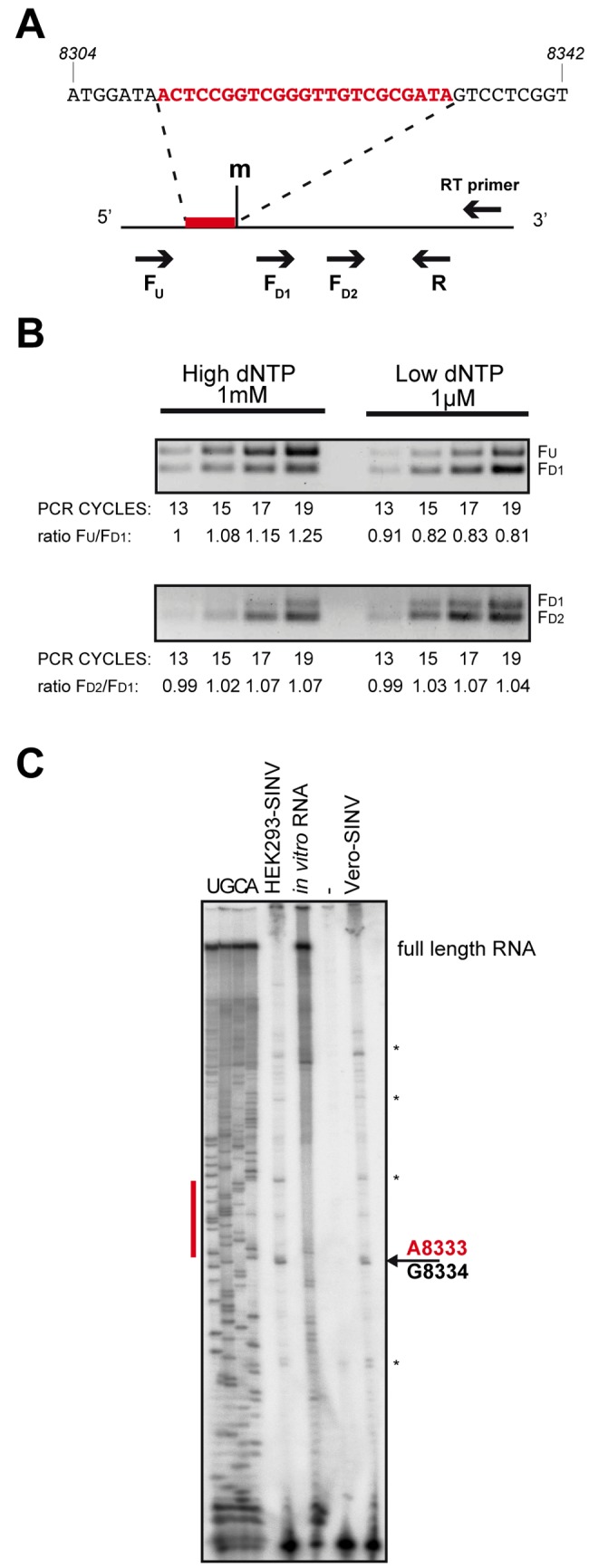
SINV genomic RNA is modified. (A) Schematic diagram of the primer design for RTL-P to detect the presence of 2′-O-methylated nucleotides (m) on the viral genome. SvsRNA-1 is indicated in red. (B) Detection of methylation by RTL-P under both high and low dNTP concentrations and PCR cycles on SINV-infected HEK293 cells. The ratio of PCR signal intensity is indicated beneath each lane. (C) Primer extension mapping of 2′-O-methylated nucleotides. Lanes U, G, C, and A represent dideoxy sequencing reactions performed on the in vitro transcript amplified for the same viral region. Asterisks indicate stops of the reverse transcriptase.
To further strengthen this observation and to identify the precise position of the nucleotide modification, we performed primer extension assays on RNA isolated from either SINV-infected HEK293 or Vero cells (Fig. 4C). 2′-O-methylation can cause reverse transcriptase to pause one nucleotide before and/or at the O-methylated nucleotide. Indeed, we identified two bands (RT stops) that correspond to nucleotides G8334 and A8333, the latter corresponding precisely to the 3′ end of the SvsRNA-1 sequence (Fig. 4C).
These data imply that the 3′-end modification of SvsRNA-1 was likely already present on the viral genome and might be a 2′-O-methylation.
3′-end modification is a hallmark of SvsRNAs.
Taken together, our observations indicate that the SINV genomic RNA is modified, and are therefore suggestive of a potential modification of SvsRNAs. We then decided to investigate to what extent reads mapping to the SINV genome were modified at the last nucleotide position. Small RNA libraries were prepared from noninfected or SINV-infected HEK293 cells using total RNA treated (or not) with NaIO4. This treatment specifically impairs the 3′ adapter ligation to RNA species with unmodified 3′ ends (2′ and 3′ OH), while RNAs bearing a modification at one of these positions are resistant to oxidation, and can therefore be ligated. Overnight ligation of the 3′ adapter at 16°C was performed in order to enhance the cloning of 2′-O modified small RNAs (39).
After deep sequencing of these libraries, we mapped the total number of reads against both the human and SINV genome (Table 1). As expected, in both noninfected and infected cells, the NaIO4 treatment drastically reduced the number of clonable miRNA sequences (see also Fig. S9A in the supplemental material). In contrast, modified small RNAs (such as tRNA fragments) were preferentially cloned over miRNAs upon NaIO4 treatment (Fig. S9B). Strikingly, almost 75% of the viral reads were kept after oxidation (Table 1). The oxidation treatment did not dramatically change the genomic distribution of the viral reads compared to the nontreated sample (Fig. 5A). However, deep sequencing revealed some new peaks (Fig. 5A, arrows) that we could also validate by β-elimination and Northern blot analysis (see SvsRNA-15 and -16 in Fig. S8).
TABLE 1 .
Deep sequencing results of NalO4-treated and nontreated small RNA librariesa
| Categoryb | HEK293-NI-NT |
HEK293-NI-NalO4 |
HEK293-SINV-NT |
HEK293-SINV-NalO4 |
||||
|---|---|---|---|---|---|---|---|---|
| Read count | Read % | Read count | Read % | Read count | Read % | Read count | Read % | |
| Raw data | 21,631,645 | 34,131,455 | 20,753,238 | 28,263,345 | ||||
| Filtered out | 92,556 | 3,589,935 | 146,674 | 5,425,669 | ||||
| Unmapped | 840,747 | 7,135,376 | 1,276,025 | 5,143,518 | ||||
| Mapped | 20,698,342 | 100.00 | 23,406,144 | 100.00 | 19,330,539 | 100.00 | 17,694,158 | 100.00 |
| hsa miRNAs | 18,298,024 | 88.40 | 7,325,147 | 31.30 | 14,850,144 | 76.82 | 4,138,513 | 23.39 |
| hsa rRNAs | 66,614 | 0.32 | 1,509,137 | 6.45 | 337,226 | 1.74 | 850,630 | 4.81 |
| hsa tRNAs | 878,642 | 4.24 | 6,260,105 | 26.75 | 1,680,819 | 8.70 | 5,915,996 | 33.43 |
| hsa snRNAs | 8,956 | 0.04 | 13,719 | 0.06 | 8,913 | 0.05 | 6,996 | 0.04 |
| hsa snoRNAs | 91,453 | 0.44 | 93,919 | 0.40 | 111,048 | 0.57 | 70,898 | 0.40 |
| hsa miscRNAs | 59,911 | 0.29 | 36,831 | 0.16 | 283,636 | 1.47 | 98,472 | 0.56 |
| Only viral RNA | 491 | 0.00 | 37 | 0.00 | 400,252 | 2.07 | 262,916 | 1.49 |
| Unannotated RNA | 1,294,251 | 6.25 | 8,167,249 | 34.89 | 1,658,501 | 8.58 | 6,349,737 | 35.89 |
HEK293 cells were infected with SINV or not infected (NI) and treated with sodium periodate (NalO4) or not treated with sodium periodate (NT).
Abbreviations: hsa, homo sapiens; miscRNAs, miscellaneous RNAs.
FIG 5 .
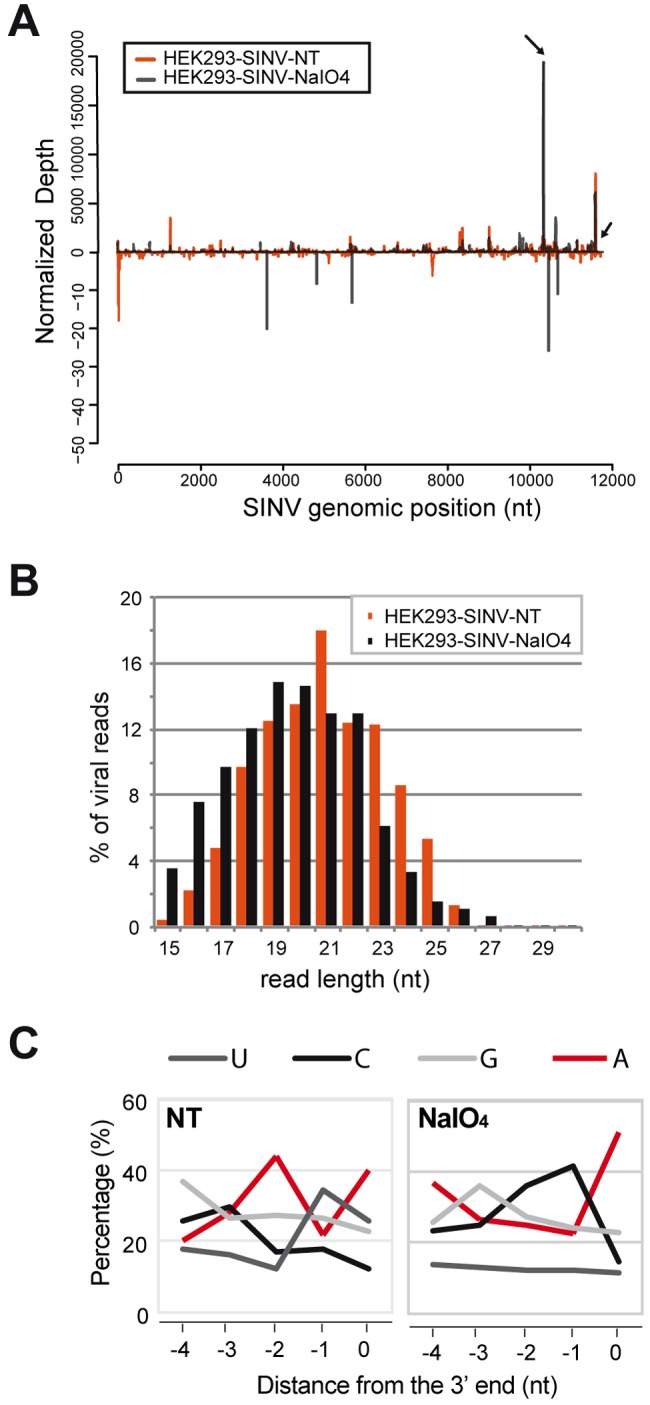
Deep sequencing upon periodate oxidation in SINV-infected HEK293 cells. (A) Distribution of viral reads mapping to the SINV genome. The abundance of small RNAs was calculated and plotted as the sum of normalized reads (per 105 viral reads) in each single-nucleotide sliding window along the SINV RNA. The small black arrows indicate the peaks chosen for Northern blot validation. (B) Size distribution of SINV-derived small RNA populations. The percentage of viral sRNAs deriving from the genomic sense strand in each size class is shown as a percentage of the total viral small RNA reads for this strand. Data from SINV-infected HEK293 cell samples treated with NaIO4 or not treated with NaIO4 (NT) are shown. (C) Relative nucleotide frequency per position. The viral reads with 0 mismatch to the SINV positive strand were considered for this analysis. Only the last five nucleotides from the 3′ end of each sequence are displayed.
The size distribution of the viral reads remained unchanged before and after NaIO4 treatment, indicating that SINV-derived modified RNAs belonged to each length (Fig. 5B). Finally, we analyzed the nucleotide composition of the 3′ end of SINV small RNAs and observed that we had enrichment in adenosine (A) at the 3′ end after oxidation, which was not clearly discernible in the nontreated sample (Fig. 5C). This is in line with the specific 2′-O-methylation occurring on internal adenosines recently identified on the genome of dengue virus (36).
DISCUSSION
Mammals have evolved a very sophisticated innate immune response to ward off pathogens that is based on the sensing of pathogen-associated molecular patterns (PAMPs) by dedicated pattern recognition receptors (PRRs). When it comes to viral infection, one of the most potent PAMPs is dsRNA. The recognition of dsRNA by PRRs triggers a signaling cascade that ultimately leads to the activation of interferon-sensitive genes (ISGs) (40). RNA silencing is another type of antiviral innate immunity, which, as of now, has mostly been demonstrated in plant and insect organisms (1). Nonetheless, a role for RNA silencing as a host defense system against mammalian viruses could reasonably be considered. Indeed, Dicer, the key enzyme for dsRNA cleavage is conserved in higher vertebrates, and although its main substrate in vivo is pre-miRNA molecules, it is able to process long dsRNA into short RNAs in vitro (41). However, the question as to which other types of dsRNA molecules mammalian Dicer can cleave in vivo has been the matter of intense research, and especially so when considering RNAs of viral origin. Here, we wanted to make our contribution to the field by studying the prototypical arbovirus, Sindbis virus. This type of virus can infect both insects and mammals, and as such is a useful tool to study the conservation of antiviral mechanisms in both phyla. Intriguingly, it has been shown for some members of the alphavirus genus that their genomic RNA can be processed into both siRNAs and piRNAs in insect somatic cells (42, 43).
In this study, we generated small RNA libraries from two different mammalian cell lines infected with SINV. Although the percentage of sequences mapping to the viral genome was significant (~1.7 to ~8% in HEK293 and Vero cells, respectively), we were unable to detect either siRNA or piRNA signatures. On the contrary, the viral reads displayed a highly biased strand distribution. Although antigenomic (negative) strand-derived sequences represented a very small percentage of the total viral reads, they showed properties distinct from the genomic (positive) strand reads: their size distribution peaked at 22 nt, and their localization on the viral genome is restricted to the promoters required for the synthesis of both the genomic and subgenomic RNA. Although the cloned 22-nt sequences were not visible by Northern blot analysis, we could detect the accumulation of an ~44-nt product that in our knowledge has never been observed before. We hypothesize that these RNA species could be critical for viral replication, similarly to what has been previously reported for influenza virus (44).
Consistent with the relative abundance of the positive strand compared to the negative strand, the majority of reads were derived from the plus-strand RNA and more specifically from the region corresponding to the subgenomic RNA sequence. We could detect several hot spots of small RNA accumulation, indicating that discrete SINV-derived viral small RNA species (SvsRNAs) could be produced in infected mammalian cells. These SvsRNAs do not display any peak in size distribution and do not map to any alphavirus conserved regions. Nonetheless, we noticed that they originate from the same viral genomic positions in both infected African green monkey and human cells. In order to confirm that this was not solely due to a bias in the small RNA cloning protocol, we performed Northern blot analyses of several small RNA candidates. For some of the candidates, we were able to detect discrete bands, which increased in intensity throughout the time course of infection.
We therefore focused on the characterization of the positive-strand-derived small RNAs. One hypothesis regarding their identity could be that some of these SvsRNAs are in fact virus-encoded miRNAs. Using bioinformatics tools such as miRDeep2 (45, 46) and miRanalyzer2 (47, 48), we excluded the possibility that putative miRNA precursor structures were present in the viral genome. Nonetheless, we tested whether the miRNA machinery was required for the production of viral small RNAs by knocking down Drosha, Dicer, and Ago2. The downregulation of these factors affected neither SINV replication nor accumulation of the three most abundant identified SvsRNAs, indicating that their production must involve another RNase. We discovered that the RNase L endonuclease is implicated in SvsRNA genesis. Indeed, its downregulation causes a dramatic reduction in the accumulation of Sindbis viral small RNAs detected by Northern blot analysis; this is accompanied by an upregulation of the viral genomic RNA. In order to extend this observation, we also generated small RNA libraries from HEK293 cells treated with siRNAs against RNase L and infected with SINV. We found that, under these conditions, the global population of SINV sRNAs was downregulated. RNase L is an endoribonuclease that is present in the cell in the form of an inactive monomer. Upon type I interferon induction, and more specifically 2′-5′ oligoadenylate synthetase activation (6), it can dimerize and then act upon its viral RNA targets. RNase L is known to play an antiviral role against viruses such as West Nile virus (49), hepatitis C virus (50), and SINV (51). We also looked at the involvement of exoribonucleases in the production of SvsRNAs and found that, indeed, the accumulation of these small RNAs is counteracted by the cellular 5′-3′ exonuclease Xrn1. This suggests that SvsRNAs represent stable viral products of cellular decay enzymes. We therefore hypothesize that RNase L is the enzyme primarily involved in the generation of small RNAs and that the further processing operated by other uncharacterized enzymes could give rise to the final 20- to 30-nt-long viral RNAs. It also remains possible that some of the fragments we detected are direct products of RNase L but are further modified by kinases and dephosphorylases, which would allow their cloning.
The fact that RNA fragments generated by RNase L are detected implies that some of them must be stabilized. Indeed, we discovered that the viral small RNA candidates appear to be modified on the 3′ proximal nucleotide. We could extend this peculiarity to the global small RNA population by cloning and sequencing small RNAs after an oxidation treatment designed to prevent ligation of nonmodified RNAs. Our data indicate that this modification is most likely a 2′-O-methylation and seems to occur at the level of the viral genomic RNA, preferentially on adenosines. Thanks to a double approach of RTL-P and primer extension, we were able to map such a modification on position A8333 of the SINV viral genome. Moreover, our sequencing data indicate that there are most likely many others given the enrichment in A residues at the 3′ end of small RNAs cloned after oxidative treatment.
The main protective mechanism against 3′-5′ degradation is a 2′-O-methylation on the 3′ proximal ribose, which prevents degradation by exoribonucleases and thus helps to stabilize small RNAs such as miRNAs and siRNAs in plants, piRNAs in animals, and siRNAs in Drosophila melanogaster (52). This suggests that RNA modification could have evolved as a response to protect the anti-pathogen small RNAs during infections. Posttranscriptional RNA modifications represent an important but not yet fully understood system to regulate transcripts. Cellular RNAs contain many distinct posttranscriptional modifications at thousands of sites. In the same way, RNA viruses take advantage of nucleoside modifications on the base or on the ribose of their genomic RNA (36, 53) as a host mimicry strategy to abrogate innate immune signaling (54).
The enzyme responsible for the methylation of SINV RNA remains unknown. However, it has been reported that the alphavirus nsP2 protein contains a putative methyltransferase (MT)-like domain, similar to dengue virus NS5 MTase (55). Although the activity of this domain has not been verified yet, nsP2-MTase mutant viruses exhibit a strong phenotype: negative-strand synthesis is continuous, and unstable viral replication complexes are produced. Surprisingly, RNase L-deficient cells infected with SINV display the same phenotype, suggesting that the two proteins function in similar events (55). For all these reasons, we propose that RNase L-dependent, 3′-modified small RNAs from SINV could be involved as intermediates in this mechanism.
Altogether, our findings show a link between the host degradation pathway and stabilization of modified viral small RNAs. Clearly, not all the small RNAs identified by our deep sequencing approach correspond to RNase L downstream products. Nonetheless, the fact that at least some of these small RNAs are protected and detected might implicate them in signaling events, maybe upstream of the infection front, which might prove difficult to assess in cell culture. We hypothesized that by removing RNase L we might uncover signatures corresponding to other types of small RNAs, such as siRNAs or piRNAs, but it did not turn out to be the case. This could be due to the fact that other enzymes might also be involved in the production of RNA fragments and is an indication that it will make the detection of antiviral RNAi complicated in differentiated mammalian cells. An alternate hypothesis could also be that SINV encodes an RNA silencing suppressor that blocks Dicer activity but has no effect on RNase L. Finally, it is also possible that the antiviral RNAi response might be detected only in specific subtypes of cells in mammals, which would be different from the differentiated somatic cells we used for our study. Further investigation will be needed to answer these questions and to decipher the roles played by this type of virus-derived small RNAs, as well as their potential implication in the design of new antiviral therapeutic strategies.
MATERIALS AND METHODS
Viral stocks, cell culture, and virus infection.
Plasmid carrying wild-type (WT) and green fluorescent protein (GFP)-SINV genomic sequence (kindly provided by M. C. Saleh) was linearized with XhoI and used as a substrate for in vitro transcription using mMESSAGE mMACHINE capped RNA transcription kit (Ambion) following the manufacturer’s instructions. Sindbis viral stock was prepared in BHK21 hamster kidney cells, and titers were measured by plaque assay. HEK293 and Vero cells were maintained in Dulbecco’s modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Clontech) in a humidified atmosphere of 5% CO2 at 37°C. Cells were infected with SINV at an MOI of 0.01. Samples were harvested at 0, 4, 8, 12, 16, 24, and 48 h postinfection (hpi).
RNAi-mediated protein depletion.
For siRNA transfection, 0.4 × 105 HEK293 cells were seeded in 6-well plates. Transfections were carried out for 72 h using 20 nM ON-TARGET plus Smart Pool siRNAs (Dharmacon) for one-step depletion (human RNase L and Xrn1) or 100 nM ON-TARGET plus Smart Pool siRNAs (Dharmacon) for two-step depletion (human Drosha, Dicer, and Ago2) with Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. The ON-TARGET plus Nontargeting Pool siRNAs (Dharmacon) was used as a negative control. Cells were infected with SINV and harvested after 16 h for RNA and protein analysis.
Small RNA cloning and sequencing.
RNA was extracted from noninfected and SINV-infected HEK293 and Vero cells 16 hpi. Small RNA cloning was conducted with 10 or 20 µg of total RNA as previously described (14, 29). Small RNA libraries from SINV-infected HEK293 cells treated with control or RNase L-specific siRNAs were generated following the same protocol with some modifications. Mainly, small RNAs were ligated using degenerate 5′ and 3′ adapters to limit biases in the ligation step (56). This procedure was also used to prepare small RNA libraries after NaIO4 treatment of 100 µg of total RNA, as previously described (57), except that the 3′ ligation was performed at 16°C overnight, in 12% polyethylene glycol 8000 (PEG 8000) to favor the cloning of modified small RNAs. Sequencing was performed at the IGBMC Microarray and Sequencing platform, Illkirch, France, using different Illumina instruments (Genome Analyzer IIx, HiSeq 2000, and HiSeq 2500) with a read length of 36 or 50 bp.
A detailed description of the deep sequencing data analysis is available in Text S1 in the supplemental material.
Microarray data accession number.
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (58) and are accessible through GEO Series accession number GSE48831 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48831).
SUPPLEMENTAL MATERIAL
Supplemental Materials and Methods, supplemental references, and Table S1 (primers and RNA oligonucleotides used in this study) Download
Time course of SINV infection in HEK293 and Vero cells. (A) HEK293 cells (left panel) or Vero cells (right panel) were infected with SINV-GFP at an MOI of 0.01 and were harvested at 4, 8, 16, 24, and 48 h for FACS analysis. Noninfected (NI) cells served as a negative control. Dot plot examples of a representative fluorescence-activated cell sorting (FACS) analysis of GFP and propidium iodide (PI) levels are shown. The uninfected control cells are present in the lower left quadrant. The GFP-positive SINV-infected living cells are shown in the lower right quadrant. Percentages of cells are indicated for each quadrant. (B and C) The percentages of GFP-positive (SINV-infected) (B) and living cells (C) were obtained from the FACS analysis during the time course of infection and represented in the graphs for HEK293 (in red) and Vero (in green) cells, respectively. At 16 hpi, the majority of the cells are infected (GFP positive) and alive. Download
Small RNA accumulation from SINV negative strand. (A) Distribution of SINV reads mapping to the viral antigenome. The abundance of small RNAs was calculated and plotted as the sum of normalized reads (per 105 viral reads) in each single-nucleotide sliding window along the viral antigenome. Arrows indicate the peaks chosen for Northern blot validation (see Fig. S3A, SvsRNA-12 and -14). (B) Size distribution of the SINV reads derived from the antigenomic strand in each size class is shown as a percentage of the total viral small RNA reads for this strand. Data from HEK293 and Vero cells are represented in black and orange, respectively. (C) The secondary structure of the 3′-end region of SINV antigenome was predicted using the Mfold algorithm (59). The negative-strand sequence is shown from 5′ to 3′, and the positions of the nucleotides in the structure are represented. The viral sequence identified in our small RNA library (see Fig. S3A, SvsRNA-14) is depicted in red. (D) Northern blot analysis using a probe complementary to the aforementioned sequence was performed in HEK293 and Vero cells. Asterisks indicate the expected sizes in nucleotides. Download
Validation of SINV candidates identified by deep sequencing. (A) Genomic position, strand, length, and number of reads in both SINV-infected HEK293 and Vero cells are shown for the chosen candidates. (B) Northern blot analysis of several SvsRNAs in infected HEK293 cells (16 hpi) is shown. Bands corresponding to small RNAs between 20 and 30 nt in size are highlighted. (C) Northern blot analysis in SINV-infected HEK293 and Vero cells. Probes corresponding to SvsRNA-1, -2, and -3 and U6 (normalizer) were used. Download
Drosha, Dicer, and Ago2 do not affect SvsRNA-1, -2, and -3 accumulation. (A to C) Drosha, Dicer, and Ago2 protein levels were verified by Western blotting upon KD in SINV-infected HEK293 cells. Tubulin was used as a normalizer. (D to F) SINV genomic RNA was quantified by RT-qPCR before and after specific KD of Drosha, Dicer, and Ago2. (G) Northern blot analysis of total RNA from infected HEK293 cells before and after Drosha, Dicer, and Ago2 KD. Specific DNA oligonucleotides were used as probes for detection of SvsRNA-1, -2, and -3. U6 was used as a normalizer. (H) Northern blot analysis of RNA purified after Ago2 RNA immunoprecipitation (IP) (left panel) and Pan-Ago RNA-IP (right panel) in SINV-infected HEK293 cells. Bromodeoxyuridine (BrdU) or IgG antibodies were used as negative controls. Specific DNA oligonucleotides were used as probes for detection of SvsRNA-1, -2, and -3 or hsa-miR-16. Download
Deep sequencing upon RNase L KD in SINV-infected HEK293 cells. (A) Virus-specific small RNA reads were recovered from each library and were mapped on both positive and negative viral strands. (B) Size distribution of SINV-derived small RNA populations. The percentage of viral sRNAs deriving from the genome and antigenome in each size class is shown relative to the total viral small RNA reads for either strand. Data from sictrl-treated and siRNase L-treated HEK293 cells are represented in black and orange, respectively. (C) Distribution of viral reads mapping to the SINV genome. The abundance of small RNAs was calculated and plotted as the sum of normalized reads (per 105 viral reads) in each single-nucleotide sliding window along the viral RNA. Download
Effects of RNase L, Dicer, and Xrn1 single and double knockdown on viral RNAs in HEK293 cells infected with SINV-GFP. (A and B) Small RNA Northern blot (A) and high-molecular-weight Northern blot (B) analyses were performed on total RNA from infected HEK293 cells before and after single knockdown of RNase L, Dicer, and Xrn1 or double knockdown of RNase L/Dicer and RNase L/Xrn1. Specific DNA oligonucleotides were used as probes for detection of SvsRNA-1 and U6 (A) or genome/subgenome and GAPDH mRNA (B). (C) RNase L and Xrn1 protein levels were verified by Western blotting upon single or double KD in SINV-infected HEK293 cells. Tubulin was used as a normalizer. Download
Characterization of the 3′ extremities of SvsRNA-1 precursors. β-elimination on both an in vitro transcript of ~60 nt (positive control) and total RNA of SINV-infected HEK293 cells followed by Northern blotting of SvsRNA-1 after migration on long 8% polyacrylamide gel. Asterisks denote putative precursors of SvsRNA-1. Lanes M, 10- to 100-nt marker. Download
Validation of 3′-end protection by β-elimination. β-elimination on total RNA extracted from SINV-infected HEK293 cells was followed by Northern blot analysis. The electrophoretic mobilities of SvsRNA-2, -3, -15, and -16 remain unchanged, indicating the presence of a modification on the 2′-O of the terminal ribose. Download
Periodate oxidation affects miRNA cloning. (A) Relative abundance of human miRNAs in HEK293 samples. Oxidation drastically reduces the number of clonable microRNA sequences, independently of the infection. (B) Relative abundance of human tRNA fragments in HEK293 samples. Oxidation preserves the number of clonable tRNA fragments. miRNA and tRNA read counts are normalized per million mapped reads (RPM). Download
ACKNOWLEDGMENTS
This work was supported by an ATIP starting grant from CNRS, Agence Nationale pour la Recherche sur le Sida et les Hépatites Virales (ANRS-2009-136), Agence Nationale pour la Recherche (labex netRNA, ANR-10-LABX-36), and the European Research Council (ERC starting grant ncRNAVIR 260767).
We are grateful to M. C. Saleh for the gift of the plasmids for SINV preparation and G. Meister for the anti-Ago2 antibody. We thank Frédéric Gros for technical help with the fluorescence-activated cell sorting (FACS) analyses. We acknowledge Valérie Cognat and the BioImage facility (IBMP, Strasbourg, France) for help with the initial analysis of the deep sequencing data. We also thank the IGBMC Microarray and Sequencing platform, member of the France Genomique program, for the sequencing of our libraries. We thank all members of the Pfeffer laboratory, especially Lee Tuddenham for critically reading the manuscript and Gabrielle Haas for fruitful discussions.
Footnotes
Citation Girardi E, Chane-Woon-Ming B, Messmer M, Kaukinen P, Pfeffer S. 2013. Identification of RNase L-dependent, 3′-end-modified, viral small RNAs in Sindbis virus-infected mammalian cells. mBio 4(6):e00698-13. doi:10.1128/mBio.00698-13.
REFERENCES
- 1. Ding SW, Voinnet O. 2007. Antiviral immunity directed by small RNAs. Cell 130:413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 3. Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801 [DOI] [PubMed] [Google Scholar]
- 4. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6:975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U. S. A. 95:15623–15628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silverman RH. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81:12720–12729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parameswaran P, Sklan E, Wilkins C, Burgon T, Samuel MA, Lu R, Ansel KM, Heissmeyer V, Einav S, Jackson W, Doukas T, Paranjape S, Polacek C, dos Santos FB, Jalili R, Babrzadeh F, Gharizadeh B, Grimm D, Kay M, Koike S, Sarnow P, Ronaghi M, Ding SW, Harris E, Chow M, Diamond MS, Kirkegaard K, Glenn JS, Fire AZ. 2010. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog. 6:e1000764. 10.1371/journal.ppat.1000764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. 2001. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293:834–838 [DOI] [PubMed] [Google Scholar]
- 9. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, Kim VN. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419 [DOI] [PubMed] [Google Scholar]
- 10. Krol J, Loedige I, Filipowicz W. 2010. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11:597–610 [DOI] [PubMed] [Google Scholar]
- 11. Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. 2007. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449:919–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saïb A, Voinnet O. 2005. A cellular microRNA mediates antiviral defense in human cells. Science 308:557–560 [DOI] [PubMed] [Google Scholar]
- 13. Pfeffer S, Zavolan M, Grässer FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, Tuschl T. 2004. Identification of virus-encoded microRNAs. Science 304:734–736 [DOI] [PubMed] [Google Scholar]
- 14. Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grässer FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. 2005. Identification of microRNAs of the herpesvirus family. Nat. Methods 2:269–276 [DOI] [PubMed] [Google Scholar]
- 15. Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. 2005. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:5570–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Samols MA, Hu J, Skalsky RL, Renne R. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 79:9301–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tuddenham L, Jung JS, Chane-Woon-Ming B, Dölken L, Pfeffer S. 2012. Small RNA deep sequencing identifies microRNAs and other small noncoding RNAs from human herpesvirus 6B. J. Virol. 86:1638–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kincaid RP, Burke JM, Sullivan CS. 2012. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl. Acad. Sci. U. S. A. 109:3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cullen BR. 2006. Is RNA interference involved in intrinsic antiviral immunity in mammals? Nat. Immunol. 7:563–567 [DOI] [PubMed] [Google Scholar]
- 20. Shapiro JS, Varble A, Pham AM, tenOever BR. 2010. Noncanonical cytoplasmic processing of viral microRNAs. RNA 16:2068–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shapiro JS, Langlois RA, Pham AM, tenOever BR. 2012. Evidence for a cytoplasmic microprocessor of pri-miRNAs. RNA 18:1338–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Griffin DE. 2007. Alphaviruses, p 1023–1068 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 23. Akhrymuk I, Kulemzin SV, Frolova EI. 2012. Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 86:7180–7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jose J, Snyder JE, Kuhn RJ. 2009. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4:837–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Desmyter J, Melnick JL, Rawls WE. 1968. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 2:955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li Z, Ender C, Meister G, Moore PS, Chang Y, John B. 2012. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 40:6787–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Floyd-Smith G, Slattery E, Lengyel P. 1981. Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate dependent endonuclease. Science 212:1030–1032 [DOI] [PubMed] [Google Scholar]
- 29. Pfeffer S. 2007. Identification of virally encoded microRNAs. Methods Enzymol. 427:51–63 [DOI] [PubMed] [Google Scholar]
- 30. Xhemalce B, Robson SC, Kouzarides T. 2012. Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell 151:278–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Behm-Ansmant I, Helm M, Motorin Y. 2011. Use of specific chemical reagents for detection of modified nucleotides in RNA. J. Nucleic Acids 2011:408053. 10.4061/2011/408053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ni J, Tien AL, Fournier MJ. 1997. Small nucleolar RNAs direct site-specific synthesis of pseudouridine in ribosomal RNA. Cell 89:565–573 [DOI] [PubMed] [Google Scholar]
- 33. Kiss T. 2004. Biogenesis of small nuclear RNPs. J. Cell Sci. 117:5949–5951 [DOI] [PubMed] [Google Scholar]
- 34. Kirino Y, Mourelatos Z. 2007. Mouse Piwi-interacting RNAs are 2′-O-methylated at their 3′ termini. Nat. Struct. Mol. Biol. 14:347–348 [DOI] [PubMed] [Google Scholar]
- 35. Ohara T, Sakaguchi Y, Suzuki T, Ueda H, Miyauchi K, Suzuki T. 2007. The 3′ termini of mouse Piwi-interacting RNAs are 2′-O-methylated. Nat. Struct. Mol. Biol. 14:349–350 [DOI] [PubMed] [Google Scholar]
- 36. Dong H, Chang DC, Hua MHC, Lim SP, Chionh YH, Hia F, Lee YH, Kukkaro P, Lok S-M, Dedon PC, Shi P-Y. 2012. 2′-O methylation of internal adenosine by flavivirus NS5 methyltransferase. PLoS Pathog. 8:e1002642. 10.1371/journal.ppat.1002642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Y, Su C, Ke M, Jin X, Xu L, Zhang Z, Wu A, Sun Y, Yang Z, Tien P, Ahola T, Liang Y, Liu X, Guo D. 2011. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2′-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 7:e1002294. 10.1371/journal.ppat.1002294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dong ZW, Shao P, Diao LT, Zhou H, Yu CH, Qu LH. 2012. RTL-P: a sensitive approach for detecting sites of 2′-O-methylation in RNA molecules. Nucleic Acids Res. 40:e157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Munafó DB, Robb GB. 2010. Optimization of enzymatic reaction conditions for generating representative pools of cDNA from small RNA. RNA 16:2537–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beutler B, Eidenschenk C, Crozat K, Imler JL, Takeuchi O, Hoffmann JA, Akira S. 2007. Genetic analysis of resistance to viral infection. Nat. Rev. Immunol. 7:753–766 [DOI] [PubMed] [Google Scholar]
- 41. Hammond SM. 2005. Dicing and slicing: the core machinery of the RNA interference pathway. FEBS Lett. 579:5822–5829 [DOI] [PubMed] [Google Scholar]
- 42. Morazzani EM, Wiley MR, Murreddu MG, Adelman ZN, Myles KM. 2012. Production of virus-derived ping-pong-dependent piRNA-like small RNAs in the mosquito soma. PLoS Pathog. 8:e1002470. 10.1371/journal.ppat.1002470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vodovar N, Bronkhorst AW, van Cleef KW, Miesen P, Blanc H, van Rij RP, Saleh MC. 2012. Arbovirus-derived piRNAs exhibit a ping-pong signature in mosquito cells. PLoS One 7:e30861. 10.1371/journal.pone.0030861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Umbach JL, Yen HL, Poon LL, Cullen BR. 2010. Influenza A virus expresses high levels of an unusual class of small viral leader RNAs in infected cells. mBio 1(4):e00204-10. 10.1128/mBio.00204-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Friedländer MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. 2008. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 26:407–415 [DOI] [PubMed] [Google Scholar]
- 46. Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40:37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hackenberg M, Sturm M, Langenberger D, Falcón-Pérez JM, Aransay AM. 2009. miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res. 37:W68–W76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hackenberg M, Rodríguez-Ezpeleta N, Aransay AM. 2011. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 39:W132–W138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scherbik SV, Paranjape JM, Stockman BM, Silverman RH, Brinton MA. 2006. RNase L plays a role in the antiviral response to West Nile virus. J. Virol. 80:2987–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Malathi K, Saito T, Crochet N, Barton DJ, Gale M, Silverman RH. 2010. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 16:2108–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sawicki DL, Silverman RH, Williams BR, Sawicki SG. 2003. Alphavirus minus-strand synthesis and persistence in mouse embryo fibroblasts derived from mice lacking RNase L and protein kinase R. J. Virol. 77:1801–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ji L, Chen X. 2012. Regulation of small RNA stability: methylation and beyond. Cell Res. 22:624–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Judge A, MacLachlan I. 2008. Overcoming the innate immune response to small interfering RNA. Hum. Gene Ther. 19:111–124 [DOI] [PubMed] [Google Scholar]
- 55. Mayuri, Geders TW, Smith JL, Kuhn RJ. 2008. Role for conserved residues of Sindbis virus nonstructural protein 2 methyltransferase-like domain in regulation of minus-strand synthesis and development of cytopathic infection. J. Virol. 82:7284–7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sorefan K, Pais H, Hall AE, Kozomara A, Griffiths-Jones S, Moulton V, Dalmay T. 2012. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Akbergenov R, Si-Ammour A, Blevins T, Amin I, Kutter C, Vanderschuren H, Zhang P, Gruissem W, Meins F, Hohn T, Pooggin MM. 2006. Molecular characterization of geminivirus-derived small RNAs in different plant species. Nucleic Acids Res. 34:462–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30:207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials and Methods, supplemental references, and Table S1 (primers and RNA oligonucleotides used in this study) Download
Time course of SINV infection in HEK293 and Vero cells. (A) HEK293 cells (left panel) or Vero cells (right panel) were infected with SINV-GFP at an MOI of 0.01 and were harvested at 4, 8, 16, 24, and 48 h for FACS analysis. Noninfected (NI) cells served as a negative control. Dot plot examples of a representative fluorescence-activated cell sorting (FACS) analysis of GFP and propidium iodide (PI) levels are shown. The uninfected control cells are present in the lower left quadrant. The GFP-positive SINV-infected living cells are shown in the lower right quadrant. Percentages of cells are indicated for each quadrant. (B and C) The percentages of GFP-positive (SINV-infected) (B) and living cells (C) were obtained from the FACS analysis during the time course of infection and represented in the graphs for HEK293 (in red) and Vero (in green) cells, respectively. At 16 hpi, the majority of the cells are infected (GFP positive) and alive. Download
Small RNA accumulation from SINV negative strand. (A) Distribution of SINV reads mapping to the viral antigenome. The abundance of small RNAs was calculated and plotted as the sum of normalized reads (per 105 viral reads) in each single-nucleotide sliding window along the viral antigenome. Arrows indicate the peaks chosen for Northern blot validation (see Fig. S3A, SvsRNA-12 and -14). (B) Size distribution of the SINV reads derived from the antigenomic strand in each size class is shown as a percentage of the total viral small RNA reads for this strand. Data from HEK293 and Vero cells are represented in black and orange, respectively. (C) The secondary structure of the 3′-end region of SINV antigenome was predicted using the Mfold algorithm (59). The negative-strand sequence is shown from 5′ to 3′, and the positions of the nucleotides in the structure are represented. The viral sequence identified in our small RNA library (see Fig. S3A, SvsRNA-14) is depicted in red. (D) Northern blot analysis using a probe complementary to the aforementioned sequence was performed in HEK293 and Vero cells. Asterisks indicate the expected sizes in nucleotides. Download
Validation of SINV candidates identified by deep sequencing. (A) Genomic position, strand, length, and number of reads in both SINV-infected HEK293 and Vero cells are shown for the chosen candidates. (B) Northern blot analysis of several SvsRNAs in infected HEK293 cells (16 hpi) is shown. Bands corresponding to small RNAs between 20 and 30 nt in size are highlighted. (C) Northern blot analysis in SINV-infected HEK293 and Vero cells. Probes corresponding to SvsRNA-1, -2, and -3 and U6 (normalizer) were used. Download
Drosha, Dicer, and Ago2 do not affect SvsRNA-1, -2, and -3 accumulation. (A to C) Drosha, Dicer, and Ago2 protein levels were verified by Western blotting upon KD in SINV-infected HEK293 cells. Tubulin was used as a normalizer. (D to F) SINV genomic RNA was quantified by RT-qPCR before and after specific KD of Drosha, Dicer, and Ago2. (G) Northern blot analysis of total RNA from infected HEK293 cells before and after Drosha, Dicer, and Ago2 KD. Specific DNA oligonucleotides were used as probes for detection of SvsRNA-1, -2, and -3. U6 was used as a normalizer. (H) Northern blot analysis of RNA purified after Ago2 RNA immunoprecipitation (IP) (left panel) and Pan-Ago RNA-IP (right panel) in SINV-infected HEK293 cells. Bromodeoxyuridine (BrdU) or IgG antibodies were used as negative controls. Specific DNA oligonucleotides were used as probes for detection of SvsRNA-1, -2, and -3 or hsa-miR-16. Download
Deep sequencing upon RNase L KD in SINV-infected HEK293 cells. (A) Virus-specific small RNA reads were recovered from each library and were mapped on both positive and negative viral strands. (B) Size distribution of SINV-derived small RNA populations. The percentage of viral sRNAs deriving from the genome and antigenome in each size class is shown relative to the total viral small RNA reads for either strand. Data from sictrl-treated and siRNase L-treated HEK293 cells are represented in black and orange, respectively. (C) Distribution of viral reads mapping to the SINV genome. The abundance of small RNAs was calculated and plotted as the sum of normalized reads (per 105 viral reads) in each single-nucleotide sliding window along the viral RNA. Download
Effects of RNase L, Dicer, and Xrn1 single and double knockdown on viral RNAs in HEK293 cells infected with SINV-GFP. (A and B) Small RNA Northern blot (A) and high-molecular-weight Northern blot (B) analyses were performed on total RNA from infected HEK293 cells before and after single knockdown of RNase L, Dicer, and Xrn1 or double knockdown of RNase L/Dicer and RNase L/Xrn1. Specific DNA oligonucleotides were used as probes for detection of SvsRNA-1 and U6 (A) or genome/subgenome and GAPDH mRNA (B). (C) RNase L and Xrn1 protein levels were verified by Western blotting upon single or double KD in SINV-infected HEK293 cells. Tubulin was used as a normalizer. Download
Characterization of the 3′ extremities of SvsRNA-1 precursors. β-elimination on both an in vitro transcript of ~60 nt (positive control) and total RNA of SINV-infected HEK293 cells followed by Northern blotting of SvsRNA-1 after migration on long 8% polyacrylamide gel. Asterisks denote putative precursors of SvsRNA-1. Lanes M, 10- to 100-nt marker. Download
Validation of 3′-end protection by β-elimination. β-elimination on total RNA extracted from SINV-infected HEK293 cells was followed by Northern blot analysis. The electrophoretic mobilities of SvsRNA-2, -3, -15, and -16 remain unchanged, indicating the presence of a modification on the 2′-O of the terminal ribose. Download
Periodate oxidation affects miRNA cloning. (A) Relative abundance of human miRNAs in HEK293 samples. Oxidation drastically reduces the number of clonable microRNA sequences, independently of the infection. (B) Relative abundance of human tRNA fragments in HEK293 samples. Oxidation preserves the number of clonable tRNA fragments. miRNA and tRNA read counts are normalized per million mapped reads (RPM). Download