

ABSTRACT
A positive blood culture is a critical result that requires prompt identification of the causative agent. This article describes a simple method to identify microorganisms from positive blood culture broth within the time taken to perform a Gram stain (<20 min). The method is based on intrinsic fluorescence spectroscopy (IFS) of whole cells and required development of a selective lysis buffer, aqueous density cushion, optical microcentrifuge tube, and reference database. A total of 1,121 monomicrobial-positive broth samples from 751 strains were analyzed to build a database representing 37 of the most commonly encountered species in bloodstream infections or present as contaminants. A multistage algorithm correctly classified 99.6% of unknown samples to the Gram level, 99.3% to the family level, and 96.5% to the species level. There were no incorrect results given at the Gram or family classification levels, while 0.8% of results were discordant at the species level. In 8/9 incorrect species results, the misidentified isolate was assigned to a species of the same genus. This unique combination of selective lysis, density centrifugation, and IFS can rapidly identify the most common microbial species present in positive blood cultures. Faster identification of the etiologic agent may benefit the clinical management of sepsis. Further evaluation is now warranted to determine the performance of the method using clinical blood culture specimens.
IMPORTANCE
Physicians often require the identity of the infective agent in order to make life-saving adjustments to empirical therapy or to switch to less expensive and/or more targeted antimicrobials. However, standard identification procedures take up to 2 days after a blood culture is signaled positive, and even most rapid molecular techniques take several hours to provide a result. Other techniques are faster (e.g., matrix-assisted laser desorption ionization–time of flight [MALDI-TOF] mass spectrometry) but require time-consuming manual processing steps and expensive equipment. There remains a clear need for a simple, inexpensive method to rapidly identify microorganisms directly from positive blood cultures. The promising new method described in this research article can identify microorganisms in minutes by optical spectroscopy, thus permitting the lab to simultaneously report the presence of a positive blood culture and the organism’s identity.
INTRODUCTION
Bacteremia and fungemia are potentially life-threatening medical conditions that require aggressive management. Typically, broad-spectrum multidrug therapy is initiated based on clinical symptoms, patient demographics, and clinical guidelines or antimicrobial stewardship programs. Yet despite the best intentions, empirical therapy is not universally effective, and a delay in receiving appropriate therapy is associated with increased mortality (1, 2), particularly for patients in septic shock (3).
Gram staining of a positive blood culture provides the first laboratory evidence of a possible bloodstream infection and influences the selection of antimicrobial therapy more often than receipt of full antimicrobial susceptibility testing (AST) results (4). Additionally, prompt reporting of Gram stain results has been shown to reduce patient mortality (5). However, the ability to quickly identify the etiologic agent to the species level would also help establish the clinical relevance of the organism, enable empirical therapy to be more specifically tailored (targeted, safer, or less expensive agents), and reduce the risk of contributing to the development of resistance against broad-spectrum antibiotics, all central tenets of antibiotic stewardship programs (6). For example, early identification of an isolate as “Pseudomonas aeruginosa” rather than a “Gram-negative rod” may reduce the indiscriminate use of carbapenems and lower (7) or at least prevent further increase in (8) the level of carbapenem-resistant isolates. In a recent blood culture study, early reporting of a species identification (ID) resulted in modifications to empirical therapy in 35% of cases, compared to 21% for the Gram stain result alone (9). Finally, while specific outcomes were not always consistent between studies, several groups have reported that earlier provision of clinically relevant blood culture results generated benefits in terms of either patient outcome, length of hospitalization, antimicrobial utilization, or overall cost of medical care (10–14).
Continuously monitored cultivation of microorganisms in blood specimens with liquid media is currently the “gold standard” for the detection of bacteremia/fungemia and/or sepsis, and notification of a positive blood culture demands immediate attention. Standard protocol calls for the preparation of a Gram stain and the subculture of organisms in the broth onto supportive media for subsequent phenotypic, molecular, or mass spectrometry (MS)-based ID and full AST, a process that can take two or more days to complete.
Given that positive blood culture broth contains an abundant population of microorganisms, some laboratories have obtained faster results by using growth-based phenotypic ID systems loaded with microorganisms recovered directly from positive culture bottles (15–17). However, these “direct-from-the-bottle” phenotypic tests require manual manipulation of large volumes of blood culture broth, are not appropriate for all microorganism types, are not validated by the test manufacturers, and generally take 3 to 8 h to generate an ID.
Over the past decade, many molecular tests have become commercially available that decrease the time to identification. These tests are now being used in conjunction with positive blood cultures to identify a limited number of specific pathogens and resistance mechanisms. Examples include probe-based tests, such as peptide nucleic acid fluorescence in situ hybridization (PNA-FISH) (18–20), and nucleic acid amplification-based tests, such as PCR (21–23). The probe-based tests require multiple manual processing steps, while the PCR-based assays are relatively expensive, and most still require 1 to 3 h to perform.
Matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) is fast becoming an established method for the rapid identification of microorganisms, and commercial systems are on the market and in the process of regulatory approval (24). However, blood and culture medium components also interfere with this technology and must be manually removed prior to the direct analysis of positive blood cultures to obtain acceptable sensitivity (9, 25–34). Recently, an evaluation of three processing methods for blood culture media was published (33). While MALDI-TOF MS technology is accurate, fast, and relatively inexpensive, it shares with many other rapid technologies the disadvantage of requiring a significant number of manual processing steps to prepare the sample prior to analysis.
Optical spectroscopy methods, such as Raman spectroscopy, Fourier transform infrared (FTIR) spectroscopy, and intrinsic fluorescence spectroscopy (IFS), can identify microorganisms very quickly. These methods require little preanalytical work, as intact cells are directly measured. Prior extraction of proteins or nucleic acids is not required. Both Raman spectroscopy and FTIR spectroscopy have been used to identify microcolonies (35) and even single microbial cells (36–38). IFS measures naturally occurring cell components that are capable of fluorescence, such as tryptophan, NADH, flavin adenine dinucleotide (FAD), and porphyrins. Methods that identify microorganisms based on analysis of their intrinsic fluorescence have been described in the literature for over 25 years (39–45). Recently, the potential of IFS to identify colonies growing on agar has been demonstrated (46, 47). However, all optical spectroscopy methods also encounter interference from the highly fluorescent and absorptive compounds present in liquid microbiological culture media, in container materials, and in blood-containing clinical samples.
Simple, safe, and reliable methods are needed in the microbiology lab to isolate microorganisms present in blood culture broth that are free of interfering materials and suitable for rapid identification technologies. In addition, fast and inexpensive technologies are urgently needed to identify the isolate within the time frame for performing a Gram stain. This article describes a novel method fulfilling both needs, combining a one-step purification of microorganisms with rapid identification using in situ measurements of microbial intrinsic fluorescence.
RESULTS
Positive blood culture broth samples from 751 isolates representing 37 of the most commonly isolated microbial species from blood cultures were tested. A list of species present in the database and their relative abundance are shown in Table 1. These species represent >90% of positive blood cultures found globally (48).
TABLE 1 .
List of species and numbers of strains present in the classification model
| Species in model | Total no. of strains |
No. of known blood culture isolatesa |
|---|---|---|
| Staphylococcus aureus | 55 | 11 |
| Escherichia coli | 53 | 4 |
| Staphylococcus epidermidis | 42 | 12 |
| Streptococcus pneumoniae | 39 | 6 |
| Klebsiella pneumoniae | 38 | 6 |
| Enterococcus faecalis | 31 | 3 |
| Candida albicans | 30 | 0 |
| Candida tropicalis | 29 | 0 |
| Streptococcus mitis/Streptococcus oralis | 28 | 0 |
| Pseudomonas aeruginosa | 27 | 0 |
| Candida parapsilosis | 27 | 0 |
| Enterococcus faecium | 26 | 2 |
| Enterobacter cloacae Cpx | 25 | 3 |
| Enterobacter aerogenes | 24 | 1 |
| Citrobacter freundii | 23 | 0 |
| Salmonella enterica serovar Enteritidis | 20 | 0 |
| Providencia stuartii | 18 | 2 |
| Staphylococcus lugdunensis | 15 | 1 |
| Stenotrophomonas maltophilia | 14 | 1 |
| Staphylococcus hominis | 14 | 4 |
| Haemophilus influenzae | 12 | 0 |
| Klebsiella oxytoca | 12 | 0 |
| Proteus mirabilis | 12 | 1 |
| Listeria monocytogenes | 12 | 0 |
| Candida krusei | 12 | 0 |
| Morganella morganii | 11 | 0 |
| Neisseria meningitidis | 11 | 0 |
| Proteus vulgaris | 11 | 0 |
| Streptococcus pyogenes | 11 | 0 |
| Acinetobacter baumannii | 10 | 0 |
| Staphylococcus capitis | 10 | 0 |
| Staphylococcus warneri | 10 | 0 |
| Streptococcus agalactiae | 10 | 0 |
| Streptococcus infantarius subsp. coli | 10 | 0 |
| Candida glabrata | 10 | 0 |
| Serratia marcescens | 9 | 0 |
Number of strains isolated from clinical blood cultures.
A summary of the cross-validated, multilevel classification algorithm results is given in Table 2. In brief, when an identification result was given, 100% were correct to the Gram and family classification levels, and 99.2% were correct to the species level. Of the 1,121 tests completed, 99.6% were assigned a correct, one-choice Gram result. Of the 1,117 tests correctly identifying the isolate to the Gram level, 99.3% were correctly assigned a one-choice family result. Similarly, of the 1,109 tests correctly identifying the isolate to the family level, 1,080 (97.4%) were correctly assigned to the species level. Compared to the number of initial tests (1,121), 96.5% of samples gave correct species-level identification, 2.7% gave no identification, and 0.8% gave a discordant result.
TABLE 2 .
Multilevel microbial classification using intrinsic fluorescence
| Level | Microbial group |
No. (%) of tests |
||||
|---|---|---|---|---|---|---|
| Total | One-choice agreement |
Low-discrimination agreement |
Incorrect ID | No ID | ||
| 1 | Gram type | 1,121 | 1,117 (99.6) | 0 (0) | 0 (0) | 4 (0.4) |
| 2 | Family | 1,117 | 1,109 (99.3) | 2 (0.2) | 0 (0) | 6 (0.5) |
| 3 | Species | 1,109 | 1,048 (94.5) | 32 (2.9) | 9 (0.8) | 20 (1.8) |
The species-level results are shown in Table 3. There were 1,048 one-choice (94.5%) and 32 low-discrimination (2.9%) species-level results, for an overall accuracy of 97.4%. Excellent results were obtained for all three major Gram groups: 96.3% of 519 Gram-negative broth samples, 97.7% of 430 Gram-positive broth samples, and 100% of 160 yeast-containing broth samples were assigned to the correct species. Many of the low-discrimination calls were to species of the same genus. Five of these cases were Klebsiella pneumoniae-Klebsiella oxytoca pairings, four were Enterobacter cloacae Cpx–Enterobacter aerogenes, two were Proteus mirabilis-Proteus vulgaris, and the final case was Staphylococcus warneri-Staphylococcus capitis. There were 20 test samples that were not identified at the species level (1.8%); however, all were correctly identified at the family level. Sixteen were identified as members of the Enterobacteriaceae family (5 as Providencia stuartii, 1 as Proteus mirabilis, 2 as Morganella morganii, 3 as Klebsiella pneumoniae, 1 as Escherichia coli, 2 as Enterobacter cloacae Cpx, and 2 as Citrobacter freundii), and the remaining four were identified as members of the Staphylococcaceae family (1 each as Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus hominis, and Staphylococcus capitis). Nine isolates were incorrectly identified. Apart from an Escherichia coli isolate that was classified as an Enterobacter cloacae Cpx, the remaining eight misidentified isolates were assigned to species of the same genus (see the footnote to Table 3).
TABLE 3.
Details of species-level classification by intrinsic fluorescence spectroscopy
| Species | No. (%) of tests |
||||
|---|---|---|---|---|---|
| Total | One-choice ID | Low-discrimination ID | Incorrect IDa | No ID | |
| Gram negative | |||||
| Citrobacter freundii | 33 | 30 (90.9) | 1 (3.0) | 0 (0) | 2 (6.1) |
| Enterobacter aerogenes | 34 | 27 (79.4) | 7 (20.6) | 0 (0) | 0 (0) |
| Enterobacter cloacae Cpx | 35 | 28 (80.0) | 3 (8.6) | 2 (5.7) | 2 (5.7) |
| Escherichia coli | 76 | 71 (93.4) | 3 (3.9) | 1 (1.3) | 1 (1.3) |
| Klebsiella oxytoca | 24 | 22 (91.7) | 2 (8.3) | 0 (0) | 0 (0) |
| Klebsiella pneumoniae | 53 | 43 (81.1) | 7 (13.2) | 0 (0) | 3 (5.7) |
| Morganella morganii | 22 | 20 (90.9) | 0 (0) | 0 (0) | 2 (9.1) |
| Proteus mirabilis | 23 | 19 (82.6) | 3 (13.0) | 0 (0) | 1 (4.3) |
| Proteus vulgaris | 20 | 20 (100) | 0 (0) | 0 (0) | 0 (0) |
| Providencia stuartii | 18 | 13 (72.2) | 0 (0) | 0 (0) | 5 (27.8) |
| Salmonella enterica serovar Enteritidis | 31 | 29 (93.5) | 2 (6.5) | 0 (0) | 0 (0) |
| Serratia marcescens | 18 | 18 (100) | 0 (0) | 0 (0) | 0 (0) |
| Acinetobacter baumanii | 20 | 20 (100) | 0 (0) | 0 (0) | 0 (0) |
| Neisseria meningitidis | 22 | 22 (100) | 0 (0) | 0 (0) | 0 (0) |
| Haemophilus influenzae | 21 | 21 (100) | 0 (0) | 0 (0) | 0 (0) |
| Pseudomonas aeruginosa | 47 | 47 (100) | 0 (0) | 0 (0) | 0 (0) |
| Stenotrophomonas maltophilia | 22 | 22 (100) | 0 (0) | 0 (0) | 0 (0) |
| Total | 519 | 472 (90.9) | 28 (5.4) | 3 (0.6) | 16 (3.1) |
| Gram positive | |||||
| Enterococcus faecalis | 45 | 45 (100) | 0 (0) | 0 (0) | 0 (0) |
| Enterococcus faecium | 39 | 39 (100) | 0 (0) | 0 (0) | 0 (0) |
| Listeria monocytogenes | 12 | 12 (100) | 0 (0) | 0 (0) | 0 (0) |
| Staphylococcus aureus | 85 | 82 (96.5) | 1 (1.2) | 1 (1.2) | 1 (1.2) |
| Staphylococcus capitis | 10 | 9 (90) | 0 (0) | 0 (0) | 1 (10.0) |
| Staphylococcus epidermidis | 65 | 63 (96.9) | 0 (0) | 1 (1.5) | 1 (1.5) |
| Staphylococcus hominis | 14 | 13 (92.9) | 0 (0) | 0 (0) | 1 (7.1) |
| Staphylococcus lugdunensis | 15 | 15 (100) | 0 (0) | 0 (0) | 0 (0) |
| Staphylococcus warneri | 9 | 7 (77.8) | 2 (22.2) | 0 (0) | 0 (0) |
| Streptococcus agalactiae | 20 | 20 (100) | 0 (0) | 0 (0) | 0 (0) |
| Streptococcus infantarius subsp. coli | 10 | 7 (70) | 1 (10.0) | 2 (20) | 0 (0) |
| Streptococcus mitis-Streptococcus oralis | 35 | 35 (100) | 0 (0) | 0 (0) | 0 (0) |
| Streptococcus pneumoniae | 49 | 47 (95.9) | 0 (0) | 2 (4.1) | 0 (0) |
| Streptococcus pyogenes | 22 | 22 (100) | 0 (0) | 0 (0) | 0 (0) |
| Total | 430 | 416 (96.7) | 4 (0.9) | 6 (1.4) | 4 (0.9) |
| Yeast | |||||
| Candida albicans | 40 | 40 (100) | 0 (0) | 0 (0) | 0 (0) |
| Candida glabrata | 20 | 20 (100) | 0 (0) | 0 (0) | 0 (0) |
| Candida krusei | 24 | 24 (100) | 0 (0) | 0 (0) | 0 (0) |
| Candida parapsilosis | 37 | 37 (100) | 0 (0) | 0 (0) | 0 (0) |
| Candida tropicalis | 39 | 39 (100) | 0 (0) | 0 (0) | 0 (0) |
| Total | 160 | 160 (100) | 0 (0) | 0 (0) | 0 (0) |
Two E. cloacae Cpx isolates were misidentified as E. aerogenes, one E. coli isolate as E. cloacae Cpx, one S. aureus isolate as S. lugdunensis, one S. epidermidis isolate as S. warneri, two S. pneumoniae isolates as S. mitis-S. oralis and S. agalactiae, and two S. infantarius subsp. coli isolates as S. mitis-S. oralis and S. agalactiae.
DISCUSSION
In this article, we describe a simple method to purify microorganisms from positive blood culture broth and concomitantly identify them within a closed centrifuge tube by using IFS. Although it requires some customized components, the method is well suited to immediate or stat testing, as an ID result is available within 20 min of collection of the positive broth sample. Microbial classification was performed using a series of algorithms that assigned results sequentially to the Gram, family, and species levels. Only tests that were assigned a mutually consistent one-choice result at the Gram and family groups were analyzed in the species algorithm. This multilevel approach minimizes the risk of misclassification and makes it easier to diagnose processing errors or atypical measurements. The method correctly identified 96.5% of all test samples to the species level. No species identification was given for 2.7% of the samples, but the majority (67%) were correctly identified to the family level, which still provides more clinically relevant information about the isolate than the conventional Gram stain result.
In a recent study, it was demonstrated that 65% of the analytical errors made in a clinical microbiology laboratory involved misinterpretation of the Gram stain (50). The IFS method delivered no incorrect results at either the Gram or family level from a total of 1,121 tests and gave confident one-choice Gram and family results in 99.6% and 99.3% of tests, respectively (Table 2). The IFS results compare favorably with a published study on the accuracy of the Gram stain report (51), which demonstrated that even highly skilled technicians can report incorrect results, particularly for nonhemolytic streptococci and Gram-positive rods. In other laboratories with less skilled staff, the accuracy of the Gram report was quite poor (52).
The immediate isolation of purified microorganisms from positive blood cultures is not a trivial task. Commonly employed methods for recovering microorganisms directly from blood culture broth include differential centrifugation (15, 25, 31), centrifugation within a serum separator tube (16, 17), or a combination of the two (26, 53). For some direct testing applications, mild (27) or harsh (28, 30, 32) cell lytic agents are used at neutral pH, followed by several washing steps. Others have used hypotonic solutions in combination with washing (29). However, all of these methods have drawbacks. The resultant microbial preparations often contain contaminating red blood cells, incompletely solubilized blood cell membranes, precipitated proteins, platelets, lipid particles, plasma enzymes, and hemoglobin, which can lead to poor results. Some of these processing methods use harsh conditions that may damage microorganisms. They are also very labor-intensive and potentially unsafe due to steps that can result in aerosol exposure to lab technicians.
In recent years, rapid microbial identification obtained by the processing of positive blood cultures for use with MALDI-TOF MS systems has been more successful with samples containing Gram-negative rather than Gram-positive microorganisms. Typically, the species-level identification rate is 20 to 50% lower for the Gram-positive group (25, 29–31, 33, 34). We believe this is partly related to the significantly higher cell concentration of the most common Gram-negative species found in positive blood cultures (unpublished data), which results in further dilution of contaminating nonmicrobial debris, relative to the microbial cells, in the suspensions used for analysis. Additionally, the sample processing conditions also play a role in determining the final outcome. The brief alkaline detergent lysis method described here reduces the level of nonmicrobial debris effectively enough to allow unencumbered measurement of intact cell fluorescence and thereby generate a highly accurate species ID for both cultures containing Gram-positive organisms (97.7%) as well as those containing Gram-negative organisms (96.3%).
The novel lysis buffer of this method combines a high-pH CAPS (N-cyclohexyl-3-aminopropanesulfonic acid) buffer with a mild nonionic polyoxyethylene class detergent (Brij-O10 [formerly Brij-97]) to rapidly solubilize human blood cells while leaving microorganisms intact. The selective lysis conditions, including detergent, buffer, sample dilution, temperature, and contact time, were chosen to minimize any alteration in intrinsic fluorophores and maximize viability of the most common microorganisms found in blood cultures.
The salt-based aqueous density cushion provides two important functions. First, passage of microorganisms through the high-density salt solution is a very efficient wash step, removing interfering plasma and fluorescent or fluorescence-attenuating medium-derived compounds while rapidly transferring the cells from an alkaline to a physiological pH environment. Second, the high ionic strength of this solution causes microbial cells to shrink, increase in buoyant density, and sediment more rapidly to the base of the optical centrifuge tube, where they can be interrogated by front-face IFS. Such a mechanism has been described for E. coli (54). This property is useful when separating microbial species with low buoyant densities, such as Streptococcus pneumoniae and some Gram-negative rods.
Simplicity and enhanced safety are other features of the method. Once the lysate has been prepared and loaded into the optical centrifuge tube, the remaining steps of the method can be performed using general lab equipment without the risk of aerosol generation. The sealed tube is centrifuged, transferred to a spectrofluorimeter for reading, and then disposed of. No manual suspension, washing, or extraction of microbial cell pellets is required, rendering the method potentially useful for the direct and safe identification of mycobacteria as well.
The use of intrinsic fluorescence for the identification of microorganisms has been described in the literature for many years, and there is renewed interest in the applications of fluorescence spectroscopy in the field of medical microbiology (55). These studies employed either time-resolved fluorescence (56), steady-state fluorescence (39–47), or steady-state fluorescence in combination with specific ligands (57). Although promising, they lacked the strain numbers or species diversity to draw any firm conclusions about the potential of IFS to become a robust ID method for clinical use. During our initial evaluation of IFS using a diverse range of clinically and industrially relevant microorganisms (J. Hyman and J. Walsh, unpublished data), like other investigators, we found that when suspensions of microbial cells were examined by standard right-angle fluorescence optics, tryptophan was the predominant fluorophore measured. Surprisingly, when microbial cells were firmly packed into the UV-transparent centrifuge tubes used in this study and then examined by front-face fluorescence, many additional cellular fluorophores were evident, and more importantly, the spectral distribution of these fluorophores enabled a significant improvement in classification accuracy.
We chose to use a nontargeted approach in analyzing the EEM data presented in this report and found that the species-resolving power of intrinsic fluorescence measurements of intact microbial cells was not limited to the analysis of a few well-known fluorophores, such as tryptophan or NADH, but rather to a combination of fluorophores existing in multiple physical states and environments within viable, metabolically active cells. This finding is hardly surprising as the fluorescence of many intrinsic fluorophores is dependent upon their physical environment within a cell, and it is well known that the interaction of cell fluorophores and proteins alters their fluorescence properties (58). For example, the fluorescence intensity of NADH increases when bound to cellular proteins, and the excitation and emission maxima are blue-shifted (59). The emission maximum of another intrinsic fluorophore, pyridoxal, is also blue-shifted when bound to proteins such as phosphorylase (58). Furthermore, both NADH and FAD act as electron donors and acceptors and play critical roles in cell energy metabolism (60), and their relative levels provide an estimate of the redox state of a cell (61). Additional cellular fluorophores include a variety of porphyrins (intermediates in heme biosynthesis), riboflavin, pterins, lipofuscin, pyridoxamine, and structural compounds, such as elastin and collagen (49). ATP has also been shown to autofluoresce, albeit under specific conditions (62). Furthermore, some fluorophores are produced only by microorganisms. For example, P. aeruginosa cells contain a characteristic yellow-green fluorescent siderophore called pyoverdin.
Many rapid identification methods measure physical properties of unknown cells, such as nucleic acid sequences, protein masses, or the vibration of various chemical bonds. However, a nontargeted analysis of intact microbial cell intrinsic fluorescence provides a snapshot of both structural and metabolic components of viable cells, without the use of specific or expensive reagents. This method has the potential to provide species-level identification for the most commonly encountered sepsis-associated organisms. The availability of an ID result within 20 min of detection of a positive blood culture would result in a single call being made to report both a positive culture and the organism’s identity, something not always possible with other rapid technologies. The more concise reporting provided by this method will result in improved efficiency, more timely management of empirical therapy, a reduction in laboratory and hospital costs, and, potentially, a reduction in patient mortality.
MATERIALS AND METHODS
Material.
BacT/ALERT SA blood culture bottles, tryptic soy broth (TSB), tryptic soy agar with 5% sheep blood, and Sabouraud dextrose agar were from bioMérieux, Inc. (Durham, NC). CAPS buffer (N-cyclohexyl-3-aminopropanesulfonic acid) was purchased from Alpha Aesar, HEPES buffer from JT Baker, Brij-O10 (formerly Brij-97) detergent from Sigma-Aldrich, Pluronic F-108 from BASF, cesium chloride from MP Biomedicals, and polypropylene balls from CIC, Ball Company. The optical microcentrifuge tube used for this study was designed by bioMérieux and built under contract by an external manufacturer. This screw-cap tube was constructed of a low-fluorescence, UV-transparent acrylic (Acrylite H15-012; Cyro Industries). The tube was centrifuged in an Eppendorf 5417R microcentrifuge with a standard A-8-11 swing-out rotor and buckets. Small inserts were placed in the buckets to support the base of the optical tube during centrifugation. The fluorescence spectrophotometer used was a FluoroLog-3 2T2 system, built by Horiba-Jobin Yvon. The fiber optic probe (6 excitation around 1 emission bundle of 300-µm diameter core fibers) was supplied by Ocean Optics.
Blood cultures.
Samples of positive blood culture broth were generated in the following manner. BacT/ALERT SA culture bottles were inoculated with 10 ml of freshly collected sodium polyanethol sulfonate (SPS)-anticoagulated human blood from over 50 healthy donors, followed by 50 to 500 CFU of each test strain suspended in approximately 0.5 ml TSB. Inoculated bottles were then incubated at 35 to 37°C in the BacT/ALERT 3D microbial detection system.
A total of 751 bacterial and fungal strains were tested in this study. Ninety-two percent of the strains were selected from bioMérieux’s culture collection in St. Louis, while the remaining 8% were known blood culture isolates obtained from deidentified clinical blood cultures collected in 2009. Of the culture collection isolates with known sources of origin (80%), 60% were collected from multiple clinical institutions within the United States, 27% were obtained from other countries, and 13% were stock strains from ATCC or the CDC. Where possible, ATCC type strains from each species were included. Furthermore, the strains selected were deposited into the culture collection spanning the period 1984 to 2009. The list of species and their relative abundance in the database are shown in Table 1.
Test strains were recovered from frozen stock and subcultured three times on the appropriate solid media before use. The identity of each strain was confirmed with Vitek 2 cards and MALDI-TOF MS using the Axima assurance system (Shimadzu Corp.) and SARAMIS database (bioMérieux). To ensure that isolated microorganisms were always in the logarithmic growth phase, bottles were removed from the BacT/ALERT 3D microbial detection system within 5 min of generation of a positive signal and processed within 15 min of removal, unless otherwise specified.
Rapid ID procedure.
An overall schematic of the simple three-step process (lyse-spin-read) is given in Fig. 1. Briefly, a 2.0-ml sample of warm (35 to 37°C) positive broth is removed from the test blood culture bottle and added to 1.0 ml of warm (35 to 37°C) selective lysis buffer (0.45% [wt/vol] Brij-O10 in 0.3 M CAPS [pH 11.7]) contained in a 15-ml screw-cap polypropylene tube. The mixture was vortexed for 5 s at maximum speed and then placed in a 35 to 37°C water bath for 60 s. After an additional 1 to 2s vortex, 1.0 ml of the lysate was removed and layered onto a single 5/16-in.-diameter polypropylene ball (CIC, Ball Co.) floating on the surface of 0.5 ml of a solution of 24% (wt/vol) cesium chloride plus 0.005% (wt/vol) Pluronic F-108 plus 10 mM HEPES (pH 7.4) contained within an optical microcentrifuge tube. The polypropylene ball was used to control the layering process and create an undisturbed interface. The tube was sealed with a screw cap and centrifuged for 2 min at 10,000 rpm at 20 to 25°C in a microcentrifuge with an A-8-11 swing-out rotor. Photographs of a tube loaded with S. lugdunensis-positive broth taken before and after the centrifugation step are shown in Fig. 2. Following centrifugation, the tube was seated upon the end of the fiber optic probe in a custom-built holder (Fig. 3A) within a light-tight sample compartment. Fluorescence of the pelleted microorganisms was then collected through the 0.5-mm-thick bottom section of the tube using the FluoroLog-3 system with the following excitation-emission matrix (EEM) parameters. The excitation scan range was every 5 nm from 260 to 800 nm (109 discrete wavelengths), with a slit width of 5 nm. Emission was collected every 5 nm from 260 to 1,100 nm, with a slit width of 5 nm. The run time for a full EEM scan was 13.6 min per sample, however, fewer than 10% of the excitation wavelengths measured were required for the classification model. A 450-W xenon lamp and double-grating monochromator provided the specific excitation energy. Fluorescence was collected using an F3000 mirror adapter connected to the 300-µm-diameter fiber optic probe and measured using a Triax 320 spectrometer with a 600-line/mm grating (500-nm blaze) and a Symphony charge-coupled device (CCD) array. A schematic of the fiber optic probe interface to the spectrofluorimeter is given in Fig. 3B. Fluorescence signals were normalized for lamp intensity (S/R) and output to Excel spreadsheets for analysis (8,945 data points per sample). Characteristic EEMs from S. aureus, S. epidermidis, E. coli, and Candida tropicalis recovered directly from positive blood cultures are shown in Fig. 4A to D, respectively, along with the location of some known cellular fluorophores (49). Following the scan, the optical microcentrifuge tubes containing purified microbial pellets were stored frozen at −70°C for later assessment of discrepant or unidentified test results. The purity of the test broth sample was also confirmed by subculture. To better understand the stability of intrinsic microbial fluorophores to low-temperature storage conditions, 5-ml samples of positive broth from 370 of the 751 isolates were stored at 2 to 8°C for 4 to 6 h. Broth was warmed prior to testing by placing the tube in a 35 to 37°C water bath for 5 min.
FIG 1 .
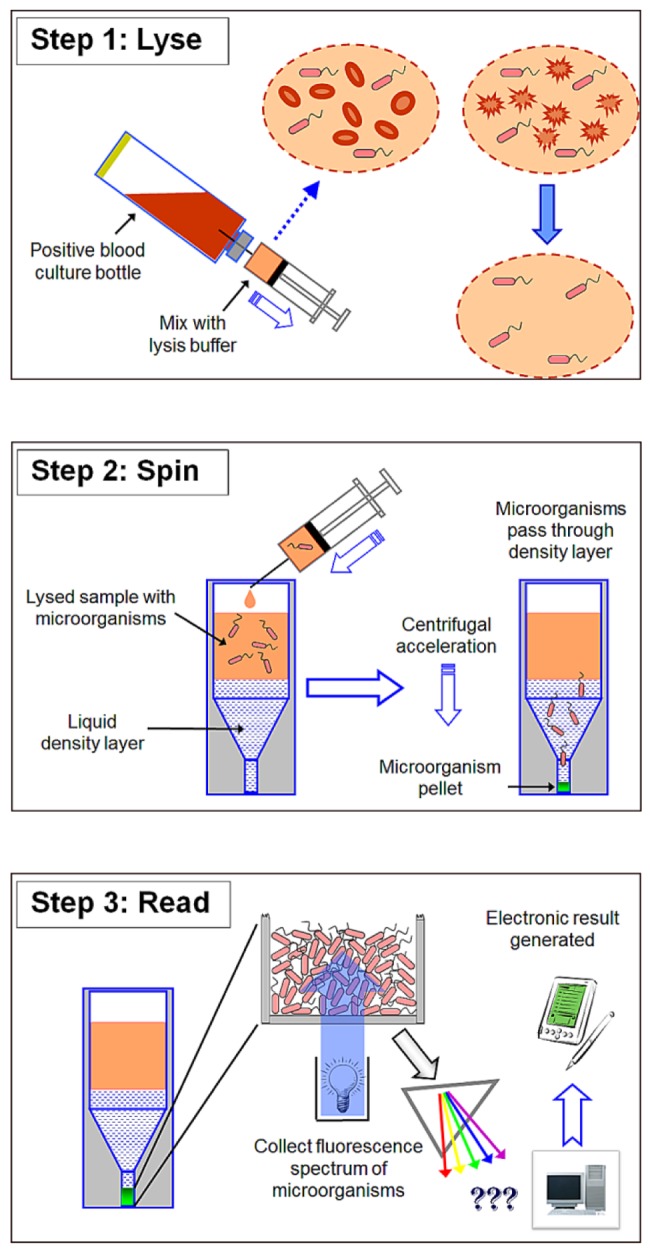
Schematic of the lysis centrifugation-intrinsic fluorescence (LC-IF) method. (Step 1) Mix 2 parts of broth sample with 1 part of lysis buffer and incubate for 1 min. The illustration shows the combining of the two liquids with a syringe. In practice, a separate tube is used so that the mixture can be vortexed. (Step 2) Layer the resultant lysate over a density cushion and centrifuge for 2 min. (Step 3) Place the centrifuge tube in the spectrofluorimeter and measure the intrinsic fluorescence of the packed isolated microorganisms.
FIG 2 .

Optical centrifuge tubes loaded with lysed positive broth. The tube shown on the left was not centrifuged, while the replicate tube on the right was centrifuged for 2 min at 10,000 rpm (10,600 × g). A pellet of sedimented Staphylococcus lugdunensis cells is indicated by the black arrow.
FIG 3 .
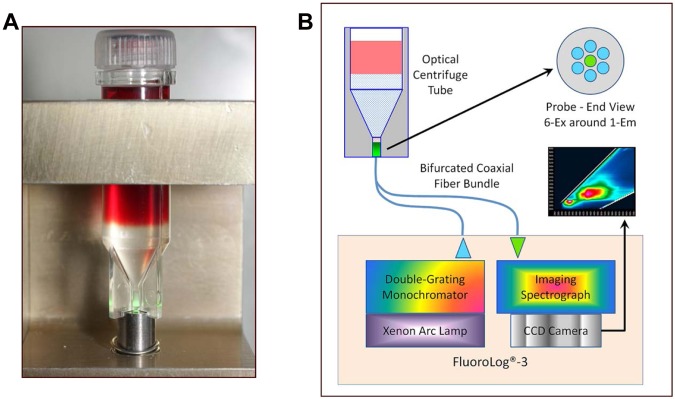
Optical interface for isolated microorganisms. (A) Holder for coupling the centrifuged optical tube to the fiber optic probe. Notches built into the base of the tube center it directly over the bundle of 300-μm-diameter excitation and emission fibers. The microbial pellet is shown here illuminated with a green laser. (B) Schematic of the interface between the optical centrifuge tube and the FluoroLog-3 system.
FIG 4 .

Examples of microbial excitation-emission matrices (EEMs). (A) Isolated cells from a Staphylococcus aureus-containing positive broth sample. The peak positions of the common fluorophores tryptophan and NADH are indicated by the arrows. (B) Isolated cells from a Staphylococcus epidermidis-containing positive broth sample. (C) Isolated cells from an Escherichia coli-containing positive broth sample. The flavin peak is shown by the arrow. (D) Isolated cells from a Candida tropicalis-containing positive broth sample. The positions of two porphyrin peaks are indicated.
Data analysis.
A classification database was built using the EEMs of both fresh and stored positive broth samples, for a total of 1,121 scans. The classification method was based on a mathematical distance calculation. For a given scan from the test set, the distance from the centroid was calculated for each possible classification group. If a distance was within a prespecified percentage of the shortest distance, the test result was considered a low-discrimination agreement between the two classification groups. However, if a second distance was not within this prespecified percentage, the group associated with the shortest distance was called a “one-choice agreement.” If none of the distances were less than a maximum distance, the test was given a “no identification” result. An incorrect result was obtained if a test scan was classified into the wrong group. Data were transformed by first converting fluorescence signals to a natural log and then calculating the first derivative. A limited set of nine excitation wavelengths (285, 300, 315, 330, 345, 400, 415, 430, and 445 nm) were selected for the ability to discriminate between the species represented in the model and were not limited to an evaluation of known fluorophores. Classification was performed using a multilevel algorithm that assigned results to the Gram, family, and then species level. For the Gram level to be considered correct, independent analyses to the Gram and family group levels had to agree. Data with confident Gram classification (1st level) were then further analyzed into family-level groups (2nd level) and finally to the species level (3rd level). All results were cross-validated using a 10-fold venetian blind approach. In this method, 9/10 of the data were used to train the model, and the remaining set of data was used as a test or challenge set. Each combination of groups was analyzed, and the results were pooled to provide an overall estimate of performance.
Instrument QC.
As a daily internal quality control (QC) for the reproducibility of the custom optical separation tube and the fiber optic interface with the spectrofluorimeter, positive broth from a BacT/ALERT SA bottle inoculated with 400 CFU of E. coli ATCC 8739 (no blood) was overlaid directly onto the cesium chloride density cushion and spun, and the EEMs were collected.
ACKNOWLEDGMENTS
We acknowledge Alex Urdanetta for expert technical assistance.
J.D.W. and J.M.H. conceived and developed the method, designed the experiments, and contributed equally to this project. J.D.W. wrote the manuscript, with contributions from J.M.H., M.U., A.v.B., and W.M.D. J.D.W., J.M.H., L.B., A.B., and C.M. conducted the laboratory work. M.U. and E.M. analyzed the data. C.R., J.L., M.W., and B.C. provided engineering support to build the optical centrifuge tubes and associated optical fixtures. R.R., T.T., A.v.B., and W.M.D. provided advice, resources, and financial support for the project.
Footnotes
Citation Walsh JD, Hyman JM, Borzhemskaya L, Bowen A, McKellar C, Ullery M, Mathias E, Ronsick C, Link J, Wilson M, Clay B, Robinson R, Thorpe T, van Belkum A, Dunne WM, Jr. 2013. Rapid intrinsic fluorescence method for direct identification of pathogens in blood cultures. mBio 4(6):e00865-13. doi:10.1128/mBio.00865-13.
REFERENCES
- 1. Vallés J, Rello J, Ochagavía A, Garnacho J, Alcalá M. 2003. Community-acquired bloodstream infection in critically ill adult patients: impact of shock and inappropriate antibiotic therapy on survival. Chest 123:1615–1624 [DOI] [PubMed] [Google Scholar]
- 2. Paul M, Shani V, Muchtar E, Kariv G, Robenshtok E, Leibovici L. 2010. Systematic review and meta-analysis of the efficacy of appropriate empiric antibiotic therapy for sepsis. Antimicrob. Agents Chemother. 54:4851–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar A, Roberts D, Wood KE, Light B, Parillo J, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Kumar A, Cheang M. 2006. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 34:1589–1596 [DOI] [PubMed] [Google Scholar]
- 4. Munson EL, Diekema DJ, Beekmann SE, Chapin KC, Doern GV. 2003. Detection and treatment of bloodstream infection: laboratory reporting and antimicrobial management. J. Clin. Microbiol. 41:495–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barenfanger J, Graham DR, Kolluri L, Sangwan G, Lawhorn J, Drake CA, Verhulst SJ, Peterson R, Moja LB, Ertmoed MM, Moja AB, Shevlin DW, Vautrain R, Callahan CD. 2008. Decreased mortality associated with prompt Gram staining of blood cultures. Am. J. Clin. Pathol. 130:870–876 [DOI] [PubMed] [Google Scholar]
- 6. Dellit TH, Owens RC, McGowan JE, Gerding DN, Weinstein RA, Burke JP, Huskins WC, Paterson DL, Fishman NO, Carpenter CF, Brennan PJ, Billeter M, Hooton TM, Infectious Diseases Society of America. Society for Healthcare Epidemiology of America 2007. Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America guidelines for developing an institutional program to enhance antimicrobial stewardship. Clin. Infect. Dis. 44:159–177 [DOI] [PubMed] [Google Scholar]
- 7. Pakyz AL, Oinonen M, Polk RE. 2009. Relationship of carbapenem restriction in 22 university teaching hospitals to carbapenem use and carbapenem-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53:1983–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Willmann M, Marschal M, Hölzl F, Schröppel K, Autenrieth IB, Peter S. 2013. Time series analysis as a tool to predict the impact of antimicrobial restriction in antibiotic stewardship programs using the example of multidrug-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57:1797–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clerc O, Prod’hom G, Vogne C, Bizzini A, Calandra T, Greub G. 2013. Impact of matrix-assisted laser desorption ionization time-of-flight mass spectrometry on the clinical management of patients with gram-negative bacteremia: a prospective observational study. Clin. Infect. Dis. 56:1101–1107 [DOI] [PubMed] [Google Scholar]
- 10. Doern GV, Vautour R, Gaudet M, Levy B. 1994. Clinical impact of rapid in vitro susceptibility testing and bacterial identification. J. Clin. Microbiol. 32:1757–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cunney RJ, McNamara EB, Alansari N, Loo B, Smyth EG. 1997. The impact of blood culture reporting and clinical liaison on the empiric treatment of bacteraemia. J. Clin. Pathol. 50:1010–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barenfanger J, Drake C, Kacich G. 1999. Clinical and financial benefits of rapid bacterial identification and antimicrobial susceptibility testing. J. Clin. Microbiol. 37:1415–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beekmann SE, Diekema DJ, Chapin KC, Doern GV. 2003. Effects of rapid detection of bloodstream infections on length of hospitalization and hospital charges. J. Clin. Microbiol. 41:3119–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kerremans JJ, Verboom P, Stijnen T, Hakkaart-van Roijen L, Goessens W, Verbrugh HA, Vos MC. 2008. Rapid identification and antimicrobial susceptibility testing reduce antibiotic use and accelerate pathogen-directed antibiotic use. J. Antimicrob. Chemother. 61:428–435 [DOI] [PubMed] [Google Scholar]
- 15. Moore DF, Hamada SS, Marso E, Martin WJ. 1981. Rapid identification and antimicrobial susceptibility testing of gram-negative bacilli from blood cultures by the AutoMicrobic system. J. Clin. Microbiol. 13:934–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kerremans JJ, Goessens WH, Verbrugh HA, Vos MC. 2004. Accuracy of identification and susceptibility results by direct inoculation of Vitek 2 cards from positive BACTEC cultures. Eur. J. Clin. Microbiol. Infect. Dis. 23:892–898 [DOI] [PubMed] [Google Scholar]
- 17. Bruins MJ, Bloembergen P, Ruijs GJ, Wolfhagen MJ. 2004. Identification and susceptibility testing of Enterobacteriaceae and Pseudomonas aeruginosa by direct inoculation from positive BACTEC blood culture bottles into Vitek 2. J. Clin. Microbiol. 42:7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oliveira K, Procop GW, Wilson D, Coull J, Stender H. 2002. Rapid identification of Staphylococcus aureus directly from blood cultures by fluorescence in situ hybridization with peptide nucleic acid probes. J. Clin. Microbiol. 40:247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forrest GN, Mankes K, Jabra-Rizk MA, Weekes E, Johnson JK, Lincalis DP, Venezia RA. 2006. Peptide nucleic acid fluorescence in situ hybridization-based identification of Candida albicans and its impact on mortality and antifungal therapy costs. J. Clin. Microbiol. 44:3381–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deck MK, Anderson ES, Buckner RJ, Colasante G, Coull JM, Crystal B, Della Latta P, Fuchs M, Fuller D, Harris W, Hazen K, Klimas LL, Lindao D, Meltzer MC, Morgan M, Shepard J, Stevens S, Wu F, Fiandaca MJ. 2012. Multicenter evaluation of the Staphylococcus QuickFISH method for simultaneous identification of Staphylococcus aureus and coagulase-negative staphylococci directly from blood culture bottles in less than 30 minutes. J. Clin. Microbiol. 50:1994–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gebert S, Siegel D, Wellinghausen N. 2008. Rapid detection of pathogens in blood culture bottles by real-time PCR in conjunction with the pre-analytic tool MolYsis. J. Infect. 57:307–316 [DOI] [PubMed] [Google Scholar]
- 22. Blaschke AJ, Heyrend C, Byington CL, Fisher MA, Barker E, Garrone NF, Thatcher SA, Pavia AT, Barney T, Alger GD, Daly JA, Ririe KM, Ota I, Poritz MA. 2012. Rapid identification of pathogens from positive blood cultures by multiplex polymerase chain reaction using the FilmArray system. Diagn. Microbiol. Infect. Dis. 74:349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wojewoda CM, Sercia L, Navas M, Tuohy M, Wilson D, Hall GS, Procop GW, Richter SS. 2013. Evaluation of the Verigene Gram-positive blood culture nucleic acid test for rapid detection of bacteria and resistance determinants. J. Clin. Microbiol. 51:2072–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Belkum A, Welker M, Erhard M, Chatellier S. 2012. Biomedical mass spectrometry in today’s and tomorrow’s clinical microbiology laboratories. J. Clin. Microbiol. 50:1513–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. La Scola B, Raoult D. 2009. Direct identification of bacteria in positive blood culture bottles by matrix-assisted laser desorption ionisation time-of-flight mass spectrometry. PLoS One 4:e8041. 10.1371/journal.pone.0008041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stevenson LG, Drake SK, Murray PR. 2010. Rapid identification of bacteria in positive blood culture broths by matrix-assisted laser desorption ionization–time of flight mass spectrometry. J. Clin. Microbiol. 48:444–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferroni A, Suarez S, Beretti JL, Dauphin B, Bille E, Meyer J, Bougnoux ME, Alanio A, Berche P, Nassif X. 2010. Real-time identification of bacteria and Candida species in positive blood culture broths by matrix-assisted laser desorption ionization–time of flight mass spectrometry. J. Clin. Microbiol. 48:1542–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marinach-Patrice C, Fekkar A, Atanasova R, Gomes J, Djamdjian L, Brossas JY, Meyer I, Buffet P, Snounou G, Datry A, Hennequin C, Golmard JL, Mazier D. 2010. Rapid species diagnosis for invasive candidiasis using mass spectrometry. PLoS One 5:e8862. 10.1371/journal.pone.0008862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prod’hom G, Bizzini A, Durussel C, Bille J, Greub G. 2010. Matrix-assisted laser desorption ionization–time of flight mass spectrometry for direct bacterial identification from positive blood culture pellets. J. Clin. Microbiol. 48:1481–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kok J, Thomas LC, Olma T, Chen SC, Iredell JR. 2011. Identification of bacteria in blood culture broths using matrix-assisted laser desorption-ionization Sepsityper and time of flight mass spectrometry. PLoS One 6:e23285. 10.1371/journal.pone.0023285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vlek AL, Bonten MJ, Boel CH. 2012. Direct matrix-assisted laser desorption ionization time-of-flight mass spectrometry improves appropriateness of antibiotic treatment of bacteremia. PLoS One 7:e32589. 10.1371/journal.pone.0032589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lagacé-Wiens PR, Adam HJ, Karlowsky JA, Nichol KA, Pang PF, Guenther J, Webb AA, Miller C, Alfa MJ. 2012. Identification of blood culture isolates directly from positive blood cultures by use of matrix-assisted laser desorption ionization–time of flight mass spectrometry and a commercial extraction system: analysis of performance, cost, and turnaround time. J. Clin. Microbiol. 50:3324–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Loonen AJ, Jansz AR, Stalpers J, Wolffs PF, van den Brule AJ. 2012. An evaluation of three processing methods and the effect of reduced culture times for faster direct identification of pathogens from BacT/ALERT blood cultures by MALDI-TOF MS. Eur. J. Clin. Microbiol. Infect. Dis. 31:1575–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferreira L, Sánchez-Juanes F, Porras-Guerra I, García-García MI, García-Sánchez JE, González-Buitrago JM, Muñoz-Bellido JL. 2011. Microorganisms direct identification from blood culture by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Microbiol. Infect. 17:546–551 [DOI] [PubMed] [Google Scholar]
- 35. Maquelin K, Kirschner C, Choo-Smith LP, Ngo-Thi NA, van Vreeswijk T, Stämmler M, Endtz HP, Bruining HA, Naumann D, Puppels GJ. 2003. Prospective study of the performance of vibrational spectroscopies for rapid identification of bacterial and fungal pathogens recovered from blood cultures. J. Clin. Microbiol. 41:324–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuster KC, Reese I, Urlaub E, Gapes JR, Lendl B. 2000. Multidimensional information on the chemical composition of single bacterial cells by confocal Raman microspectroscopy. Anal. Chem. 72:5529–5534 [DOI] [PubMed] [Google Scholar]
- 37. Xie C, Yong-Qing L. 2003. Confocal micro-Raman spectroscopy of single biological cells using optical trapping and shifted excitation difference techniques. J. Appl. Physiol. 93:2982–2986 [Google Scholar]
- 38. Rösch P, Harz M, Schmitt M, Peschke KD, Ronneberger O, Burkhardt H, Motzkus HW, Lankers M, Hofer S, Thiele H, Popp J. 2005. Chemotaxonomic identification of single bacteria by micro-Raman spectroscopy: application to clean-room-relevant biological contaminations. Appl. Environ. Microbiol. 71:1626–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dalterio RA, Nelson WH, Britt D, Sperry JF, Tanguay JF, Suib SL. 1987. The steady-state and decay characteristics of primary fluorescence from live bacteria. Appl. Spectrosc. 41:234–241 [Google Scholar]
- 40. Sorrell MJ, Tribble J, Reinisch L, Werkhaven JA, Ossoff RH. 1993. Bacteria identification of otitis media with fIuorescence spectroscopy. Lasers Surg. Med. 14:155–163 [DOI] [PubMed] [Google Scholar]
- 41. Leblanc L, Dufour E. 2002. Monitoring the identity of bacteria using their intrinsic fluorescence. FEMS Microbiol. Lett. 211:147–153 [DOI] [PubMed] [Google Scholar]
- 42. Giana HE, Silveira L, Jr, Zangaro RA, Pacheco MTT. 2003. Rapid identification of bacterial species by fluorescence spectroscopy and classification through principal components analysis. J. Fluoresc. 13:489–493 [Google Scholar]
- 43. Bhatta H, Goldys EM, Learmonth RP. 2006. Use of fluorescence spectroscopy to differentiate yeast and bacterial cells. Appl. Microbiol. Biotechnol. 71:121–126 [DOI] [PubMed] [Google Scholar]
- 44. Ammor MS. 2007. Recent advances in the use of intrinsic fluorescence for bacterial identification and characterization. J. Fluoresc. 17:455–459 [DOI] [PubMed] [Google Scholar]
- 45. Sohn M, Himmelsbach DS, Barton FE, Fedorka-Cray PJ. 2009. Fluorescence spectroscopy for rapid detection and classification of bacterial pathogens. Appl. Spectrosc. 63:1251–1255 [DOI] [PubMed] [Google Scholar]
- 46. Hyman JM, Walsh JD, Thorpe T. February 2011. US patent 2011/0033847 A1
- 47. Tourkya B, Karoui R, Berdague JL, Boubellouta T, Dufour E, Leriche F. 2011. Optical fiber-based synchronous fluorescence spectroscopy for bacterial discrimination directly from colonies on agar plates. Anal. Methods 3:133–143 [DOI] [PubMed] [Google Scholar]
- 48. Biedenbach DJ, Moet GJ, Jones RN. 2004. Occurrence and antimicrobial resistance pattern comparisons among bloodstream infection isolates from the SENTRY Antimicrobial Surveillance Program (1997–2002). Diagn. Microbiol. Infect. Dis. 50:59–69 [DOI] [PubMed] [Google Scholar]
- 49. Monici M. 2006. Cell and tissue autofluorescence research and diagnostic applications. Biotechnol. Annu. Rev. 11:227–256 [DOI] [PubMed] [Google Scholar]
- 50. Yuan S, Astion ML, Schapiro J, Limaye AP. 2005. Clinical impact associated with corrected results in clinical microbiology testing. J. Clin. Microbiol. 43:2188–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Søgaard M, Nørgaard M, Schønheyder HC. 2007. First notification of positive blood cultures and the high accuracy of the Gram stain report. J. Clin. Microbiol. 45:1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Munson E, Block T, Basile J, Hryciuk JE, Schell RF. 2007. Mechanisms to assess Gram-stain interpretation proficiency of technologists at satellite laboratories. J. Clin. Microbiol. 45:3754–3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. de Cueto M, Ceballos E, Martinez-Martinez L, Perea EJ, Pascual A. 2004. Use of positive blood cultures for direct identification and susceptibility testing with the Vitek 2 system. J. Clin. Microbiol. 42:3734–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baldwin WW, Sheu MJ, Bankston PW, Woldringh CL. 1988. Changes in buoyant density and cell size of Escherichia coli in response to osmotic shocks. J. Bacteriol. 170:452–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shahzad A, Köhler G, Knapp M, Gaubitzer E, Puchinger M, Edetsberger M. 2009. Emerging applications of fluorescence spectroscopy in medical microbiology field. J. Transl. Med. 7:99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brahma SK, Baek MP, Gaskill D, Force RK, Nelson WH, Sperry J. 1985. The rapid identification of bacteria using time-resolved fluorescence and fluorescence excitation spectral methods. Appl. Spectrosc. 39:869–872 [Google Scholar]
- 57. Mason HY, Lloyd C, Dice M, Sinclair R, Ellis W, Jr, Powers L. 2003. Taxonomic identification of microorganisms by capture and intrinsic fluorescence detection. Biosens. Bioelectron. 18:521–527 [DOI] [PubMed] [Google Scholar]
- 58. Lakowicz JR. 1999, Principles of fluorescence spectroscopy, 2nd ed. Kluwer Publishers Academic/Plenum Publishers, New York, NY. [Google Scholar]
- 59. Salmon JM, Kohen E, Viallet P, Hirschberg JP, Wouters AW, Kohen C, Thorell B. 1982. Microspectrofluorometric approach to the study of free/bound NAD(P)H ratio as metabolic indicator in various cell types. Photochem. Photobiol. 36:585–593 [DOI] [PubMed] [Google Scholar]
- 60. Stryer L. 1988. Biochemistry, 34th ed. W. H. Freeman, New York, NY. [Google Scholar]
- 61. Chance B, Schoener B, Oshino R, Itshak F, Nakase Y. 1979. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples: NADH and flavoprotein fluorescence signals. J. Biol. Chem. 254:4764–4771 [PubMed] [Google Scholar]
- 62. Amat A, Rigau J, Waynant RW, Ilev IK, Tomas J, Anders JJ. 2005. Modification of the intrinsic fluorescence and the biochemical behavior of ATP after irradiation with visible and near-infrared laser light. J. Photochem. Photobiol. B 81:26–32 [DOI] [PubMed] [Google Scholar]