

ABSTRACT
Nitrogen is one of the major nutrients limiting microbial productivity in the ocean, and as a result, most marine microorganisms have evolved systems for responding to nitrogen stress. The highly abundant alphaproteobacterium “Candidatus Pelagibacter ubique,” a cultured member of the order Pelagibacterales (SAR11), lacks the canonical GlnB, GlnD, GlnK, and NtrB/NtrC genes for regulating nitrogen assimilation, raising questions about how these organisms respond to nitrogen limitation. A survey of 266 Alphaproteobacteria genomes found these five regulatory genes nearly universally conserved, absent only in intracellular parasites and members of the order Pelagibacterales, including “Ca. Pelagibacter ubique.” Global differences in mRNA and protein expression between nitrogen-limited and nitrogen-replete cultures were measured to identify nitrogen stress responses in “Ca. Pelagibacter ubique” strain HTCC1062. Transporters for ammonium (AmtB), taurine (TauA), amino acids (YhdW), and opines (OccT) were all elevated in nitrogen-limited cells, indicating that they devote increased resources to the assimilation of nitrogenous organic compounds. Enzymes for assimilating amine into glutamine (GlnA), glutamate (GltBD), and glycine (AspC) were similarly upregulated. Differential regulation of the transcriptional regulator NtrX in the two-component signaling system NtrY/NtrX was also observed, implicating it in control of the nitrogen starvation response. Comparisons of the transcriptome and proteome supported previous observations of uncoupling between transcription and translation in nutrient-deprived “Ca. Pelagibacter ubique” cells. Overall, these data reveal a streamlined, PII-independent response to nitrogen stress in “Ca. Pelagibacter ubique,” and likely other Pelagibacterales, and show that they respond to nitrogen stress by allocating more resources to the assimilation of nitrogen-rich organic compounds.
IMPORTANCE
Pelagibacterales are extraordinarily abundant and play a pivotal role in marine geochemical cycles, as one of the major recyclers of labile dissolved organic matter. They are also models for understanding how streamlining selection can reshape chemoheterotroph metabolism. Streamlining and its broad importance to environmental microbiology are emerging slowly from studies that reveal the complete genomes of uncultured organisms. Here, we report another remarkable example of streamlined metabolism in Pelagibacterales, this time in systems that control nitrogen assimilation. Pelagibacterales are major contributors to metatranscriptomes and metaproteomes from ocean systems, where patterns of gene expression are used to gain insight into ocean conditions and geochemical cycles. The data presented here supply background that is essential to interpreting data from field studies.
INTRODUCTION
Identifying nutrients that limit microbial productivity in the oceans has been one of the key missions of biological oceanographers for over half a century. Using a combination of nutrient fertilization and direct measurement of dissolved nutrient concentrations, studies have alternately found nitrogen (1–10), phosphate (11–13), iron (14–17), or silica (18–21) to limit the productivity in seawater. Meta-analyses that coalesced experimental results across hundreds of studies found that anthropogenic contamination, geographic features, and time scales influenced the limiting nutrient, with nitrogen more often limiting in pelagic marine environments, polluted coastal waters, and short-term steady-state systems (19, 22–24). Recently, individual studies and meta-analyses have found more than one nutrient—often nitrogen and phosphate—to be colimiting (22, 25–27), due to shifts in overall N:P stoichiometry of bacterial communities according to nutrient availability (28, 29).
Biosynthesis of nitrogenous compounds such as DNA, RNA, and proteins is dependent on maintaining intracellular pools of glutamine and glutamate. In nearly all bacteria, these two compounds are synthesized by glutamine synthetase (GS) and glutamate synthetase (glutamine-2-oxoglutarate-amidotransferase [GOGAT]). These two enzymes work in concert to first condense ammonia and glutamate via GS to form glutamine, followed by the GOGAT-mediated transfer of an amine group from glutamine onto 2-oxoglutarate to yield two molecules of glutamate (30–33). The activity of these enzymes in many Alphaproteobacteria is regulated by the PII protein GlnB, which is alternatively uridylylated/deuridylylated by GlnD based on the 2-oxoglutarate/glutamine ratio within the cell (30–33). The two-component signaling system NtrB/NtrC transduces the uridylylation state of GlnB into transcriptional inhibition/activation of GS and other nitrogen assimilation genes (34). Unuridylylated GlnB also stimulates adenylylation of GS, thereby inhibiting GS activity when glutamine is sufficient (35, 36). Uridylylated GlnB activates adenylremovase activity to restore activity of GlnB. A second PII protein, GlnK, is commonly cotranscribed with the ammonium transporter amtB and posttranslationally reversibly inhibits AmtB’s transport activity within seconds of micromolar changes in ammonium levels (31, 37–40). Altogether, this posttranslational signaling cascade is believed to enable the cell to quickly inhibit ammonia uptake and glutamine synthesis when exposed to pulses of high concentrations of ammonia (41–43), thereby preventing toxic buildup of intracellular ammonia and depletion of the tricarboxylic acid (TCA) cycle intermediate 2-oxoglutarate. One of the few studies of PII transcription and translation showed a 50-fold to 100-fold increase in mRNA abundance of five PII genes and a 72-fold to 115-fold increase in the abundance of two PII protein products in response to nitrogen limitation in the nitrogen-fixing bacterium Dehalococcoides ethenogenes (44). A thorough review and biochemical diagram of this pathway have been compiled by Arcondéguy et al. (30).
When the first representative species of the SAR11 clade was sequenced in 2005, only two genes for regulating the assimilation of nitrogen were identified: ntrY and ntrX (45). Although the sequence similarity between ntrY/X and ntrB/C may suggest a shared evolutionary pathway for these two-component signaling systems, structural and functional studies indicate that NtrY/X cannot substitute for NtrB/C in cellular regulatory pathways. Unlike NtrB/C, which responds to fluctuations in intracellular glutamine, research on NtrY/X suggests that this two-component system is involved in sensing the concentration of extracellular nitrate (46) and has been postulated to connect nitrogen control to the redox state of the cell through interactions with the RegB/RegA two-component system (47).
The elemental composition of microorganisms is shaped in large part by nutrient availability (48–54). A survey of metagenomic sequences and the genomes of marine bacteria, including SAR11, concluded that competition for nitrogen in the marine environment has selected for genomes high in AT and proteomes low in nitrogenous amino acids (10). Additionally, microorganisms with the lowest mass in this environment have a distinct advantage—not only because their absolute nutrient requirements for growth and division are minimal, but also because the increased surface-to-volume ratio of small cells favors nutrient acquisition (10, 55). Nitrogen and phosphate limitation on evolutionary time scales is hypothesized to drive genome streamlining, in which loss of nonessential DNA results in an increase in fitness due to decreased costs of genome replication (45, 56, 57). Oligotrophic marine environments are numerically dominated by members of the Prochlorococcus genus and SAR11 that are well adapted to surviving in nitrogen-limited environments through the abovementioned adaptations (10, 45, 58). A cultured representative of the SAR11 clade (proposed order Pelagibacterales [59]), “Candidatus Pelagibacter ubique” HTCC1062, was found to have one of the smallest genomes (1.3 Mbp), lowest GC contents (33%), and smallest cell sizes (0.025 to 0.045 µm3) among free-living heterotrophs (45, 60). Assuming that these characteristics are the result of extreme selective pressure to optimize nitrogen utilization, we are led to question if the regulation, acquisition, or assimilation of nitrogen has also been radically remodeled in this bacterium.
The objective of this study was to understand how “Ca. Pelagibacter ubique” and other SAR11 bacteria respond to nitrogen limitation. Previous work had shown that many SAR11 metabolic systems have been reduced in complexity by streamlining selection, resulting in noncanonical pathways with fewer enzymatic reactions. Genome streamlining, essentially reduced instruction sets for cells, in the past have been linked to tradeoffs that make SAR11 more sensitive to environmental variation and explain why these bacteria are challenging to culture. Our experimental strategy was to measure both the transcriptome and the proteome because, in a previous study of iron limitation in this bacterium, we observed that the responses of the proteome and transcriptome are largely uncoupled, an apparent consequence of an integrated regulatory response that includes both transcriptional and posttranscriptional mechanisms of regulation (61). The data that we present here demonstrate that “Ca. Pelagibacter ubique” has a robust response to nitrogen limitation that is independent of the PII regulatory systems. Faced with nitrogen limitation, these bacteria devote increased resources to assimilating amine groups from organic compounds. The streamlined regulatory response of “Ca. Pelagibacter ubique” to nitrogen limitation is simpler than any described previously in the Alphaproteobacteria. The analysis that we present also raises questions about the role of riboswitches and other posttranscriptional control mechanisms in the integrated metabolic response of these cells to changes in their environment.
RESULTS AND DISCUSSION
Nitrogen requirement.
As reported previously, the genome of “Ca. Pelagibacter ubique” HTCC1062 encodes transporters and enzymes for assimilating nitrogen from ammonium and nitrogenous organic compounds but lacks pathways for reducing and assimilating nitrite, nitrate, and dinitrogen (45, 62). Achieving nitrogen limitation in culture was complicated by the fact that “Ca. Pelagibacter ubique” cells are conditional auxotrophs for glycine or serine and normally do not grow well in the absence of these compounds or their precursors, such as glycine betaine (63, 64). The requirement for glycine or glycine precursors presented an experimental design problem because “Ca. Pelagibacter ubique” contains genes encoding the glycine cleavage complex (gcvTHP), which degrades glycine to ammonium, potentially supplying a source of ammonium from glycine. Working with an artificial seawater (ASW) medium in which ammonium (NH4) was the sole nitrogen source, we found that “Ca. Pelagibacter ubique” cultures achieved relatively high growth rates when provided with high levels of oxaloacetate (500 µM) (Fig. 1; see Table S1 in the supplemental material). Titration of ammonium into “Ca. Pelagibacter ubique” cultures growing on this medium revealed an apparent requirement of 338 attomoles of N per cell (Fig. 2). Although the pathway of glycine biosynthesis under these conditions is uncertain, we propose that, when provided at high concentrations, oxaloacetate is converted to glyoxylate via the activity of isocitrate lyase (AceA), followed by transamination to glycine by AspC (Fig. 3). Previously, based on its position in the genome adjacent to genes encoding glycolate oxidase (glcDEF), it was proposed that the gene annotated as aspartate aminotransferase (aspC, or glutamic-oxaloacetic transaminase) is instead an aminotransferase that produces glycine from glyoxylate (64).
FIG 1 .
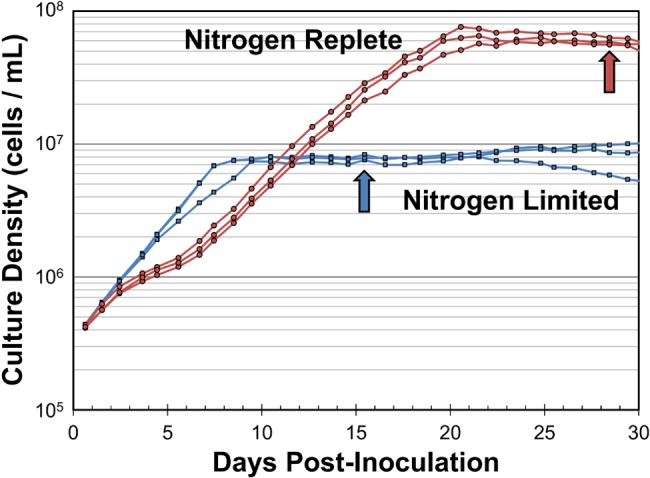
Growth curves of nitrogen-replete (red) and nitrogen-limited (blue) batch cultures of “Ca. Pelagibacter ubique.” Artificial seawater medium contained 500 µM oxaloacetate, 10 µM DMSP, 500 µM pyruvate, and trace nutrients (see Table S1 in the supplemental material). Arrows indicate time points where samples for proteomic and microarray analysis were collected.
FIG 2 .
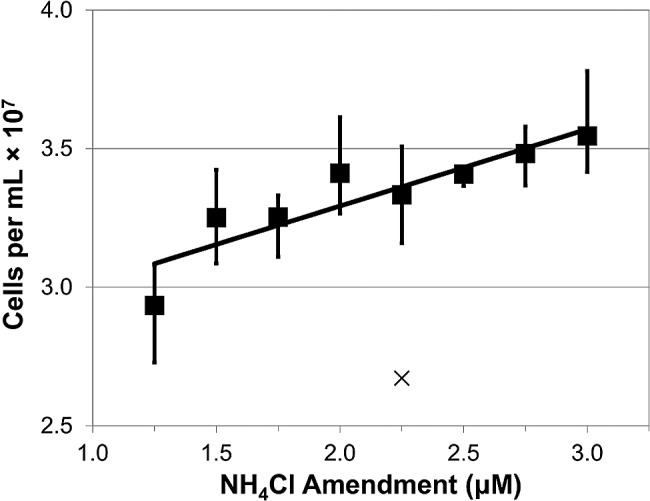
The maximum cell density achieved for batch cultures grown in the dark at 16°C is plotted against addition of NH4Cl in the presence of excess nutrients (average, n = 3 for each data point; R2 = 0.80; error bars show complete range excluding one outlier shown with ×). The culturing medium is described fully in Table S1 in the supplemental material. The trend line is given by the equation y = 0.28x + 2.7, indicating a requirement of 338 attomoles of nitrogen per cell.
FIG 3 .
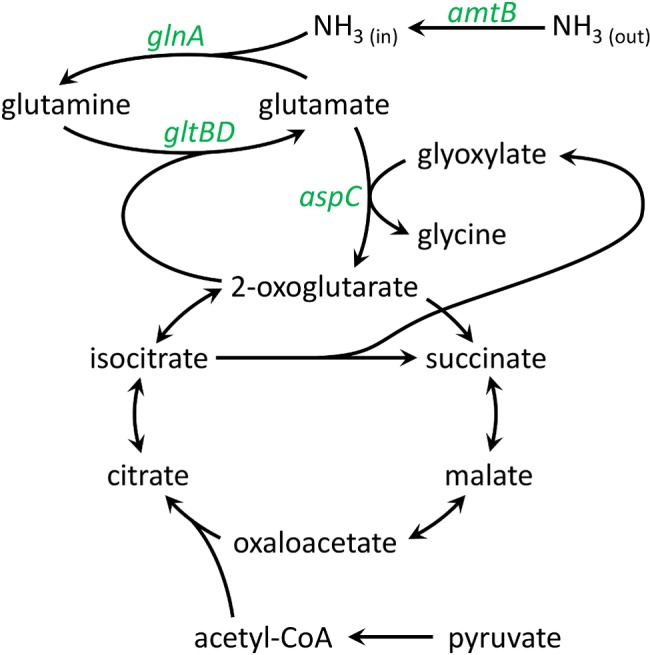
Proposed pathway for the assimilation of NH3. Labeled genes were detected at higher protein abundances under nitrogen-limiting stationary phase, compared to nitrogen-replete stationary phase (quantified in Table 1).
Absence of the PII regulatory network.
The canonical PII nitrogen assimilation regulatory network present in all other free-living Alphaproteobacteria that we examined is absent from “Ca. Pelagibacter ubique” (Fig. 4). The absence of glnB, glnD, glnK, ntrB, and ntrC is not unique to strain HTCC1062; examination of eight available genomes from the SAR11 clade (HTCC1062, HTCC1002, HTCC9565, HTCC7211, HIMB5, HIMB114, IMCC9063, and HIMB59) suggests that the absence of these five genes is a common characteristic of subclade Ia organisms, although glnB, glnD, and glnK are present in the highly divergent subclade Va organism HIMB59 (59, 65) and the subclade IIIa organism IMCC9063 has a putative homolog of glnK. The distribution of these genes across the Alphaproteobacteria confirmed that only a subset of obligate and facultative intracellular organisms were similarly deficient in PII regulatory genes (Fig. 4).
FIG 4 .

Distribution of nitrogen assimilation genes in the Alphaproteobacteria. Maximum likelihood tree constructed with concatenated 16S-23S rRNA genes. Most members of the SAR11 clade (highlighted in red) and some Alphaproteobacteria that are intracellular parasites (blue) are missing the PII genes glnB and glnK. Monophyletic groups in which all members had the same pattern of genes present and absent were collapsed, except for SAR11. Bootstrap values (n = 1,000) are indicated at the nodes. The scale bar indicates 0.1 change per position. “+” indicates at least one homolog present in the genome, while “−” indicates that no homolog was identified.
The missing PII genes glnB, glnD, glnK, ntrB, and ntrC are often observed in other bacteria within widely conserved gene neighborhoods: glnB-glnA-ntrB-ntrC, map-glnD-dapA, and glnK-amtB (33). We therefore examined the gene neighborhoods of the SAR11 genes glnA, map, dapA, and amtB in each of the SAR11 genomes and in metagenomic data as a complementary method to confirm the absence of the PII genes from the alphaproteobacterial subgroup containing “Ca. Pelagibacter ubique.” Manual inspection of whole-genome alignments constructed with progressiveMauve (66) revealed that glnB, glnD, glnK, ntrB, and ntrC were indeed absent from the canonical loci (above) in most strains, with a few exceptions: HIMB59, which possessed glnB, glnD, and glnK, exhibited the typical glnB-glnA and glnK-amtB arrangements. However, the predicted HIMB59 glnD was not carried adjacent to map or dapA, and the putative glnK in IMCC9063 was not near amtB.
To assess the abundance of the PII genes glnB, glnD, glnK, ntrB, and ntrC in natural populations of SAR11, the global ocean sampling (GOS) environmental data set (67) was queried for three known PII operons (33), glnB-glnA-ntrB-ntrC, map-glnD-dapA, and glnK-amtB, using SAR11 glnA, map, dapA, or amtB as a query sequence to find fragments in which the greatest similarity was to a SAR11 strain. Among 287 GOS sequences retrieved that contained amtB and at least one additional gene, only three contained the glnK-amtB pair (3/287). Queries with glnA, map, and dapA found no adjacent PII genes, yielding 0/137, 0/15, and 0/74, respectively, for multigene fragments. Thus, metagenomic data supported the conclusion that the PII regulatory network is widely absent from the SAR11 clade. These findings suggested that “Ca. Pelagibacter ubique” might use an alternative mechanism to modulate the expression and activity of nitrogen assimilation genes—possibly one which does not rely on the relative concentrations of glutamine and 2-oxoglutarate.
Response to nitrogen limitation.
“Ca. Pelagibacter ubique” responded to nitrogen limitation by increasing the expression of proteins responsible for transporting nitrogenous molecules into the cell and for transferring amine groups to 2-oxoglutarate or glutamate to form glutamate or glutamine, respectively (Table 1; Fig. 3). The two most highly upregulated genes—an ammonium transporter and a sarcosine oxidase—appear to be expressed only in nitrogen-limited cells. A diverse collection of organic molecule transporters, as well as aminotransferases, was also observed at elevated abundances in nitrogen-limited cells, suggesting that “Ca. Pelagibacter ubique” may be able to utilize a broad range of nitrogenous compounds to satisfy its nitrogen requirement. This observation is in agreement with previous genome investigations (45), in vivo protein expression assays (68), and autoradiography measurements of substrate uptake (69), which indicated a heavy reliance on broad-specificity ABC transporters to acquire amino acids, glycine betaine, ammonia, urea, spermidine, putrescine, and other nitrogenous compounds. Interestingly, many proteins predicted to be involved in nitrogen metabolism, including glutamine synthetase, glutamate synthase, and three of the four putative ammonium transporters encoded in the “Ca. Pelagibacter ubique” genome, were either downregulated or unchanged by nitrogen limitation (Table 2). We speculate that these proteins either are constitutively expressed or are modulated in response to cell growth rate, rather than nitrogen availability.
TABLE 1 .
Transcripts and proteins that were more abundant during nitrogen-limited stationary phasec
| Locus ID | Gene | Description | Fold change |
|
|---|---|---|---|---|
| mRNA | Protein | |||
| SAR11_0818 | amtB | Ammonium transporter | 2.72 | +INF |
| SAR11_1304 | sox | Monomeric sarcosine oxidase | 2.31 | +INF |
| SAR11_0747 | glnA | Glutamine synthetase | 1.91 | 5.16b |
| SAR11_0460 | acuC | Histone deacetylase-like | 1.08 | 2.65a |
| SAR11_0953 | yhdW | Amino acid transporter: periplasmic | 1.19 | 2.35b |
| SAR11_0279 | aspC | Aspartate transaminase | 1.42 | 2.04b |
| SAR11_0433 | gltD | Glutamate synthase, small subunit | 0.44 | 2.03b |
| SAR11_0032 | ftsZ | Cell division protein | 1.48 | 1.84b |
| SAR11_1365 | msrB | Peptide methionine sulfoxide reductase | 1.15 | 1.77b |
| SAR11_0434 | gltB | Glutamate synthase, large subunit | 0.90 | 1.74b |
| SAR11_1210 | occT | Octopine/nopaline transporter: periplasmic | 1.35 | 1.61b |
| SAR11_0061 | pilP | Type 4 fimbrial biogenesis protein | 1.23 | 1.58b |
| SAR11_1166 | Protein of unknown function | 1.12 | 1.57b | |
| SAR11_0359 | ompU | Outer membrane porin | 0.67 | 1.55b |
| SAR11_0733 | prfB | Bacterial peptide chain release factor 2 (RF-2) | 1.04 | 1.55a |
| SAR11_0321 | phhB | Pterin-4-alpha-carbinolamine dehydratase | 0.91 | 1.52a |
| SAR11_0807 | tauA | Sulfonate/nitrate/taurine transporter: periplasmic | 1.30 | 1.52b |
| SAR11_0134 | cox1 | Cytochrome c oxidase | 1.21 | 1.51a |
| SAR11_0183 | Xanthine/uracil/vitamin C permease | 1.59 | 1.45 | |
| SAR11_0162 | groEL | Chaperonin GroEL | 2.55 | 1.42b |
| SAR11_1285 | fhs | Formate-tetrahydrofolate ligase | 1.80 | 1.20b |
| SAR11_0267 | msmX | Multiple sugar transporter: ATP binding | 1.54 | 1.19b |
| SAR11_0008 | hflK | Integral membrane proteinase | 2.36 | 1.13a |
| SAR11_0334 | hslU | ATP-dependent protease | 1.53 | 1.08a |
| SAR11_0077 | trxB | Thioredoxin reductase | 3.00 | 0.83b |
| SAR11_0142 | Pirin; protein of unknown function | 1.69 | 0.82a | |
| SAR11_0304 | Short-chain dehydrogenase | 1.88 | 0.79a | |
| SAR11_0750 | mmuM | Homocysteine S-methyltransferase | 1.62 | 0.76b |
| SAR11_0504 | cycM | Cytochrome c | 2.30 | 0.74a |
| SAR11_0680 | fdsB | Formate dehydrogenase | 1.58 | 0.64b |
| SAR11_1143 | grlA | Glutaredoxin | 1.82 | 0.61b |
| SAR11_0171 | Rhodanese-related sulfur transferase | 1.62 | 0.57b | |
q value of ≤0.05. No changes in mRNA abundance were supported by a q value of ≤0.05.
q value of ≤1e−5.
The 32 genes in this table showed a minimum fold change of 1.5 supported by a P value of less than 0.05 in either mRNA or protein. Numbers in bold indicate a change in expression supported by a P value of less than 0.05. +INF (positive infinity) is used to denote instances where the protein was detected solely in the nitrogen-limited treatment.
TABLE 2 .
Selected genes related to nitrogen assimilation that either decreased or remained constant in response to nitrogen limitationd
| Locus ID | Gene | Description | Fold change |
|
|---|---|---|---|---|
| mRNA | Protein | |||
| SAR11_0040 | carA | Carbamoylphosphate synthase, small subunit | 1.02 | 1.09 |
| SAR11_0049 | amt | Ammonium transporter | 1.21 | 0.95 |
| SAR11_0050 | amt | Ammonium transporter | 1.05 | 0.88a |
| SAR11_0080 | aatA | Aspartate transaminase | 1.04 | 0.60b |
| SAR11_0086 | ilvE | Branched-chain amino acid aminotransferase | 0.71 | 0.56b |
| SAR11_0216 | hisC | Histidinolphosphate aminotransferase | 1.04 | 0.46 |
| SAR11_0502 | argD | Acetylornithine aminotransferase | 1.03 | 0.99a |
| SAR11_0534 | Aminotransferase | 1.09 | 0.64a | |
| SAR11_0655 | braC | Leucine/isoleucine/valine transporter: periplasmic | 1.08 | 1.02a |
| SAR11_0699 | purF | Amidophosphoribosyltransferase | 1.32 | 1.11a |
| SAR11_0809 | ald | Alanine dehydrogenase | 1.03 | 0.83b |
| SAR11_0829 | metC | Cystathionine beta-lyase | 1.31 | 0.60b |
| SAR11_0946 | ntrY | Nitrogen regulation protein | 1.00 | 1.04 |
| SAR11_0948 | ntrX | Nitrogen assimilation regulatory protein | 0.91 | 0.43b |
| SAR11_1151 | glmS | Glutamine-fructose-6-phosphate transaminase | 1.10 | 0.70a |
| SAR11_1187 | gdhA | Glutamate dehydrogenase | 0.53 | 0.57b |
| SAR11_1305 | glnT | Glutamine synthetase | 0.94 | 0.67b |
| SAR11_1308 | gltB2 | Glutamate synthase, large subunit | 1.29 | 0.66b |
| SAR11_1310 | amt | Ammonium transporter | 1.01 | 0.00c |
| SAR11_1313 | glxB | Glutamine amidotransferase | 1.08 | 0.70b |
| SAR11_1315 | glxD | Glutamate synthase, large subunit | 0.82 | 0.83a |
| SAR11_1316 | glnT | Glutamine synthetase | 0.87 | 0.72b |
| SAR11_1317 | ygeX | Diaminopropionate ammonia-lyase | 0.78 | 0.73b |
| SAR11_1346 | livJ | Leucine/isoleucine/valine transporter: periplasmic | 0.76 | 0.79b |
| SAR11_1361 | livJ2 | Leucine/isoleucine/valine transporter: periplasmic | 1.41 | 0.65b |
| SAR11_1368 | dadA | d-Amino-acid dehydrogenase, small chain | 1.16 | 0.73b |
q value of ≤ 0.05. No changes in mRNA abundance were supported by a q value of ≤0.05.
q value of ≤1e−5.
Peptides for SAR11_1310 were detected in control cultures but not in nitrogen-limited cultures.
Fold change values in bold indicate that differential expression is supported by a P value of 0.05 or less.
Comparison of mRNA versus protein expression.
Significant differences in the relative abundances of individual proteins were observed between nitrogen-limited and nitrogen-replete stationary-phase cultures, but no statistically significant changes in mRNA abundance were found at an alpha value of 0.05 when using a Student t test corrected for multiple comparisons. In general, changes in the abundances of mRNAs and their corresponding proteins were not correlated, as is evident in Fig. 5, Table 1, and Table 2. Overall, the range of change in mRNAs between nitrogen-limited and nitrogen-replete stationary-phase cultures was less than 3-fold, which is lower than what is typically observed in studies of similar design. A consistent observation in similar studies is that large changes in mRNA abundance are more likely to be reflected by corresponding changes in protein abundance. Likewise, in other studies, in plots analogous to Fig. 5 that compare changes in mRNA levels to changes in protein abundance, correlations are typically low for mRNA changes of less than 3-fold (log10 = ±0.5). Therefore, because all mRNA changes that we observed in response to nitrogen limitation were ≤3-fold (Table 1), the most straightforward interpretation of our observation that there was little correlation between transcripts and protein is that there was little transcriptional regulation in response to nitrogen limitation. Below, we consider an alternative explanation, that pulses of transcription were missed.
FIG 5.
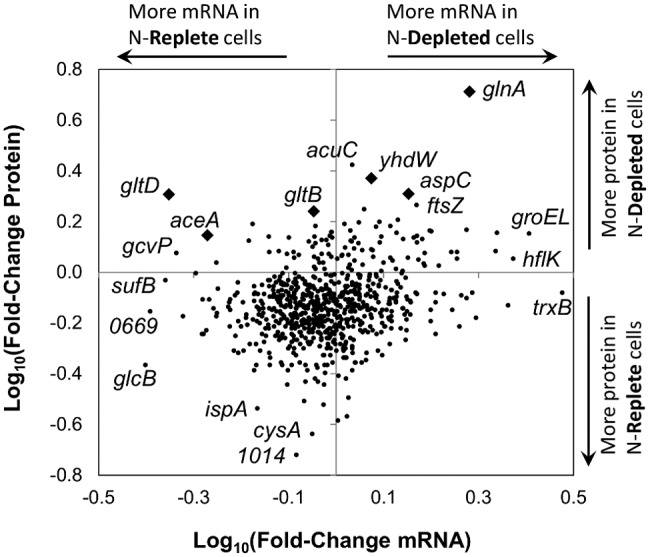
Ratios for nitrogen-depleted relative to nitrogen-replete stationary phase cells for mRNA abundance and protein abundance. Genes represented by a diamond are discussed in the text. The absence of a linear relationship between proteins and their corresponding mRNAs indicates that transcription and translation are uncoupled for a majority of proteins. Though highly upregulated, amtB and sox are absent from the plot because peptides for these gene products were not detected in the nitrogen-replete cultures.
A potential explanation for the lack of statistical support for changes in mRNA abundances in Table 1 was illustrated by Taniguchi et al. (70), who observed that half-lives for mRNA transcripts in Escherichia coli average only a few minutes. Because protein molecules are much more stable, often staying intact over the entire cell cycle (70), highly significant changes in protein abundances observed in our experiment could have been driven by discontinuous pulses of mRNA transcription. Examples of temporal variation in mRNA transcription have previously been observed in phosphate-limited Synechococcus cells (71) and iron-limited “Ca. Pelagibacter ubique” (61). We considered the possibility that discontinuous pulses of mRNA transcription could account for our observations by examining data from additional samples collected from these nitrogen-limited cultures at ~5 × 106 cells/ml, at the onset of stationary phase (Fig. 1), and from excess-nitrogen cultures midway through exponential phase. The additional data did not provide support for the hypothesis that missed pulses of transcription could account for the lack of congruence between mRNA expression data and protein abundance seen in Fig. 5. Samples collected at the onset of stationary phase exhibited trends in differential expression of mRNA and protein for glnA, gltB, and gltD that were nearly identical to cultures 6 to 8 days into stationary phase. Although the GlnA, GltB, and GltD proteins were already elevated by the onset of stationary phase in nitrogen-limited cultures, statistically significant changes in mRNA expression (q value, ≤0.05) were not observed for any gene in the same comparison (see Table S2 in the supplemental material).
A second hypothesis that can explain incongruence between mRNA and protein pools is differences in protein expression arising from posttranscriptional regulation of the mRNA pool, or differential protein stability (70, 72–74). Steady-state mRNA/protein ratios are driven by the synthesis and degradation rates of both the mRNA and the corresponding protein molecules, which are influenced by both properties of the RNA, such as transcription factor binding site efficiency and codon bias, as well as regulatory activity, for example, targeted degradation of proteins in response to specific stimuli (72–79). Recently, many riboswitches that had previously been identified in other bacteria, and a number of novel riboswitch-like structures, were identified in Pelagibacterales genomes (80). Riboswitches are composed of stem-loop structures in the 5′ untranscribed region (UTR) of mRNA transcripts and function by binding to low-molecular-weight metabolites and conditionally inhibiting translation of RNA transcripts based on metabolite availability (63, 80). Two different, novel riboswitch-like structures are located upstream of amt and amtB (80), two genes with protein products observed to be highly responsive to nitrogen limitation in this study; AmtB (SAR11_0818) was detected only in nitrogen-limited cultures, while Amt (SAR11_1310) was detected only in nitrogen-replete cultures (Tables 1 and 2). To explain uncoupling between transcription and translation previously observed in iron-limited “Ca. Pelagibacter ubique” cells, Smith et al. proposed a mechanism that could broadly suppress translation of transcripts via the expression of members of the cold shock family of RNA-binding proteins, CspE and CspL (61). However, CspE and CspL were not among the proteins that changed in abundance in response to nitrogen limitation.
In this study, in our previous work examining the effect of iron limitation on “Ca. Pelagibacter ubique,” and in another unpublished study of sulfur limitation (81), we observed little or no coupling between mRNA and protein levels. In other studies, where good accord between transcriptomes and proteomes has been observed, the focus was the cell’s response to changing external growth factors, such as metabolites (82), rather than a shift from growth to starvation, as in our studies. It is possible that the differences in mRNA/protein coupling between our studies and others are due to our focus on macronutrient limitation, rather than to intrinsic differences between the regulatory systems of different cell types. Studying Prochlorococcus, another streamlined organism that has a reduced suite of regulatory proteins, Hess et al. concluded that the number of noncoding RNAs (ncRNAs) relative to genome size is comparable to those in other bacteria (83). They suggested that RNA regulators likely play a major role in regulation in Prochlorococcus and speculated that regulation by ncRNAs may have been favored during selection for genome reduction because they require fewer resources than large protein regulators. Thus, it remains uncertain whether the differences in mRNA/protein coupling between our studies and others are due to our focus on macronutrient limitation or instead to fundamental differences between streamlined cells and others.
Regulatory proteins NtrY and NtrX.
Nitrogen limitation in “Ca. Pelagibacter ubique” did not have any effect on transcription or translation of the membrane sensor NtrY. However, the relative abundance of the transcriptional regulator NtrX was much lower under nitrogen-limiting conditions (Table 2). It seems likely that in “Ca. Pelagibacter ubique,” the two-component signaling system NtrY/NtrX is responsive to nitrogen limitation and plays a role in the regulation of cellular responses. However, future studies are needed to determine which, if any, proteins are regulated by NtrX. Fortunately, “Ca. Pelagibacter ubique” is an excellent model organism for studying the activity of the NtrY/NtrX system due to the absence of NtrB/NtrC, GlnB, and GlnD, which influence the activity of NtrY/NtrX in other organisms (46, 84, 85).
Ammonium transport and assimilation.
Ammonium (NH4) is an important source of nitrogen for microbial life in marine environments, and it is likely that in nature and in our experiments ammonium transport is facilitated by one or more of the four Amt paralogs found in the “Ca. Pelagibacter ubique” genome: SAR11_1310 (Amt), SAR11_0818 (AmtB), SAR11_0049, and SAR11_0050. All four of these proteins were detected in this study. The protein products of SAR11_0049 and SAR11_0050 were equally abundant across treatments, while the Amt encoded by SAR11_1310 was more abundant in nitrogen-replete cells (Table 2), and the SAR11_0818 product was more abundant in nitrogen-limited cells (Table 1).
Throughout this study, we use gene and protein names (e.g., “amt,” “amtA,” and “amtB”) as they appear in the literature and in public databases; however, the phylogeny, naming, and classification of amt genes have not yet been resolved into a consistent system. SAR11_0049, SAR11_0050, and SAR11_0818 are most closely related to genes described as amtB, although only SAR11_0818 is annotated as amtB. SAR11_1310, also annotated as amt, is a phylogenetic outlier (data not shown). AmtA transporters have been associated with the transport of methylammonium (67, 68, 86–88). In the Gram-positive actinobacterium Corynebacterium glutamicum, AmtA and AmtB were found to exhibit different substrate specificities for ammonium and methylammonium, with AmtA displaying high substrate affinity and membrane potential-dependent transport of methylammonium (86, 89). However, other work has suggested that, in Salmonella enterica serovar Typhimurium, AmtB functions by facilitating diffusion of the gas NH3 and that it may serve a role in NH3 efflux (87).
The independent regulation of “Ca. Pelagibacter ubique” Amt proteins suggests functional diversification, but there was no clear indication of the role played by each paralog. Previously, “Ca. Pelagibacter ubique” has been shown to oxidize methyl groups from trimethylamine (TMA) and trimethylamine oxide (TMAO) (86), and it may be able to use ammonium produced by demethylation of these compounds as a nitrogen source. We speculate that some of the diversified family of Amt transporters found in “Ca. Pelagibacter ubique” could be involved in transport of these compounds.
Interestingly, the two Amt proteins that were regulated in abundance, products of SAR11_1310 (amt) and SAR11_0818 (amtB), both have novel riboswitch-like structures in their 5′ UTRs (80). These putative riboswitches were first discovered in “Ca. Pelagibacter ubique,” but no function for them has been demonstrated (80). Here, we found that SAR11_1310 was equally transcribed under both nitrogen-replete and nitrogen-limited conditions, but its protein product was not detected in the nitrogen-limited treatment; conversely, both transcription of gene SAR11_0818 and the abundance of the corresponding protein increased in the nitrogen-limited treatment. Evidence for the involvement of structural RNA elements in nitrogen regulation has been increasing in recent years: a glutamine-sensing riboswitch has been described in cyanobacteria and marine metagenomic sequences (90) and a transcriptional attenuator was found to regulate expression of the PII gene for nitrogen fixation in the deltaproteobacterium Geobacter sulfurreducens (91). In light of the utilization by “Ca. Pelagibacter ubique” of riboswitches for regulating a diverse array of metabolic processes (63, 80, 92) and the absence of the usual AmtB regulators (NtrC and GlnK) in this genome, we speculate that “Ca. Pelagibacter ubique” may have acquired a novel riboswitch system for regulating its Amt paralogs, but how these paralogs function, and are regulated, is far from clear.
Metabolic reconstruction suggests that ammonium transported into cells by Amt proteins is condensed with glutamate to form glutamine, a common reaction catalyzed by glutamine synthetase (GlnA). The large (GltB) and small (GltD) subunits of GOGAT are also present and complete the metabolic pathway for regenerating glutamate from glutamine and 2-oxoglutarate. The genome of “Ca. Pelagibacter ubique” encodes three copies of glutamine synthetase, only one of which (SAR11_0747) was upregulated in response to nitrogen limitation. Protein products from the two other glutamine synthetase genes (SAR11_1305 and SAR11_1316) were less abundant during nitrogen limitation. SAR11_0747 is in class I (glnA), while SAR11_1305 and SAR11_1316 are in class III (glnT). Previous studies of GlnA and GlnT characterized these two protein classes as highly dissimilar in structure, function, and phylogenetic distribution (33); GlnA is nearly universal among bacteria, assembles into a dodecameric multiprotein complex, and has a Km of 0.2 mM for ammonium, while GlnT is clade specific, is an octamer, and has a Km of 33 mM.
It is noteworthy that in an order of organisms where paralogs are infrequent (59), members of two different paralogous gene families—ammonium transporters and glutamine synthetases—were regulated in response to nitrogen limitation. Clearly, the metabolism of nitrogenous compounds plays an important, if as yet poorly understood, role in the ecology of these cells.
Organic nitrogen transport and assimilation.
Several organic molecule transporters were observed to be more abundant in cells limited by nitrogen availability. As detailed in Table 1, these include the periplasmic components of ABC transporters which recognize a diverse collection of nitrogenous molecules, including amino acids (YhdW), taurine (TauA), and opines (OccT). The outer membrane porin protein OmpU was also more abundant in nitrogen-limited cultures and may serve to increase the diffusion rate of both ammonium and nitrogenous organic molecules into the periplasmic space. This collection of upregulated proteins is in good accord with the set of most highly detected proteins from a metaproteomic analysis of seawater from the Oregon coast, in which the environmental proteome was dominated by YhdW, TauA, polyamine transporters, and glutamine synthetase from “Ca. Pelagibacter ubique” (93), and underscores the means by which laboratory experiments can be used to validate interpretations of gene expression in the environment.
The second most highly upregulated enzyme (SAR11_1304) in nitrogen-limited cells is annotated as monomeric sarcosine oxidase. Previously, Sun et al. (89) noted the presence of a family of paralogous sox genes in “Ca. Pelagibacter ubique,” some of which were linked to the pathway for the oxidation of glycine betaine, through the intermediate sarcosine. However, it is unknown why “Ca. Pelagibacter ubique” has genes that encode multiple Sox proteins. The other two sox operons in the “Ca. Pelagibacter ubique” genome encode the subunits of multiprotein holoenzymes, whereas SAR11_1304 is presumed to encode a monomeric protein. Analysis with the program I-TASSER confirmed that SAR11_1304 is structurally related to other monomeric sarcosine oxidases and also has structural similarities to both amine demethylases and deaminases (Fig. 6). Given the high expression of the SAR11_1304 mRNA transcripts and protein in nitrogen-limiting cells, we speculate that this protein functions in making amine groups available for biosynthesis. Amine demethylases convert secondary amines (R–NH–CH3) and tertiary amines [R–N(CH3)2] to primary amines (R–NH2). Deaminases, on the other hand, liberate ammonia from compounds with primary amine groups.
FIG 6 .
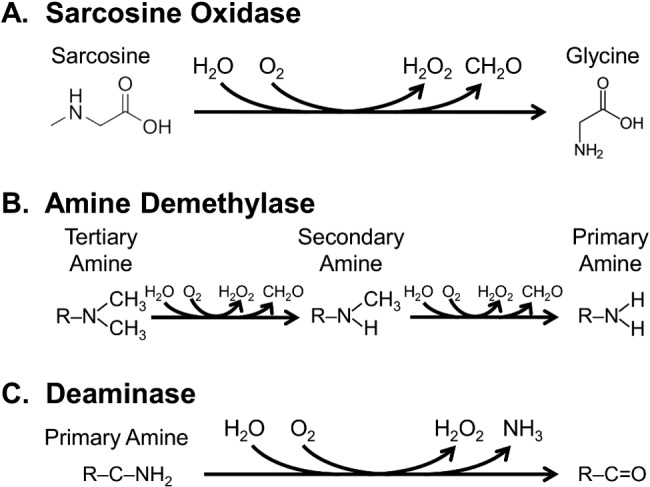
(A) Annotated function of the highly upregulated protein SAR11_1304. (B and C) Alternate functions for SAR11_1304, predicted by a metaprotein threading approach (I-TASSER).
A number of incompletely characterized pathways for the uptake and oxidation of nitrogenous organic compounds have been demonstrated or suggested in “Ca. Pelagibacter ubique.” The monomeric sarcosine oxidase SAR11_1304 is likely involved in some aspect of this metabolism, but its function remains uncertain. Previous work has shown that methyl groups from TMA and TMAO are oxidized by “Ca. Pelagibacter ubique” (89), and a TMAO oxidase has been identified in the genomes of “Ca. Pelagibacter ubique” (94). However, the genome does not contain genes for dimethylamine (DMA) monooxygenase, leaving in question the pathway used by “Ca. Pelagibacter ubique” to metabolize DMA, a product of TMAO oxidation.
Other questions are raised by the observations that, although transporters for a variety of nitrogenous compounds are upregulated by HTCC1062 during nitrogen limitation, few, if any, enzymes were concomitantly expressed for transferring amine groups from these substrates to other compounds. One potential explanation for this is that such enzymes are expressed only when specific nitrogenous substrates are present within the cell. If this is the case, the control of nitrogen metabolism contains additional regulatory elements not stimulated by the experimental conditions of this study.
Proposed regulatory model.
The PII regulatory system performs three important functions: (i) regulating GS/GOGAT activity to prevent the depletion of the TCA cycle intermediate 2-oxoglutarate, (ii) controlling ammonia transport to guard against toxic pulses, and (iii) inhibiting/stimulating transcription of genes related to nitrogen assimilation. Based on the observations in this study, as well as those from previous experiments with “Ca. Pelagibacter ubique,” we propose the following model for how this organism regulates these three processes without the usual collection of PII genes.
Maintaining an adequate pool of the intermediate 2-oxoglutarate is essential to the functions of the TCA cycle. One of the functions of the PII system is to regulate competition between the TCA cycle and GS/GOGAT for 2-oxoglutarate. In principle, competition for 2-oxoglutarate could cause energy starvation during periods of rapid N uptake. We speculate that reorganization of central metabolism in “Ca. Pelagibacter ubique” compensates for the negative consequences of removing the PII system by employing the glyoxylate bypass to replenish TCA cycle intermediates by AceA-mediated cleavage of isocitrate to succinate and glyoxylate. By this alternate pathway (Fig. 3), succinate reenters the TCA cycle directly, and glyoxylate, after condensation with acetyl coenzyme A (acetyl-CoA), reenters as malate (63). In E. coli, the glyoxylate bypass is regulated by the product of aceK, which is missing in “Ca. Pelagibacter ubique.” Thus, we propose that constitutive expression of glyoxylate bypass enzymes allows unchecked consumption of 2-oxoglutarate by GS/GOGAT without cessation of TCA cycle functions. This theory is supported by our observation that the SAR11 strain HIMB59, the genome of which encodes a PII network, is lacking the genes for the glyoxylate bypass (59).
We propose that SAR11_1310 (Amt) facilitates ammonia efflux to limit intracellular spikes in ammonia concentration. We speculate that, at toxic levels, ammonia, or one of its metabolic products, such as glutamine, interacts with a cis-acting structural RNA (80) to activate translation of the downstream SAR11_1310 gene on the mRNA transcript, thereby producing the protein product to export excess ammonia from the cell. This hypothesis is untested, in the sense that there is no direct evidence that the SAR11_1310 Amt is an exporter; however, this model is in accord with the pattern of expression observed for this gene. The SAR11_0818 product was more abundant in nitrogen-limited cells and is likely an importer for an unknown nitrogenous compound. The different riboswitch on this gene leads us to speculate that translation of this protein product is released when concentrations of an intracellular effector, possibly either ammonia or glutamine, drop.
The nitrogen-responsive transcriptional regulator in “Ca. Pelagibacter ubique” is proposed to be NtrX. This protein has been previously noted for its similarity to NtrC, the transcriptional regulator in the typical PII regulatory system, and can recognize DNA promoter regions (84, 85). Nitrogen limitation was observed to have an effect on NtrX abundance in this experiment, with nitrogen-limited cells containing less than half as much NtrX as nitrogen-replete cells. This difference in concentration, as well as potential posttranslational modifications through the NtrY/NtrX two-component system, may affect transcription of genes involved in nitrogen assimilation in a manner similar to NtrC in other alphaproteobacteria.
Summary.
“Ca. Pelagibacter ubique” and other members of the Pelagibacterales are proving to be fascinating subjects for experimental study because of their genomic and metabolic simplicity and their extraordinary success in competitive pelagic environments, which are frequently nutrient limited.
In examining the transcriptional and proteomic responses of “Ca. Pelagibacter ubique” to nitrogen limitation, we have observed strong support for the following conclusions. (i) The PII system for regulating nitrogen metabolism is conserved in all known free-living alphaproteobacteria, except for most members of the order Pelagibacterales, including “Ca. Pelagibacter ubique.” (ii) Nitrogen-limited stationary phase in “Ca. Pelagibacter ubique” is characterized by increases in genes encoding the metabolic enzymes GlnA, GltB, GltD, AspC, and Sox and the transport proteins YdhW, AmtB, OccT, and TauA. (iii) In “Ca. Pelagibacter ubique” cells limited for nitrogen, significant differences in protein expression occurring in late log phase and late stationary phase could not be accounted for solely by measured changes in transcript expression levels. Furthermore, observations from this study have led us to speculate the following. (i) In response to streamlining selection, “Ca. Pelagibacter ubique” and other members of the Pelagibacterales have evolved a noncanonical system, encoded by fewer genes, for responding to nitrogen limitation and assimilating amines. This system appears to operate under the control of the NtrY/NtrX two-component regulatory system. (ii) When nitrogen limited, “Ca. Pelagibacter ubique” increases the abundance of transporters for nitrogenous organic compounds and ammonium, suggesting that these are important sources of N for these cells in the environment. (iii) The uncoupling of transcription from translation observed for the genes SAR11_0818 (amtB) and SAR11_1310 (amt) suggests that they may be controlled independently by two different, novel riboswitches previously identified in 5′ UTRs of these genes. (iv) Increases in the SAR11_1304 (sox) gene product, and structural homology to other enzymes, suggest that this protein, which is often annotated as a “monomeric sarcosine oxidase,” participates in the production of primary amines or ammonia from environmentally available nitrogenous compounds.
MATERIALS AND METHODS
Phylogenetic analysis/PII orthologs.
The Hal software package (95) was used to generate clusters of orthologous proteins from 266 Alphaproteobacteria (IMG v350) using all-versus-all BLASTP followed by Markov clustering (MCL) at 13 inflation parameters. Clusters generated with the conservative inflation parameter of 3.0 were used to identify orthologs of nitrogen assimilation genes. The genes glnB and glnK were clustered together at the 3.0 inflation parameter (and they are considered part of the same COG-0347), so the distribution of these was curated manually for organisms with only one ortholog by comparing gene neighborhoods according to reference 32. glnB was designated if the ortholog was located near glnA, and glnK was designated if the ortholog was located next to amtB. In only two cases, these neighborhoods were not conserved, and the IMG gene neighborhood alignment function was used to determine appropriate orthology designations. The complete gene distribution for all strains is listed in Table S3 in the supplemental material and reported in Fig. 4.
A concatenated 16S-23S rRNA gene tree was created manually using rRNA gene sequences from 250 Alphaproteobacteria and six outgroups from the Beta-, Gamma-, and Deltaproteobacteria. All genome and gene identifiers for the taxa in the tree are also in Table S3 in the supplemental material. Some rRNA gene sequences from the original group of 263 taxa (above) were of sufficiently poor quality or length to be excluded from the tree or were missing altogether, although their nitrogen metabolism gene distributions are reported in Table S3. 16S and 23S rRNA genes were aligned separately with MUSCLE (96), and poorly aligned positions were curated with Gblocks (97) using the settings in reference 98. The alignments were normalized and concatenated with normalize_alignments.py and catPhylip.pl, respectively, included in the Hal package. The final alignment contained 256 taxa and 4,025 characters. The final tree was constructed with RAxML (99) using –f a –m GTRGAMMA –\# 1,000.
Searching metagenomic sequences for Pelagibacteraceae PII genes.
To investigate the frequency of PII genes in natural populations of Pelagibacteraceae, we searched the global ocean sampling (GOS) data set (67) for PII genes present on the same read as Pelagibacteraceae genes. In this search, PII genes absent from most Pelagibacteraceae genomes (PII genes) are defined as glnB, glnD, glnK, ntrB, and ntrC. Genes present in Pelagibacteraceae which are often neighbors to PII genes in other species (neighbors) are defined as glnA, map, dapA, amtB, ntrX, and ntrY. TBLASTn was used to search the GOS read data set for neighbors using an expect score cutoff of 1e−5 and a limit of 10,000 matches per neighbor. Open reading frames (ORFs) were translated from reads returned by this search and then searched against the NCBI nonredundant database using BLASTP to locate their top match. Reads were retained for further analysis if they contained at least two ORFs, where one ORF was a neighbor with a top match to a Pelagibacteraceae strain and a second ORF had a top match to a PII gene from any species. The three reads which met these criteria were examined manually and determined to have nonspurious top matches; all top matches on these reads had amino acid identities ranging from 63 to 67% over 108 to 140 residues and expect scores of 2e−33 to 2e−43.
Growth media and harvesting.
Artificial seawater (ASW) medium was made using previously established protocols (64). Water, salts, and metals were added to eight 20-liter polycarbonate carboys as detailed in Table S1 in the supplemental material and then autoclaved for 10 h. After cooling to room temperature, carboys were sparged with CO2 for 20 h and brought up to 20 liters using sterile water to compensate for evaporation due to autoclaving. Vitamins, nutrients, and ammonium were added to each carboy from a filter-sterilized stock solution and then sparged overnight on air while cooling to 16°C. Final nutrient concentrations were as follows: 10 µM dimethylsulfoniopropionate (DMSP), 500 µM oxaloacetate, 500 µM pyruvate, and (for the nitrogen-replete cultures only) 20 µM ammonium. Nitrogen-limited cultures received no ammonium amendment but were expected to grow to a lower density on trace contaminants containing nitrogen. The medium was then inoculated with an ASW-adapted culture of “Ca. Pelagibacter ubique” HTCC1062 (64) growing exponentially in nitrogen-limited medium. Following inoculation, cultures were incubated at 16°C with constant air sparging.
Culture growth was tracked daily by staining cells with SYBR Green and counting on a Guava EasyCyte flow cytometer. Two cultures, one from each treatment, failed to attain the density necessary for analysis and were not harvested. Samples for microarray and proteomic analysis were taken from the six remaining cultures during stationary phase. At these time points, samples were collected using the protocol developed previously (61), that is, 5 × 1010 cells were removed to a separate vessel and amended with 10 mg chloramphenicol, 100 µl 500 mM EDTA, and 100 µl 100× Halt protease inhibitor cocktail (Thermo Scientific catalog no. 78438) per liter of culture. Tangential flow filtration for 20 to 30 min against a Pellicon 2 Mini 30-K membrane (Millipore catalog no. P2C030C01) reduced the volume of culture to <150 ml, which was subsequently centrifuged for 1 h at 20,000 rpm and 0°C. The pelleted cells were resuspended in 1 ml Tris-EDTA (TE) and centrifuged again in a single 1.5-ml tube for 40 min at 40,000 rpm and 10°C. After the supernatant was decanted, the pellet was stored at −80°C until proteomic analysis. Microarray samples, which require less biomass, were harvested through a separate protocol simultaneously. Eighty milliliters of culture was collected and centrifuged for 1 h at 20,000 rpm and 0°C. Cells were resuspended in 1 ml of RNAprotect bacterial reagent (Qiagen catalog no. 76506) and then centrifuged again in a 1.5-ml tube for 40 min at 40,000 rpm and 0°C. After the supernatant was decanted, microarray samples were placed at −80°C until analysis.
mRNA preparation.
Each cell pellet was resuspended in 100 µl TE (1 mM Tris, 1 mM EDTA, pH 8) containing 40 µg lysozyme and incubated at room temperature for 5 min. Total RNA was extracted using an RNeasy MinElute cleanup kit (Qiagen catalog no. 74204) according to the manufacturer’s instructions, with the exception of an additional wash step with 700 µl buffer RW1 (Qiagen catalog no. 1053394) immediately prior to the prescribed washes with RPE buffer. Eluted RNA was quantified using a NanoDrop spectrophotometer (Thermo Scientific ND-1000) and found to vary over the range of 243 to 837 ng. One hundred nanograms of total RNA from each sample was amplified and labeled using a MessageAmp II bacterial RNA amplification kit (Ambion catalog no. AM1790) and biotin-11-UTP (Ambion catalog no. AM8451) according to the MessageAmp Improved Protocol handbook. Polyadenylation, reverse transcription, second-strand synthesis, and in vitro transcription reactions were performed in the recommended volume, and reaction mixtures were incubated for 19 h rather than 14 h. NanoDrop measurements of amplified RNA showed recovery of 93 to 147 µg per sample.
RNA labeling and microarray processing.
Protocols for microarray analysis were the same as previously described (61): RNA integrity screening, probe synthesis, hybridization, and scanning were conducted by the CGRB Core Laboratories at Oregon State University, Corvallis, OR. Five micrograms of total RNA was used to generate biotinylated cRNA for each treatment group using the One-Cycle target labeling protocol (Affymetrix, Santa Clara, CA) from the GeneChip Expression Analysis Technical Manual (701021 rev. 5). In short, isolated total RNA was checked for integrity and concentration using the RNA 6000 Nano LabChip kit on the Agilent Bioanalyzer 2100 (Agilent Technologies, Inc., Palo Alto, CA). Poly(A) RNA control kit RNA and “Ca. Pelagibacter ubique” RNA were reverse transcribed using a T7-(dT)24 primer and Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA), and double-stranded cDNA was synthesized and purified with GeneChip sample cleanup modules (Affymetrix, Santa Clara, CA). Biotinylated cRNA was synthesized from the double-stranded cDNA using T7 RNA polymerase and a biotin-conjugated pseudouridine-containing nucleotide mixture provided in the IVT labeling kit (Affymetrix, Santa Clara, CA). Prior to hybridization, the cRNA was purified with GeneChip sample cleanup modules (Affymetrix, Santa Clara, CA) and fragmented. Ten micrograms from each experimental sample along with Affymetrix prokaryotic hybridization controls was hybridized for 16 h to “Ca. Pelagibacter ubique” genome arrays (pubiquea) in an Affymetrix GeneChip hybridization oven 640. Affymetrix GeneChip Fluidics Station 450 was used to wash and stain the arrays with streptavidin-phycoerythrin (Molecular Probes, Eugene, OR) and biotinylated antistreptavidin (Vector Laboratories, Burlingame, CA) according to the standard antibody amplification protocol for prokaryotic targets. Arrays were scanned with an Affymetrix GeneChip Scanner 3000 at 570 nm. The Affymetrix prokaryotic hybridization control kit and poly(A) RNA control kit were used to ensure efficiency of hybridization and cRNA amplification. All cRNA was synthesized at the same time. Hybridizations were conducted with one replicate of all times and treatments concurrently. Each array image was visually screened to discount for signal artifacts, scratches, or debris.
MS sample preparation.
Each pellet was brought up to 100 µl with 8 M urea (Sigma-Aldrich, St. Louis, MO) and sonicated in a water bath with ice until the pellet went into solution. The samples were briefly spun and transferred to PCT MicroTube barocycler pulse tubes with 100-µl caps (Pressure Biosciences Inc., South Easton, MA). The MicroTubes were placed in a MicroTube cartridge and barocycled for 10 cycles (20 s at 35,000 lb/in2 back down to ambient pressure for 10 s). All of the material was removed from the MicroTubes and transferred to 1.5-ml microcentrifuge tubes. A Coomassie Plus (Thermo Scientific, Rockford, IL) assay was used to determine protein concentration. Dithiothreitol (DTT) was added to each sample at a concentration of 5 mM (Sigma-Aldrich, St. Louis, MO) and incubated at 60°C for 1 h. The samples were then diluted 10-fold with 100 mM NH4HCO3, and tryptic digestion (Promega, Madison, WI) was performed at a 1:50 (wt/wt) ratio with the addition of 1 mM CaCl2 to stabilize the trypsin and reduce autolysis. The sample was incubated for 3 h and cleaned via C18 solid-phase extraction (Supelco, Bellefonte, PA). The samples were dried to 50 µl and assayed with bicinchoninic acid (Thermo Scientific, Rockford, IL) to determine the final peptide concentration and placed in vials for mass spectrometry (MS) analysis.
Capillary LC-MS proteome analysis.
Quantitative mass spectrometry proteomics measurements were obtained using high-performance liquid chromatography (HPLC) in tandem with quantitative mass spectrometry. The HPLC system consisted of a custom configuration of 65-ml Isco Model 65 D syringe pumps (Isco, Inc., Lincoln, NE), 2-position Valco valves (Valco Instruments Co., Houston, TX), and a PAL autosampler (Leap Technologies, Carrboro, NC), allowing for fully automated sample analysis across four separate HPLC columns (100). Reversed-phase capillary HPLC columns were manufactured in-house by slurry packing 3-µm Jupiter C18 stationary-phase columns (Phenomenex, Torrance, CA) into a 60-cm length of fused silica capillary tubing (360-µm outside diameter [o.d.] by 75-µm inside diameter [i.d.]) (Polymicro Technologies Inc., Phoenix, AZ) using a 1-cm sol-gel frit (unpublished Pacific Northwest National Laboratory [PNNL] variation of reference 101) for retention of the packing material. The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid-acetonitrile (B). The mobile phase was degassed by using an in-line Degassex Model DG4400 vacuum degasser (Phenomenex, Torrance, CA). The HPLC system was equilibrated at 10,000 lb/in2 with 100% mobile phase A, and then a mobile-phase selection valve was switched 50 min after injection, which created a near-exponential gradient as mobile phase B displaced phase A in a 2.5-ml active mixer. A 40-cm length of 360-µm-o.d. by 15-µm-i.d. fused silica tubing was used to split ~17 µl/min of flow before it reached the injection valve (5-µl sample loop). The split flow controlled the gradient speed under conditions of constant pressure operation (10,000 lb/in2). Flow through the capillary HPLC column when equilibrated to 100% mobile phase A was ~500 nl/min.
MS analysis was performed using an LTQ Orbitrap Velos electron transfer dissociation (ETD) mass spectrometer (Thermo Scientific, San Jose, CA) outfitted with a custom electrospray ionization interface. Electrospray emitters were custom made using 150-µm-o.d. by 20-μm-i.d. chemically etched fused silica (102). The heated capillary temperature and spray voltage were 250°C and 2.2 kV, respectively. Data were acquired for 100 min, beginning 65 min after sample injection (15 min into gradient). Orbitrap spectra (automatic gain control [AGC], 1 × 106) were collected from 400 to 2,000 m/z at a resolution of 100k followed by data-dependent ion trap collision-induced dissociation (CID) tandem MS (MS/MS) (collision energy, 35%; AGC, 1 × 104) and Orbitrap ETD MS/MS (activation time, 100 ms; AGC, 2 × 105) of the six most abundant ions. A dynamic exclusion time of 60 s was used to discriminate against previously analyzed ions.
Mass spectrometry data analysis.
Identification and quantification of the detected peptide peaks were performed utilizing the accurate mass and time (AMT) tag approach (103). Briefly, multiple in-house-developed/publicly available informatics tools were used to process LC-MS data and correlate the resulting LC-MS features with an AMT tag database containing accurate mass and LC separation elution time information for peptides previously identified by LC-MS/MS measurements from “Ca. Pelagibacter ubique” proteins (i.e., AMT tags). Among the tools used were algorithms for peak-picking and for determining isotopic distributions and charge states (104). Further downstream, data analysis incorporated all of the possible detected peptides into the visualization program VIPER (105) to correlate LC-MS features with the peptide in the AMT tag database and also provided normalized LC elution times via alignment to the database and an intensity report for all detected features.
Each of the 12 biological samples was measured with quantitative mass spectrometry in triplicate. Calculating the difference in protein abundance between two conditions was a three-step process. First, an average peptide abundance under each condition was calculated by averaging together the values for individual peptides across all samples assigned to a particular condition. Peptides with fewer than three observations in a condition were marked as “not observed” in that condition. Next, the peptide average from condition 1 was divided by the peptide average from condition 2 and then log10 transformed. Finally, all log10 peptide ratios from the same protein were averaged together.
To represent the likelihood that a protein was equally abundant in both samples, the multiple peptide measurements were combined into a single statistic as previously described (61, 106). Briefly, P values for individual peptides were calculated using a one-tailed Student t test. A two-tailed Student t test was not used because P values reflecting a large increase would be indistinguishable from P values reflecting a large decrease. Instead, peptides which changed in the opposite direction from the protein average were assigned a P value of 1 for their one-tailed Student t test. All peptide P values for a single protein were then combined into a single chi-square statistic by using Fisher’s method with a Bonferroni correction:
where n is the total number of peptides for a given protein and Pi is an individual peptide’s P value as determined by a Student t test. The χ2 value is then transformed into a P value using a chi-square probability table and 2n degrees of freedom. A Perl script for performing this computation is available at https://gist.github.com/dansmith01/6188797.
q values were calculated via the Benjamini-Hochberg method (107) to estimate the false discovery rate arising from multiple comparisons in the microarray and proteomics analyses. In this procedure, each P value was multiplied by the number of proteins, T, and then divided by that P value’s rank, k, among the set of P values (e.g., the most significant P value was divided by 1, the second most significant P value was divided by 2, etc.):
Microarray data accession number.
Microarray data generated by this experiment are available from NCBI’s GEO archive under accession number GSE37445.
Proteomic data availability.
Mass spectrometry data for this experiment are publicly available at http://omics.pnl.gov/view/publication_1089.html.
SUPPLEMENTAL MATERIAL
Conditions of limited nitrogen and excess nitrogen.
Changes in gene expression and their statistical significance.
Gene distribution in all strains.
ACKNOWLEDGMENTS
This study was supported by a Marine Microbiology Initiative investigator award (S.J.G.) from the Gordon and Betty Moore Foundation. The phylogenomics portions of this work (J.C.T.) were supported by the National Science Foundation under award no. DBI-1003269. Proteomics measurements were supported by the U.S. Department of Energy (DOE) Office of Biological and Environmental Research (OBER) Pan-omics program at Pacific Northwest National Laboratory (PNNL) and performed in the Environmental Molecular Sciences Laboratory, a DOE OBER national scientific user facility on the PNNL campus. PNNL is a multiprogram national laboratory operated by Battelle for the DOE under contract DE-AC05-76RL01830.
Footnotes
Citation Smith DP, Thrash JC, Nicora CD, Lipton MS, Burnum-Johnson KE, Carini P, Smith RD, Giovannoni SJ. 2013. Proteomic and transcriptomic analyses of “Candidatus Pelagibacter ubique” describe the first PII-independent response to nitrogen limitation in a free-living alphaproteobacterium. mBio 4(6):e00133-12. doi:10.1128/mBio.00133-12.
REFERENCES
- 1. Davey M, Tarran GA, Mills MM, Ridame C, Geider RJ, LaRoche J. 2008. Nutrient limitation of picophytoplankton photosynthesis and growth in the tropical North Atlantic. Limnol. Oceanogr. 53:1722–1733 [Google Scholar]
- 2. Elser JJ, Sterner RW, Gorokhova E, Fagan WF, Markow TA, Cotner JB, Harrison JF, Hobbie SE, Odell GM, Weider LW. 2000. Biological stoichiometry from genes to ecosystems. Ecol. Lett. 3:540–550 [Google Scholar]
- 3. Goldman J. 1976. Identification of nitrogen as a growth-limiting nutrient in wastewaters and coastal marine waters through continuous culture algal assays. Water Res. 10:97–104 [Google Scholar]
- 4. Hecky RE, Kilham P. 1988. Nutrient limitation of phytoplankton in freshwater and marine environments: a review of recent evidence on the effects of enrichment. Limnol. Oceanogr. 33:796–822 [Google Scholar]
- 5. Howarth RW, Marino R. 2006. Nitrogen as the limiting nutrient for eutrophication in coastal marine ecosystems: evolving views over three decades. Limnol. Oceanogr. 51:364–376 [Google Scholar]
- 6. Oviatt C, Doering P, Nowicki B, Reed L, Cole J, Frithsen J. 1995. An ecosystem level experiment on nutrient limitation in temperate coastal marine environments. Mar. Ecol. Prog. Ser. 116:171–179 [Google Scholar]
- 7. Ryther JH, Dunstan WM. 1971. Nitrogen, phosphorus, and eutrophication in the coastal marine environment. Science 171:1008–1013 [DOI] [PubMed] [Google Scholar]
- 8. Smith S. 1984. Phosphorus versus nitrogen limitation in the marine environment. Limnol. Oceanogr. 29:1149–1160 [Google Scholar]
- 9. Thomas WH. 1970. On nitrogen deficiency in tropical Pacific oceanic phytoplankton: photosynthetic parameters in poor and rich water. Limnol. Oceanogr. 15:380–385 [Google Scholar]
- 10. Grzymski JJ, Dussaq AM. 2011. The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 6:71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cotner J, Ammerman J, Peele E, Bentzen E. 1997. Phosphorus-limited bacterioplankton growth in the Sargasso Sea. Aquat. Microb. Ecol. 13:141–149 [Google Scholar]
- 12. Wu J, Sunda W, Boyle EA, Karl DM. 2000. Phosphate depletion in the western North Atlantic Ocean. Science 289:759–762 [DOI] [PubMed] [Google Scholar]
- 13. Wulff F, Eyre BD, Johnstone R. 2011. Nitrogen versus phosphorus limitation in a subtropical coastal embayment (Moreton Bay, Australia): implications for management. Ecol. Modell. 222:120–130 [Google Scholar]
- 14. Boyd PW, Jickells T, Law CS, Blain S, Boyle EA, Buesseler KO, Coale KH, Cullen JJ, de Baar HJ, Follows M, Harvey M, Lancelot C, Levasseur M, Owens NP, Pollard R, Rivkin RB, Sarmiento J, Schoemann V, Smetacek V, Takeda S, Tsuda A, Turner S, Watson AJ. 2007. Mesoscale iron enrichment experiments 1993–2005: synthesis and future directions. Science 315:612–617 [DOI] [PubMed] [Google Scholar]
- 15. Martin JH, Fitzwater SE. 1988. Iron deficiency limits phytoplankton growth in the north-east Pacific subarctic. Nature 331:341–343 [Google Scholar]
- 16. Martin JH, Coale KH, Johnson KS, Fitzwater SE, Gordon RM, Tanner SJ, Hunter CN, Elrod VA, Nowicki JL, Coley TL, Barber RT, Lindley S, Watson AJ, Van Scoy K, Law CS, Liddicoat MI, Ling R, Stanton T, Stockel J, Collins C, Anderson A, Bidigare R, Ondrusek M, Latasa M, Millero FJ, Lee K, Yao W, Zhang JZ, Friederich G, Sakamoto C, Chavez F, Buck K, Kolber Z, Greene R, Falkowski P, Chisholm SW, Hoge F, Swift R, Yungel J, Turner S, Nightingale P, Hatton A, Liss P, Tindale NW. 1994. Testing the iron hypothesis in ecosystems of the equatorial Pacific Ocean. Nature 371:123–129 [Google Scholar]
- 17. Price NM, Ahner BA, Morel FMM. 1994. The equatorial Pacific Ocean: grazer-controlled phytoplankton populations in an iron-limited ecosystem. Limnol. Oceanogr. 39:520–534 [Google Scholar]
- 18. Yin K, Qian P-Y, Chen JC, Hsieh DPH, Harrison PJ. 2000. Dynamics of nutrients and phytoplankton biomass in the Pearl River estuary and adjacent waters of Hong Kong during summer: preliminary evidence for phosphorus and silicon limitation. Mar. Ecol. Prog. Ser. 194:295–305 [Google Scholar]
- 19. Downing JA, Osenberg CW, Sarnelle O. 1999. Meta-analysis of marine nutrient-enrichment experiments: variation in the magnitude of nutrient limitation. Ecology 80:1157–1167 [Google Scholar]
- 20. Conley D, Schelske C, Stoermer E. 1993. Modification of the biogeochemical cycle of silica with eutrophication. Mar. Ecol. Prog. Ser. 101:179–192 [Google Scholar]
- 21. Turner RE, Qureshi N, Rabalais NN, Dortch Q, Justić D, Shaw RF, Cope J. 1998. Fluctuating silicate:nitrate ratios and coastal plankton food webs. Proc. Natl. Acad. Sci. U. S. A. 95:13048–13051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elser JJ, Bracken ME, Cleland EE, Gruner DS, Harpole WS, Hillebrand H, Ngai JT, Seabloom EW, Shurin JB, Smith JE. 2007. Global analysis of nitrogen and phosphorus limitation of primary producers in freshwater, marine and terrestrial ecosystems. Ecol. Lett. 10:1135–1142 [DOI] [PubMed] [Google Scholar]
- 23. Tyrrell T. 1999. The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 400:525–531 [Google Scholar]
- 24. Schell D, Barnett B, Vinette K. 1998. Carbon and nitrogen isotope ratios in zooplankton of the Bering, Chukchi and Beaufort seas. Mar. Ecol. Prog. Ser. 162:11–23 [Google Scholar]
- 25. Harpole WS, Ngai JT, Cleland EE, Seabloom EW, Borer ET, Bracken ME, Elser JJ, Gruner DS, Hillebrand H, Shurin JB, Smith JE. 2011. Nutrient co-limitation of primary producer communities. Ecol. Lett. 14:852–862 [DOI] [PubMed] [Google Scholar]
- 26. Allgeier JE, Rosemond AD, Layman CA. 2011. The frequency and magnitude of non-additive responses to multiple nutrient enrichment. J. Appl. Ecol. 48:96–101 [Google Scholar]
- 27. Mills MM, Ridame C, Davey M, La Roche J, Geider RJ. 2004. Iron and phosphorus co-limit nitrogen fixation in the eastern tropical North Atlantic. Nature 429:292–294 [DOI] [PubMed] [Google Scholar]
- 28. Danger M, Daufresne T, Lucas F, Pissard S, Lacroix G. 2008. Does Liebig’s law of the minimum scale up from species to communities? Oikos 117:1741–1751 [Google Scholar]
- 29. Schade JD, Espeleta JF, Klausmeier CA, McGroddy ME, Thomas SA, Zhang L. 2005. A conceptual framework for ecosystem stoichiometry: balancing resource supply and demand. Oikos 109:40–51 [Google Scholar]
- 30. Arcondéguy T, Jack R, Merrick M. 2001. P(II) signal transduction proteins, pivotal players in microbial nitrogen control. Microbiol. Mol. Biol. Rev. 65:80–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forchhammer K. 2007. Glutamine signalling in bacteria. Front. Biosci. 12:358–370 [DOI] [PubMed] [Google Scholar]
- 32. Leigh JA, Dodsworth JA. 2007. Nitrogen regulation in bacteria and archaea. Annu. Rev. Microbiol. 61:349–377 [DOI] [PubMed] [Google Scholar]
- 33. Merrick MJ, Edwards RA. 1995. Nitrogen control in bacteria. Microbiol. Rev. 59:604–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hervás AB, Canosa I, Little R, Dixon R, Santero E. 2009. NtrC-dependent regulatory network for nitrogen assimilation in Pseudomonas putida. J. Bacteriol. 191:6123–6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foor F, Janssen KA, Magasanik B. 1975. Regulation of synthesis of glutamine synthetase by adenylylated glutamine synthetase. Proc. Natl. Acad. Sci. U. S. A. 72:4844–4848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Foor F, Reuveny Z, Magasanik B. 1980. Regulation of the synthesis of glutamine synthetase by the pII protein in Klebsiella aerogenes. Proc. Natl. Acad. Sci. U. S. A. 77:2636–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Javelle A, Merrick M. 2005. Complex formation between AmtB and GlnK: an ancestral role in prokaryotic nitrogen control. Biochem. Soc. Trans. 33:170–172 [DOI] [PubMed] [Google Scholar]
- 38. Javelle A, Severi E, Thornton J, Merrick M. 2004. Ammonium sensing in Escherichia coli: role of the ammonium transporter AmtB and AmtB-GlnK complex formation. J. Biol. Chem. 279:8530–8538 [DOI] [PubMed] [Google Scholar]
- 39. Javelle A, Thomas G, Marini AM, Krämer R, Merrick M. 2005. In vivo functional characterization of the Escherichia coli ammonium channel AmtB: evidence for metabolic coupling of AmtB to glutamine synthetase. Biochem. J. 390:215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blauwkamp TA, Ninfa AJ. 2003. Antagonism of PII signalling by the AmtB protein of Escherichia coli: AmtB antagonizes PII signalling. Mol. Microbiol. 48:1017–1028 [DOI] [PubMed] [Google Scholar]
- 41. Turpin DH, Parslow JS, Harrison PJ. 1981. On limiting nutrient patchiness and phytoplankton growth: a conceptual approach. J. Plankton Res. 3:421–431 [Google Scholar]
- 42. Lehman JT, Scavia D. 1982. Microscale patchiness of nutrients in plankton communities. Science 216:729–730 [DOI] [PubMed] [Google Scholar]
- 43. Seymour JR, Marcos, Stocker R. 2009. Resource patch formation and exploitation throughout the marine microbial food web. Am. Nat. 173:E15–E29 [DOI] [PubMed] [Google Scholar]
- 44. Lee PK, Dill BD, Louie TS, Shah M, VerBerkmoes NC, Andersen GL, Zinder SH, Alvarez-Cohen L. 2012. Global transcriptomic and proteomic responses of Dehalococcoides ethenogenes strain 195 to fixed nitrogen limitation. Appl. Environ. Microbiol. 78:1424–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappé MS, Short JM, Carrington JC, Mathur EJ. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309:1242–1245 [DOI] [PubMed] [Google Scholar]
- 46. Ishida ML, Assumpção MC, Machado HB, Benelli EM, Souza EM, Pedrosa FO. 2002. Identification and characterization of the two-component NtrY/NtrX regulatory system in Azospirillum brasilense. Braz. J. Med. Biol. Res. 35:651–661 [DOI] [PubMed] [Google Scholar]
- 47. Gregor J, Zeller T, Balzer A, Haberzettl K, Klug G. 2007. Bacterial regulatory networks include direct contact of response regulator proteins: interaction of RegA and NtrX in Rhodobacter capsulatus. J. Mol. Microbiol. Biotechnol. 13:126–139 [DOI] [PubMed] [Google Scholar]
- 48. Bragg JG, Hyder CL. 2004. Nitrogen versus carbon use in prokaryotic genomes and proteomes. Proc. Biol. Sci. 271:S374–S377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lv J, Li N, Niu DK. 2008. Association between the availability of environmental resources and the atomic composition of organismal proteomes: evidence from Prochlorococcus strains living at different depths. Biochem. Biophys. Res. Commun. 375:241–246 [DOI] [PubMed] [Google Scholar]
- 50. Gilbert JD, Fagan WF. 2011. Contrasting mechanisms of proteomic nitrogen thrift in Prochlorococcus. Mol. Ecol. 20:92–104 [DOI] [PubMed] [Google Scholar]
- 51. Baudouin-Cornu P, Surdin-Kerjan Y, Marlière P, Thomas D. 2001. Molecular evolution of protein atomic composition. Science 293:297–300 [DOI] [PubMed] [Google Scholar]
- 52. Bragg JG. 2011. How Prochlorococcus bacteria use nitrogen sparingly in their proteins. Mol. Ecol. 20:27–28 [DOI] [PubMed] [Google Scholar]
- 53. Mazel D, Marlière P. 1989. Adaptive eradication of methionine and cysteine from cyanobacterial light-harvesting proteins. Nature 341:245–248 [DOI] [PubMed] [Google Scholar]
- 54. Mcewan CE, Gatherer D, Mcewan NR. 1998. Nitrogen-fixing aerobic bacteria have higher genomic GC content than non-fixing species within the same genus. Hereditas 128:173–178 [DOI] [PubMed] [Google Scholar]
- 55. Button DK. 1991. Biochemical basis for whole-cell uptake kinetics: specific affinity, oligotrophic capacity, and the meaning of the Michaelis constant. Appl. Environ. Microbiol. 57:2033–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kimura M. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 57. Dufresne A, Garczarek L, Partensky F. 2005. Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 6:R14. 10.1186/gb-2005-6-2-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Partensky F, Hess WR, Vaulot D. 1999. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63:106–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ, Rappé MS. 2012. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3(5):e00252-12. 10.1128/mBio.00252-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nicastro D, Schwartz C, Pierson J, Cho J-C, Giovannoni S, McIntosh J. 2006. Three-dimensional structure of the tiny bacterium Pelagibacter ubique studied by cryo-electron tomography. Microsc. Microanal. 12(Suppl S02):180–181 [Google Scholar]
- 61. Smith DP, Kitner JB, Norbeck AD, Clauss TR, Lipton MS, Schwalbach MS, Steindler L, Nicora CD, Smith RD, Giovannoni SJ. 2010. Transcriptional and translational regulatory responses to iron limitation in the globally distributed marine bacterium Candidatus Pelagibacter ubique. PLoS One 5:e10487. 10.1371/journal.pone.0010487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tripp HJ. 2007. Genomic-assisted determination of the natural nutrient requirements of the cosmopolitan marine bacterium “Candidatus Pelagibacter ubique.” Oregon State University, Corvallis, OR [Google Scholar]
- 63. Tripp HJ, Schwalbach MS, Meyer MM, Kitner JB, Breaker RR, Giovannoni SJ. 2009. Unique glycine-activated riboswitch linked to glycine-serine auxotrophy in SAR11. Environ. Microbiol. 11:230–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carini P, Steindler L, Beszteri S, Giovannoni SJ. 2012. Nutrient requirements for growth of the extreme oligotroph “Candidatus Pelagibacter ubique” HTCC1062 on a defined medium. ISME J. 7:592–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thrash JC, Boyd A, Huggett MJ, Grote J, Carini P, Yoder RJ, Robbertse B, Spatafora JW, Rappé MS, Giovannoni SJ. 2011. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Sci. Rep. 1:13. 10.1038/srep00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Darling AE, Mau B, Perna NT. 2010. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers YH, Falcón LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter JC. 2007. The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical pacific. PLoS Biol. 5:e77. 10.1371/journal.pbio.0050077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sowell SM, Wilhelm LJ, Norbeck AD, Lipton MS, Nicora CD, Barofsky DF, Carlson CA, Smith RD, Giovanonni SJ. 2009. Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 3:93–105 [DOI] [PubMed] [Google Scholar]
- 69. Malmstrom RR, Kiene RP, Cottrell MT, Kirchman DL. 2004. Contribution of SAR11 bacteria to dissolved dimethylsulfoniopropionate and amino acid uptake in the North Atlantic Ocean. Appl. Environ. Microbiol. 70:4129–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS. 2010. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329:533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tetu SG, Brahamsha B, Johnson DA, Tai V, Phillippy K, Palenik B, Paulsen IT. 2009. Microarray analysis of phosphate regulation in the marine cyanobacterium Synechococcus sp. WH8102. ISME J. 3:835–849 [DOI] [PubMed] [Google Scholar]
- 72. Maier T, Güell M, Serrano L. 2009. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583:3966–3973 [DOI] [PubMed] [Google Scholar]
- 73. De Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. 2009. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5:1512–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vogel C, Marcotte EM. 2012. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13:227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nie L, Wu G, Brockman FJ, Zhang W. 2006. Integrated analysis of transcriptomic and proteomic data of Desulfovibrio vulgaris: zero-inflated Poisson regression models to predict abundance of undetected proteins. Bioinformatics 22:1641–1647 [DOI] [PubMed] [Google Scholar]
- 76. Nie L, Wu G, Zhang W. 2006. Correlation of mRNA expression and protein abundance affected by multiple sequence features related to translational efficiency in Desulfovibrio vulgaris: a quantitative analysis. Genetics 174:2229–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324:255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Romeo T, Vakulskas CA, Babitzke P. 2013. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ. Microbiol. 15:313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Henkin TM. 2009. Posttranscriptional regulation, p 342–356 In Schaechter M, Encyclopedia of microbiology. Elsevier Press, Oxford, United Kingdom [Google Scholar]
- 80. Meyer MM, Ames TD, Smith DP, Weinberg Z, Schwalbach MS, Giovannoni SJ, Breaker RR. 2009. Identification of candidate structured RNAs in the marine organism “Candidatus Pelagibacter ubique.” BMC Genomics 10:268. 10.1186/1471-2164-10-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Smith DP. 2011. he proteomic and transcriptomic responses to iron, sulfur, and nitrogen limitation in the abundant marine bacterium Candidatus Pelagibacter ubique. Thesis. Oregon State University, Corvallis, OR [Google Scholar]
- 82. Pan C, Oda Y, Lankford PK, Zhang B, Samatova NF, Pelletier DA, Harwood CS, Hettich RL. 2008. Characterization of anaerobic catabolism of p-coumarate in Rhodopseudomonas palustris by integrating transcriptomics and quantitative proteomics. Mol. Cell. Proteomics 7:938–948 [DOI] [PubMed] [Google Scholar]
- 83. Steglich C, Futschik ME, Lindell D, Voss B, Chisholm SW, Hess WR. 2008. The challenge of regulation in a minimal photoautotroph: non-coding RNAs in Prochlorococcus. PLoS Genet. 4:e1000173. 10.1371/journal.pgen.1000173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Assumpção MC, de Souza EM, Yates MG, de Oliveira Pedrosa F, Benelli EM. 2007. Purification and characterisation of Azospirillum brasilense N-truncated NtrX protein. Protein Expr. Purif. 53:302–308 [DOI] [PubMed] [Google Scholar]
- 85. Pawlowski K, Klosse U, de-Bruijn FJ. 1991. Characterization of a novel Azorhizobium caulinodans ORS571 two-component regulatory system, NtrY/NtrX, involved in nitrogen fixation and metabolism. Mol. Gen. Genet. 231:124–138 [DOI] [PubMed] [Google Scholar]
- 86. Walter B, Küspert M, Ansorge D, Krämer R, Burkovski A. 2008. Dissection of ammonium uptake systems in Corynebacterium glutamicum: mechanism of action and energetics of AmtA and AmtB. J. Bacteriol. 190:2611–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Meier-Wagner J, Nolden L, Jakoby M, Siewe R, Krämer R, Burkovski A. 2001. Multiplicity of ammonium uptake systems in Corynebacterium glutamicum: role of Amt and AmtB. Microbiology 147:135–143 [DOI] [PubMed] [Google Scholar]
- 88. Soupene E, Lee H, Kustu S. 2002. Ammonium/methylammonium transport (Amt) proteins facilitate diffusion of NH3 bidirectionally. Proc. Natl. Acad. Sci. U. S. A. 99:3926–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sun J, Steindler L, Thrash JC, Halsey KH, Smith DP, Carter AE, Landry ZC, Giovannoni SJ. 2011. One carbon metabolism in SAR11 pelagic marine bacteria. PLoS One 6:e23973. 10.1371/journal.pone.0023973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ames TD, Breaker RR. 2011. Bacterial aptamers that selectively bind glutamine. RNA Biol. 8:82–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ueki T, Lovley DR. 2010. Novel regulatory cascades controlling expression of nitrogen-fixation genes in Geobacter sulfurreducens. Nucleic Acids Res. 38:7485–7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Poiata E, Meyer MM, Ames TD, Breaker RR. 2009. A variant riboswitch aptamer class for S-adenosylmethionine common in marine bacteria. RNA 15:2046–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sowell SM, Abraham PE, Shah M, Verberkmoes NC, Smith DP, Barofsky DF, Giovannoni SJ. 2011. Environmental proteomics of microbial plankton in a highly productive coastal upwelling System. ISME J. 5:856–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen Y, Patel NA, Crombie A, Scrivens JH, Murrell JC. 2011. Bacterial flavin-containing monooxygenase is trimethylamine monooxygenase. Proc. Natl. Acad. Sci. U. S. A. 108:17791–17796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Robbertse B, Yoder RJ, Boyd A, Reeves J, Spatafora JW. 2011. Hal: an automated pipeline for phylogenetic analyses of genomic data. PLoS Curr. 3:RRN1213. 10.1371/currents.RRN1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17:540–552 [DOI] [PubMed] [Google Scholar]
- 98. Sassera D, Lo N, Epis S, D’Auria G, Montagna M, Comandatore F, Horner D, Peretó J, Luciano AM, Franciosi F, Ferri E, Crotti E, Bazzocchi C, Daffonchio D, Sacchi L, Moya A, Latorre A, Bandi C. 2011. Phylogenomic evidence for the presence of a flagellum and cbb3 oxidase in the free-living mitochondrial ancestor. Mol. Biol. Evol. 28:3285–3296 [DOI] [PubMed] [Google Scholar]
- 99. Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690 [DOI] [PubMed] [Google Scholar]
- 100. Livesay EA, Tang K, Taylor BK, Buschbach MA, Hopkins DF, LaMarche BL, Zhao R, Shen Y, Orton DJ, Moore RJ, Kelly RT, Udseth HR, Smith RD. 2008. Fully automated four-column capillary LC−MS system for maximizing throughput in proteomic analyses. Anal. Chem. 80:294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Maiolica A, Borsotti D, Rappsilber J. 2005. Self-made frits for nanoscale columns in proteomics. Proteomics 5:3847–3850 [DOI] [PubMed] [Google Scholar]
- 102. Kelly RT, Page JS, Luo Q, Moore RJ, Orton DJ, Tang K, Smith RD. 2006. Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal. Chem. 78:7796–7801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zimmer JS, Monroe ME, Qian WJ, Smith RD. 2006. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 25:450–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jaitly N, Mayampurath A, Littlefield K, Adkins JN, Anderson GA, Smith RD. 2009. Decon2LS: an open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics 10:87. 10.1186/1471-2105-10-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Monroe ME, Tolic N, Jaitly N, Shaw JL, Adkins JN, Smith RD. 2007. VIPER: an advanced software package to support high-throughput LC-MS peptide identification. Bioinformatics 23:2021–2023 [DOI] [PubMed] [Google Scholar]
- 106. Hess A, Iyer H. 2007. Fisher’s combined p-value for detecting differentially expressed genes using Affymetrix expression arrays. BMC Genomics 8:96. 10.1186/1471-2164-8-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Stat. Methodol. 57:289–300 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Conditions of limited nitrogen and excess nitrogen.
Changes in gene expression and their statistical significance.
Gene distribution in all strains.