

ABSTRACT
Invasive growth of the fungal pathogen Candida albicans into tissues promotes disseminated infections in humans. The plasma membrane is essential for pathogenesis because this important barrier mediates morphogenesis and invasive growth, as well as secretion of virulence factors, cell wall synthesis, nutrient import, and other processes. Previous studies showed that the Sur7 tetraspan protein that localizes to MCC (membrane compartment occupied by Can1)/eisosome subdomains of the plasma membrane regulates a broad range of key functions, including cell wall synthesis, morphogenesis, and resistance to copper. Therefore, a distinct tetraspan protein found in MCC/eisosomes, Nce102, was investigated. Nce102 belongs to the MARVEL domain protein family, which is implicated in regulating membrane structure and function. Deletion of NCE102 did not cause the broad defects seen in sur7Δ cells. Instead, the nce102Δ mutant displayed a unique phenotype in that it was defective in forming hyphae and invading low concentrations of agar but could invade well in higher agar concentrations. This phenotype was likely due to a defect in actin organization that was observed by phalloidin staining. In support of this, the invasive growth defect of a bni1Δ mutant that mislocalizes actin due to lack of the Bni1 formin was also reversed at high agar concentrations. This suggests that a denser matrix provides a signal that compensates for the actin defects. The nce102Δ mutant displayed decreased virulence and formed abnormal hyphae in mice. These studies identify novel ways that Nce102 and the physical environment surrounding C. albicans regulate morphogenesis and pathogenesis.
IMPORTANCE
The plasma membrane promotes virulence of the human fungal pathogen Candida albicans by acting as a protective barrier around the cell and mediating dynamic activities, such as morphogenesis, cell wall synthesis, secretion of virulence factors, and nutrient uptake. To better understand how the plasma membrane contributes to virulence, we analyzed a set of eight genes encoding MARVEL family proteins that are predicted to function in membrane organization. Interestingly, deletion of one gene, NCE102, caused a strong defect in formation of invasive hyphal growth in vitro and decreased virulence in mice. The nce102Δ mutant cells showed defects in actin organization that underlie the morphogenesis defect, since mutation of a known regulator of actin organization caused a similar defect. These studies identify a novel way in which the plasma membrane regulates the actin cytoskeleton and contributes to pathogenesis.
INTRODUCTION
Candida albicans is the most common cause of mucosal and systemic fungal infections in humans and is frequently responsible for hospital-acquired bloodstream infections (1, 2). The increased use of prophylactic antibacterial antibiotics, medical interventions, such as indwelling catheters, and immunosuppressive therapies to treat various conditions have provided new opportunities for this commensal organism to invade tissues and disseminate (3). An underlying factor in the pathogenesis of C. albicans arises from its ability to switch between budding yeast-form cells, which are thought to spread more easily through the bloodstream, and a hyphal morphology that facilitates the invasion of tissues (4). The plasma membrane (PM) plays key roles in pathogenesis because this essential barrier also mediates dynamic processes, including endocytosis, cell wall synthesis, secretion of virulence factors, nutrient transport, contact signaling, and sensing of the ambient osmolarity and pH. Highlighting its critical role in virulence, most of the commonly used antifungal agents affect PM functions (e.g., amphotericin, fluconazole, and caspofungin) (5).
The architecture of the fungal PM is being investigated to better define how it carries out its various roles. Recent studies have revealed that the PM of the yeast Saccharomyces cerevisiae is organized into distinct subdomains (6–8). One large domain is referred to as the MCP because it is a membrane compartment occupied by Pma1, an H+ ATPase. Proteins in the MCP diffuse rapidly, and sites of endocytosis form within this region (9, 10). The MCC, or membrane compartment occupied by Can1, corresponds to 300-nm-long furrow-like invaginations that contain several transporters, including the Can1 arginine permease (9–11). In addition, members of the Sur7 and Nce102 families of tetraspan proteins that have four membrane-spanning domains are also present. The MCC domains are very stable and are thought to act as protected regions, since they are distinct from sites of endocytosis (10, 12, 13). Associated with the cytoplasmic side of the MCC is a complex of >20 proteins known as the eisosome (14). Two important constituents are Pil1 and Lsp1, a paralogous pair of BAR domain proteins that oligomerize and promote membrane curvature to form a furrow (15, 16). Other resident proteins include the Pkh1 and Pkh2 protein kinases, which are important for regulating cell wall integrity, actin organization, and response to heat stress (17, 18). Additional PM domains are likely to exist (19, 20); however, MCC/eisosome domains are distinct in their high degree of stability and their association with membrane furrows.
Interestingly, deletion of SUR7 in C. albicans showed that the absence of this conserved tetraspan protein from the MCC caused broad defects in cell wall synthesis, morphogenesis, a 2,000-fold increase in sensitivity to copper, poor growth in macrophages, and greatly attenuated virulence in a mouse model of hematogenously disseminated candidiasis (21–23). The sur7Δ mutant also displayed an unusual phenotype in that tubes of cell wall growth extend inwards into the cell. Sur7 is not present at sites of polarized growth, indicating that it may indirectly affect morphogenesis by influencing the organization of cell polarity proteins or lipids in the plasma membrane, such as ergosterol and phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], which has been implicated in polarized growth (24, 25).
The importance of Sur7 in C. albicans suggested that another family of tetraspan proteins, represented by Nce102, may contribute to virulence. In C. albicans, in addition to the conserved Nce102 protein, this family includes seven other proteins that are not highly conserved in other fungi. S. cerevisiae Nce102 was found to localize to MCC/eisosomes and to promote proper organization of these domains, possibly by acting as a sphingolipid sensor (26, 27). In contrast, Ashbya gossypii Nce102 was not critical for eisosome stability, highlighting the importance of investigating the role of MCC/eisosome proteins in different fungi (28). Another significant aspect of Nce102 family proteins is that they contain the four-transmembrane-helix domain termed MARVEL (MAL and related proteins for vesicle trafficking and membrane link) (29). In animal cells, the MARVEL family of tetraspan proteins includes the myelin and lymphocyte (MAL), physins, gyrins, and occludin proteins. MARVEL proteins have been suggested to function where cholesterol-rich membranes become juxtaposed, such as in the formation of transport vesicles or at tight junctions (29). Although the exact function of the MARVEL proteins is not known, they carry out important roles, because mutations have been associated with serious diseases. For example, the human MARVEL proteins synaptophysin and synaptogyrin, which localize to neuronal transport vesicles, have been linked to schizophrenia (30). Mutations affecting the tight junction protein occludin have been associated with renal dysfunction and brain calcification (31).
In light of the emerging importance of both MCC/eisosomes and MARVEL domain proteins, we mutated the eight genes encoding MARVEL domain proteins in C. albicans, including NCE102. The results demonstrated that deletion of NCE102 caused a unique defect in invasive hyphal growth and reduced virulence in mice. Additionally, NCE102 is also significant because it was recently reported to be a target of a transcriptional regulatory circuit instrumental in commensal growth and virulence (32). Therefore, analysis of Nce102 identified novel ways in which C. albicans responds to its environment to undergo invasive growth and further highlights the importance of MCC/eisosomes in C. albicans, suggesting they may serve as a new target for the development of antifungal therapy.
RESULTS
Identification of eight MARVEL domain proteins in C. albicans.
Amino acid sequence comparisons revealed seven uncharacterized C. albicans proteins in addition to Nce102 that show similarity to MARVEL domain family tetraspan proteins (Fig. 1A). The similarity is low across the family, aside from the subset of residues that are found in MARVEL proteins (29). To simplify discussion of the new MARVEL genes, we named them MRV1 to MRV8 in a manner consistent with the last digit of their systematic gene names. The similarity to MARVEL domain proteins is spread throughout the four membrane-spanning domains, as shown for Nce102 in Fig. 1B. Interestingly, six of the MARVEL genes are adjacent to each other in a cluster on chromosome 5 (orf19.3902, -3, -4, -5, -6, and -8 genes). This cluster is not conserved in other Candida species, and these proteins are not highly conserved in other fungi, as is Nce102. For example, although S. cerevisiae contains two proteins highly related to C. albicans Nce102 (Ca-Nce102) (S. cerevisiae Nce102 [Sc-Nce102] and Sc-Fhn1), consistent with a gene duplication event, the two other MARVEL family proteins present in S. cerevisiae, Ynr061c and Ydl218w, are not as highly related to the Mrv proteins in C. albicans (Fig. 1A). Furthermore, Nce102 appears to be more closely related to human synaptogyrin than to the Mrv proteins. Thus, the Nce102 family represents a distinct subset of the fungal MARVEL proteins.
FIG 1 .

MARVEL domain proteins in C. albicans. (A) Cladogram of sequence relationships for MARVEL proteins from C. albicans and representatives from other species. The protein name is followed by the initials of the species in which it occurs: Ca, C. albicans; Sc, S. cerevisiae; Yl, Yarrowia lipolytica; Af, Aspergillus fumigatus; Hc, Histoplasma capsulatum; Hs, Homo sapiens; Cn, Cryptococcus neoformans. (B) Topology model of C. albicans Nce102. Black residues are conserved in the MARVEL domain. Red residues are highly conserved specifically in Nce102 proteins from different species.
The nce102Δ mutant is defective in invasive growth.
An nce102Δ mutant was analyzed to determine if there were similarities to the phenotypes caused by mutating SUR7, which encodes a distinct type of tetraspan protein with similarity to the claudin proteins that are present at tight junctions (21–23). In contrast to sur7Δ cells, which produce altered cell walls, the nce102Δ mutant showed normal cell wall staining with calcofluor white and did not display significantly increased sensitivity to a variety of drugs and conditions that exacerbate cell wall defects. However, the nce102Δ strain was similar to the sur7Δ strain in that it displayed decreased invasion of agar (Fig. 2A). When it was spotted onto medium containing serum and 1.5% agar, hyphal growth did not emanate from the nce102Δ mutant into the surrounding agar as it did for the wild-type control strain, the mrv1Δ mutant, and a deletion mutant lacking the cluster of MRV genes on chromosome 5 (mrv2-mrv8Δ). Construction of double mutants and an octuple mutant lacking all eight MARVEL genes did not uncover other phenotypes. Thus, Nce102 plays a distinct role in invasive growth.
FIG 2 .
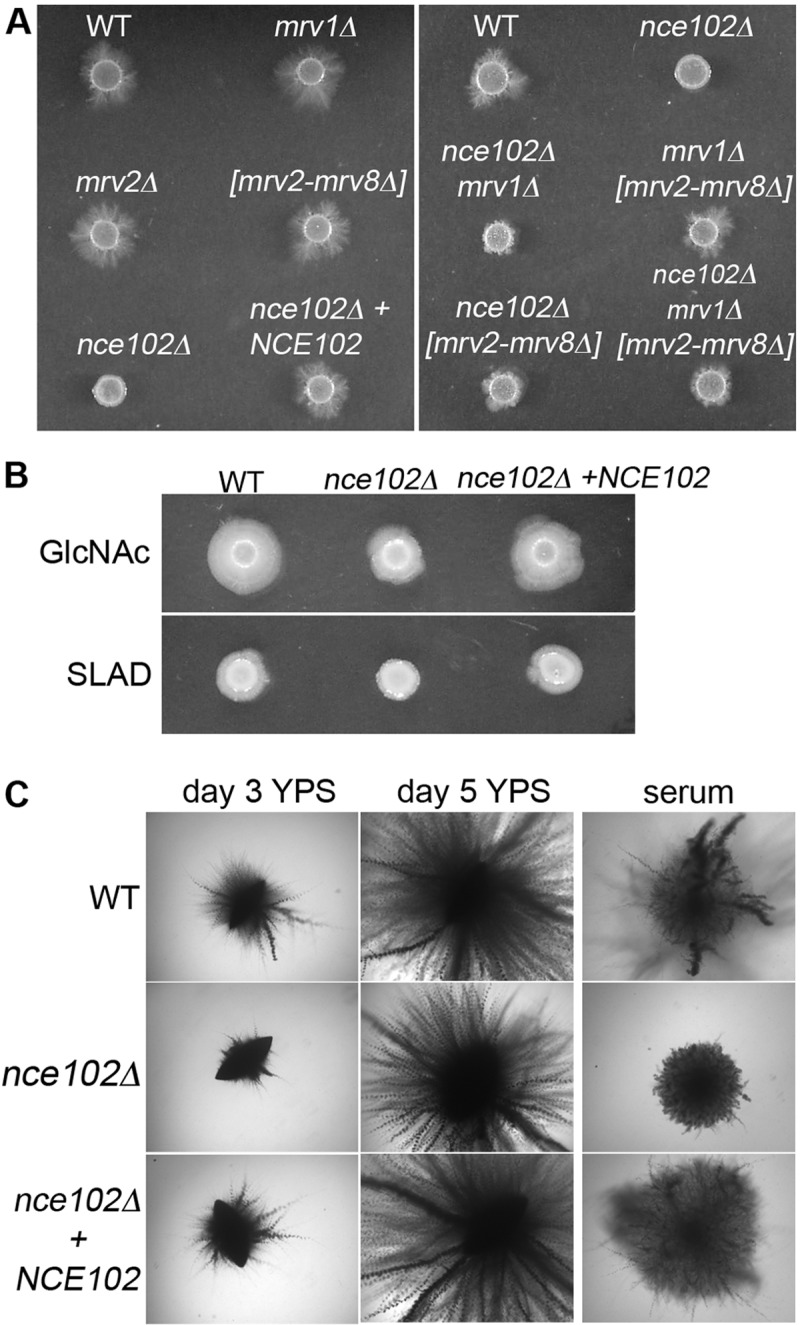
Deletion of NCE102 in C. albicans causes a defect in invasive growth under different hypha-inducing conditions. (A) C. albicans strains deleted for the indicated MARVEL family genes were spotted onto solid medium containing 4% bovine calf serum and 1.5% agar and then incubated at 37°C for 7 days. The nce102Δ strain exhibits the strongest invasion defect. (B) Strains were spotted onto 2.5 mM GlcNac and synthetic low-ammonia dextrose (SLAD) medium, both containing 1.0% agar, and incubated at 37°C for 7 days. (C) Indicated strains were embedded in yeast extract-peptone-sucrose (YPS) medium, grown for 3 or 5 days at 25°C, and compared to strains embedded in 4% bovine calf serum containing 1.5% agar. The nce102Δ strain embedded in YPS appears similar at day 5 to the wild type and the complemented strain to which a wild-type copy of NCE102 was restored. Strains used were wild-type DIC185 (WT), an nce102Δ mutant (YHXW14), an nce102Δ + NCE102 strain (YLD78-4-2-1), an orf19.4691Δ strain (YLD93-6-17-1), an orf19.3902Δ strain (YLD71-11-6-1), an orf19.3902-orf19.3908Δ strain (YLD82-4-13-1), an nce102Δ 4691Δ strain (YLD125-P), and 4691Δ 3902-3908Δ strain (YLD107-P), and an nce102Δ 3902-3908Δ 4691Δ strain (YLD124-P).
The nce102Δ mutant was also defective in invasive growth when spotted onto other types of media, including GlcNAc and low-ammonia medium (synthetic low-ammonia dextrose [SLAD]) (Fig. 2B), although the defect with serum agar appeared to be the strongest. Interestingly, the nce102Δ mutant showed only a delay in invading when embedded in yeast extract-peptone-sucrose (YPS) medium at room temperature but was strongly defective when embedded in agar containing 4% serum at 37°C (Fig. 2C).
The nce102Δ mutant invades well in a denser agar matrix.
The mutants were tested for invasion of a higher concentration of agar (4%) to determine if a stronger matrix would reveal phenotypes for the mrvΔ mutants. Previous studies with septin mutants demonstrated that their defect in invasion was worse at higher concentrations of agar, consistent with greater difficulty invading a denser matrix (33). No significant defects were detected for the mrvΔ mutants spotted onto 4% agar (Fig. 3A). Unexpectedly, the nce102Δ mutant invaded 4% agar better than it did 1.5% agar. To assess this systematically, nce102Δ and control strains were embedded in different concentrations of agar, between 0.5% and 6% (Fig. 3B). Interestingly, the nce102Δ mutant showed a strong defect when embedded in concentrations of agar below 2% but invaded well in higher concentrations of agar. Similar results were obtained with different batches of agar and also with purified agarose (not shown). Other mutants that have been reported to be defective in invasive growth did not show a similar phenotype, since they invaded serum agar well at both high and low agar concentrations (Fig. 3C). These mutants presumably failed to show a significant invasion defect with serum agar at 37°C, because they were initially identified as being defective under other conditions that are weaker inducers of invasive growth (34–37). Thus, nce102Δ displays a unique phenotype in that it is defective in invading low concentrations of agar but gets a compensating signal from a denser agar matrix that enables it to invade better.
FIG 3 .

C. albicans nce102Δ
invasive growth phenotype is suppressed at higher concentrations of agar. (A) Cultures of strains deleted for C. albicans MARVEL family genes were spotted onto 4% bovine calf serum containing 4% agar and incubated at 37 C for 7 days. (B) Indicated strains were embedded in increasing concentrations of agar containing 4% bovine calf serum and incubated at 37°C for 5 days. When embedded in 4% and 6% agar, the nce102Δ mutant appears similar to the wild-type and complemented strains. (C) Hyphal signal transduction pathway mutant strains were embedded in 4% bovine calf serum containing 1.5% agar or 6.0% agar and incubated at 37°C for 5 days. Only the nce102Δ mutant exhibited a defect in hyphal growth when embedded in 1.5% agar. Strains used included the wild-type, DIC185, nce102Δ (YHXW14), nce102Δ + NCE102 (YLD78-4-2-1), orf19.4691Δ (YLD93-6-17-1), orf19.3902Δ (YLD71-11-6-1), orf19.3902-orf19.3908Δ (YLD82-4-13-1), nce102Δ 4691Δ (YLD125-P), 4691Δ 3902-3908Δ (YLD107-P), nce102Δ 3902-3908Δ 4691Δ (YLD124-P), cph1Δ (SN180), mkc1Δ (DSY3332), and msb2Δ (CAGC056) strains.
Nce102 plays a minor role in regulating C. albicans MCC/eisosomes.
Nce102 was found to localize to MCC/eisosomes in S. cerevisiae and was proposed to affect the formation of these domains by regulating the Pkh1/2 kinases in response to sphingolipid levels (13, 27). In C. albicans cells that were grown to saturation, we found that Nce102-green fluorescent protein (GFP) localized to punctate domains that colocalized with Sur7-GFP and Lsp1-GFP (Fig. 4A and B). However, in contrast to the case with S. cerevisiae, Nce102-GFP in log-phase cells was enriched in MCC/eisosomes and was not restricted to these domains (Fig. 4C). As cells entered stationary phase, Nce102-GFP levels increased about 2-fold and Nce102-GFP was more highly restricted to punctate MCC/eisosome domains. The fact that Nce102-GFP was not restricted to MCC/eisosomes in log-phase cells made it difficult to determine whether blocking sphingolipid synthesis with myriocin would cause it to move out of MCC/eisosomes, as occurs in S. cerevisiae (27). Deletion of NCE102 from C. albicans caused about a 2-fold decrease in MCC/eisosomes (Fig. 4D), similar to the case with an Sc-nce102Δ mutant. However, the C. albicans nce102Δ cells showed typical punctate localization of Lsp1-GFP and Sur7-GFP, whereas these proteins are partially mislocalized in an Sc-nce102Δ mutant (13, 27).
FIG 4 .

Nce102-GFP localizes to MCC/eisosomes in saturated cultures. (A) Cells engineered to produce the indicated GFP fusion protein were imaged by deconvolution microscopy. Top and midsection images of the indicated cell types are shown to demonstrate the punctate pattern of MCC/eisosomes. (B) Cells showing the colocalization of Nce102-GFP with Lsp1-RFP in a top view of the cell or with Sur7-GFP in a midsection of the cell. (C) Localization patterns for the indicated GFP fusion protein in cells from log-phase (top panels) and saturated (bottom panels) cultures. Note that Nce102-GFP is more punctate in saturated cultures. (D) Punctate localization of Lsp1-GFP and Sur7-GFP in nce102Δ cells. Strains expressed the following fusion genes in a wild-type strain: NCE102-GFP (YHXW15), LSP1-GFP (YLD76-1), SUR7-GFP (YHXW4), NCE102-GFP LSP1-RFP (YLD168-4), and NCE102-GFP SUR7-RFP (YLD138-17). The nce102Δ LSP1-GFP strain was YLD84-1, and the nce102Δ SUR7-GFP strain was YHXW-16.
A potential consequence of deleting NCE102 is to alter the function of the Pkh kinases. In S. cerevisiae, Pkh1 and its redundant paralog Pkh2, homologs of the mammalian 3-phosphoinositide-dependent kinase, localize to eisosomes (27, 38). They affect diverse functions, including cell wall synthesis, since they are regulators of the Pkc1, Ypk1, and Sch9 protein kinases. C. albicans has an ortholog of Pkh1 and the more distantly related Pkh3 but not Pkh2. Deletion of PKH1 did not cause a defect in invasive growth, even though the pkh1Δ mutant cells were more sensitive to SDS and Congo red, which is typical for mutants with cell wall defects (Fig. 5A and B). It was interesting that pkh1∆ was viable, since in S. cerevisiae deletion of both PKH1 and PKH2 is lethal (39). The pkh3∆ mutant was sensitive to SDS, although the phenotype was weaker than that for pkh1∆. Deletion of one copy of PKH3 from the pkh1∆ mutant caused a further increase in sensitivity to SDS and Congo red, but this mutant still invaded well (Fig. 5A and B). We failed to isolate a pkh1∆ pkh3∆ double mutant, suggesting it may be lethal. Altogether, these results suggest that nce102Δ does not affect invasive growth by decreasing the function of the Pkh kinases.
FIG 5 .
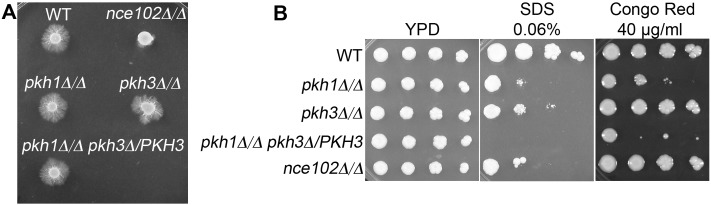
C. albicans PKH
kinase mutants do not exhibit an invasive hyphal growth defect. (A) Indicated strains were spotted onto 4% BCS containing 1.5% agar and incubated at 37°C for 7 days. (B) Serial dilutions of the indicated strains were spotted onto YPD agar medium, YPD containing 0.06% SDS, and YPD containing 40 µg/ml Congo red and then incubated at 30°C for 48 h. The pkh1Δ mutant strain carrying only one copy of PKH3 demonstrated greater sensitivity to both cell wall-damaging agents, SDS and Congo red, than either the pkh1Δ or pkh3Δ single mutant strain. Strains used included the wild type, DIC185, and the nce102Δ (YHXW14), pkh1Δ (YLD91-1-9-1), pkh3Δ (YLD95-4-5-2), and pkh1Δ pkh3Δ/PKH3 (YLD146-6-54-1) strains.
The nce102Δ mutant is defective in hyphal morphogenesis.
A time course assay to examine the nce102Δ defect in invasive growth showed that 7 h after being spotted onto a serum agar plate, the nce102Δ mutant had formed fewer hyphae than the wild type, and the hyphae that were present were shorter (Fig. 6A). After 17 h, the nce102Δ mutant had mostly reverted to budding, as only a few short hyphae were evident. The ability to form hyphae in liquid culture without an agar matrix was examined by incubating cells with different concentrations of serum. Whereas wild-type cells began forming filamentous cells with 1% serum and most cells were hyphal with 10% serum, the nce102Δ mutant did not form a significant number of hyphal cells until it was exposed to 30% serum (Fig. 6B). Since adenylyl cyclase and its regulators Ras1 and Csc25, which induce hyphal growth, are PM proteins that could potentially be affected by deletion of NCE102, we tested the ability of cAMP to directly stimulate hyphal growth (Fig. 6C). Although the cell-permeating analog dibutyryl-cAMP (db-cAMP) stimulated hyphal formation in the wild type as expected, the nce102Δ mutant was not induced to form hyphae. Since db-cAMP can bypass adenylyl cyclase to stimulate hyphal formation, these results indicate that nce102Δ affects a distinct aspect of hyphal induction. Interestingly, the nce102Δ mutant was not suppressed by overexpression of 10 different protein kinase genes that are involved in hyphal signaling, including TPK1, TPK2, CEK1, HST7, STE11, MKC1, PKC1, HOG1, CEK2, and CST20 (not shown). This overexpression approach was used previously to determine that Cek1 and Mkc1 act downstream of Rac1 in invasive growth (40). These results suggested that the invasive growth defect of nce102Δ cells was likely due to another aspect of hyphal growth.
FIG 6 .

The C. albicans nce102Δ strain exhibits defects in hyphal initiation and maintenance. (A) The wild-type, DIC185, and nce102Δ (YHXW14) strains were diluted and spotted onto the surface of a solid 4% bovine calf serum plate containing 1.5% agar. The plate was incubated at 37°C and photographed at 7 and 17 h. (B) Log-phase cultures were diluted to 1 × 106 cells/ml in 0, 1, 3, 10, and 30% bovine calf serum, incubated at 37° C for 1.75 h, and examined for hyphal development. In contrast to the wild-type (DIC185) cells, nce102Δ (YHXW14) cells exhibited hyphal growth only in 30% BCS. (C) Log-phase cells were mixed with either sterile deionized H2O or dibutyryl-cAMP (db-cAMP), final concentration of 30 mM, and incubated at 37° C for 5 h. In contrast to those of the wild type, nce102Δ cells failed to develop hyphal filaments when incubated in db-cAMP.
Deletion of NCE102 causes a defect in actin organization.
Actin localization was investigated in the nce102Δ mutant, since it is important for normal morphogenesis and it is also implicated in hyphal signaling (41, 42). Interestingly, phalloidin staining revealed that nce102Δ mutants have a defect in actin organization (Fig. 7A). There were fewer actin cables emanating from the leading edge of growth back into the mother cells, and the cables that were present appeared smaller and less distinct. Although actin patches were primarily localized to the bud in nce102Δ cells, some patches were also detected in the mother cells. The nce102Δ cells were rounder than those of the wild type (Fig. 7B), which is also an indicator of an actin defect (43, 44). In contrast, nce102Δ cells growing invasively into 4% agar showed actin organization similar to that of the wild type (Fig. 7E), as judged by analysis of strains engineered to produce the actin-binding protein LIFEACT-GFP (45).
FIG 7 .
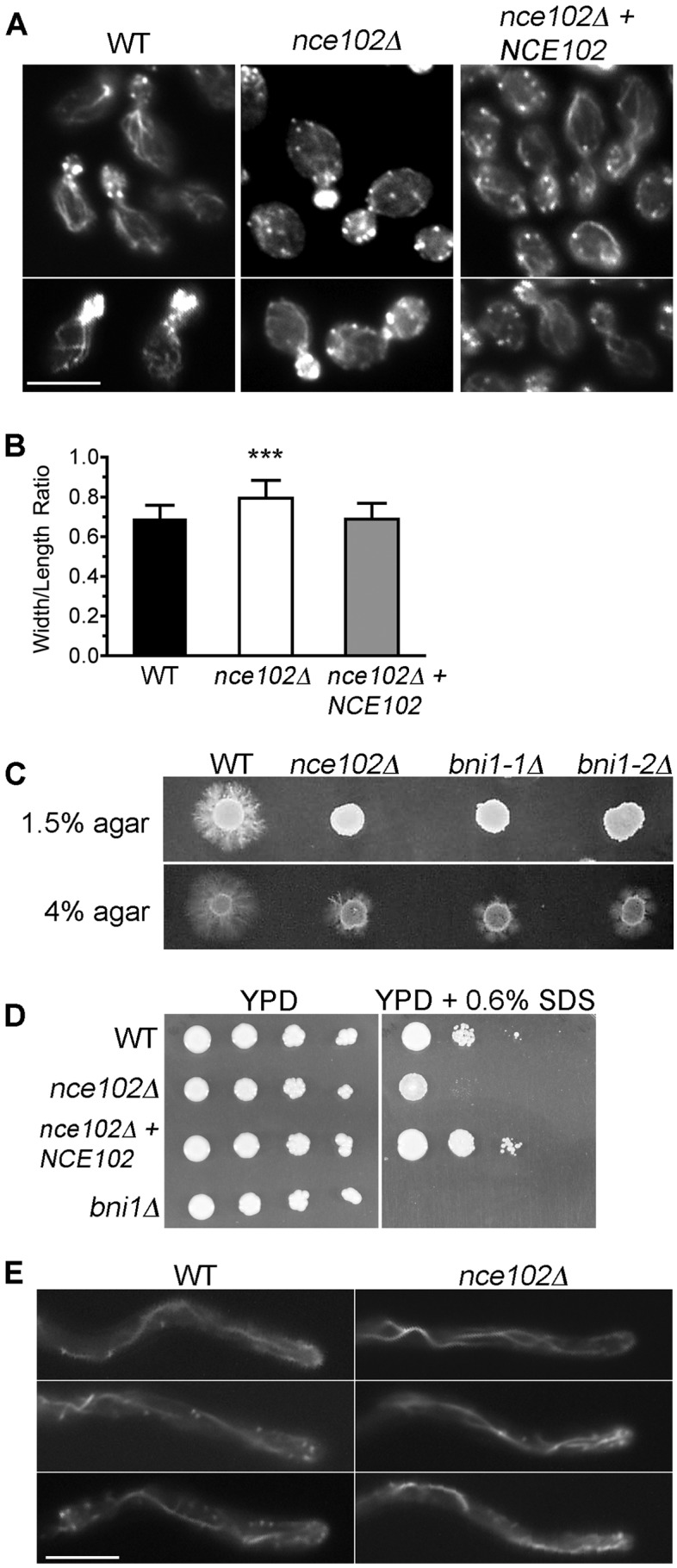
The nce102Δ mutant displays actin mislocalization and an invasive growth phenotype, similar to the bni1Δ formin mutant. (A) Log-phase cells of the indicated strains were fixed with formaldehyde and then stained with rhodamine-phalloidin to detect the localization of actin patches and cables. (B) The width/length ratio of 100 mother cells of each indicated strain was determined. A significant difference in the width/length ratio of nce102Δ yeast cells from those of the wild type and complemented strains was observed (P < 0.001). (C) Two independent bni1Δ isolates were spotted onto solid 4% bovine calf serum medium containing either 1.5% agar or 4.0% agar and incubated at 37° C for 7 days. A high agar concentration rescued the hyphal defect of both the nce102Δ and bni1Δ mutant strains. (D) Serial dilutions of the indicated strains were spotted onto YPD agar medium in the absence or presence of 0.06% SDS and then incubated at 30°C for 48 h. Strains used were the wild type (DIC185) and the nce102Δ (YHXW14), complemented nce102Δ + NCE102 (YLD78-4-2-1), and bni1Δ (YLD164-4) strains. (E) Actin localization in hyphal filaments invading 4% serum agarose. Actin was visualized by introducing LIFEACT-GFP under control of the TDH3 promoter into wild-type and nce102Δ cells.
To further assess the effects of actin mislocalization, a bni1Δ mutant, which lacks the Bni1 formin protein that promotes actin filament formation, was examined. bni1Δ mutants are similar to nce102Δ mutants in that they mislocalize actin, and they have a defect in hyphal growth that is not rescued by addition of exogenous cAMP (43). Interestingly, we found that the bni1Δ mutant was also similar to the nce102Δ mutant in failing to invade a low concentration of agar (1.5%) but invading well in a higher concentration (4%) (Fig. 7C). In addition, the bni1Δ and nce102Δ mutants were both sensitive to SDS (Fig. 7D). These similarities indicate that the defect in actin organization underlies the failure of nce102Δ mutants to undergo invasive hyphal growth into low concentrations of agar.
The nce102Δ mutant is less virulent and forms abnormal hyphae in vivo.
The nce102Δ mutant was examined using a mouse model of hematogenously disseminated candidiasis in which BALB/c mice were infected via the lateral tail vein. Mice infected with either the wild type or the complemented control strain succumbed rapidly to infection, since most were moribund by day 2 (Fig. 8A). In contrast, the median time for mice infected with the nce102Δ mutant cells to become moribund was 7 days. There was no significant difference in the fungal load in the kidneys of moribund mice, since the numbers of CFU/g were essentially the same (Fig. 8B). These data suggest that although it takes longer for disease to develop, the nce102Δ mutant can grow to high levels in vivo. However, the distribution of fungal cells in the kidney was different in the moribund mice. Histological analysis revealed that wild-type cells were disseminated in foci around the cortex of the kidney, whereas the nce102Δ mutant cells were found primarily in the region of the kidney that exits to the bladder (Fig. 8C). This difference may be due to the different length of time it takes for the infection to progress (46). Interestingly, nce102Δ cells recovered from kidney homogenates showed a wide degree of morphological variation compared to those of the wild type (Fig. 8C). Whereas the wild-type cells were found primarily as long hyphae with parallel walls, the nce102Δ cells were frequently wider and more rounded, indicating a defect in hyphal formation in vivo similar to what was seen in vitro.
FIG 8 .

Attenuated virulence of the C. albicans nce102Δ mutant in a mouse model of hematogenously disseminated candidiasis. BALB/c mice were injected via the lateral tail vein with 1 × 106 cells. (A) Survival curves of mice infected with the nce102Δ (YHXW14), wild-type (DIC185), and complemented nce102Δ + NCE102 (YLD78-4-2-1) strains. (B) Determination of CFU per gram of kidney tissue in mice that were euthanized when they became moribund or at the end of the experiment on day 28, as indicated in panel A. (C) (Left panels) Kidneys were excised from mice infected with the wild-type (DIC185) and complemented nce102Δ + NCE102 (YLD78-4-2-1) strains for 2 days, and a mouse was infected with the nce102Δ (YHXW14) strain that became moribund after 12 days. Kidney sections were stained by the periodic acid-Schiff staining (PAS) staining method, which stains the C. albicans cells a dark magenta color (see the arrows for examples). Kidney tissue infected with the wild-type or complemented strains demonstrated a high level of penetration by hyphal filaments emanating from multiple sites in the outer cortex. In contrast, nce102Δ cells were mostly restricted to the region of the kidney that exits to the bladder (right panels). Kidney tissue homogenates were stained with calcofluor white and then examined by fluorescence microscopy using a 100× objective. The nce102Δ mutant cells displayed a more pseudohyphal morphology than either the wild-type or complemented strain.
DISCUSSION
nce102Δ causes a novel invasive growth phenotype.
MCC/eisosomes are under investigation to identify novel ways in which the PM contributes to virulence. Nce102 was targeted because it localizes to these punctate domains in S. cerevisiae and because Sur7, a distinct tetraspanner that localizes to MCC/eisosomes, is important for C. albicans virulence due to its broad roles in morphogenesis, cell wall synthesis, PM organization, and resistance to copper (23). However, the phenotypes caused by deletion of NCE102 in C. albicans were distinct from those caused by deleting SUR7. The nce102Δ mutant cells displayed the novel phenotype of failing to invade low concentrations of agar but invading well in higher concentrations. Nce102 is also important for pathogenesis of C. albicans, since the nce102Δ mutant showed reduced virulence in mice (Fig. 8). The nce102Δ mutant cells formed hyphae with altered morphology in vivo, which likely diminishes their ability to grow invasively and may also make them more susceptible to recognition and attack by the immune system. Consistent with this, a separate study found that there were fewer CFU/g of the nce102Δ mutant in the kidney when the nce102Δ cells were infected as part of a pool of mutants (32). This study by Perez et al. also provided further support for the significance of Nce102 in virulence, because they identified NCE102 as a key target of a transcriptional regulatory circuit that is important for both commensal and pathogenic growth of C. albicans (32).
Actin mislocalization underlies nce102Δ invasive growth defect.
The novel invasive growth defect of nce102Δ cells is different from the phenotypes caused by mutating the signal pathways that stimulate hyphal growth (Fig. 3C). Furthermore, hyphal growth of nce102Δ cells was not rescued by addition of db-cAMP, indicating that altered function of adenylyl cyclase and its regulators in the PM was not the cause (Fig. 6C). Similarly, nce102Δ was not suppressed by overexpression of 10 different protein kinase genes that are involved in downstream aspects of hyphal signaling (40). Deletion analysis of PKH1 and PKH3, which encode two protein kinases that are localized to MCC/eisosomes in S. cerevisiae, did not implicate these important regulators in the nce102Δ phenotype (Fig. 5). These results suggested that actin might be altered in the nce102Δ cells, rather than cell signaling proteins, since actin has complex roles in hyphal induction. Actin cables are important for transporting secretory vesicles to the apex of growth in buds or hyphae to promote polarized morphogenesis. In addition, actin filaments are important for induction of hypha-specific genes (42, 47). Further evidence for a role of actin dynamics in hyphal induction comes from the discovery that G-actin forms a complex with adenylyl cyclase (Cyr1) and Srv2 that is required for activation of cAMP production (41).
Phalloidin staining revealed that nce102Δ cells display altered actin localization (Fig. 7). In particular, actin cables were not readily detected as they were in wild-type cells. Actin patches were localized primarily to the buds, but some mislocalization to the mother cells was evident. In contrast, nce102Δ hyphae growing invasively into 4% agar displayed obvious actin cables, similar to the wild type. The link between actin organization and the nce102Δ phenotype was strengthened by detection of a similar invasive growth defect caused by deletion of BNI1, which encodes a formin protein that promotes actin polymerization. The bni1Δ mutant cells failed to invade low concentrations of agar (1.5%) but invaded well in 4% agar (Fig. 7). This phenotype is indicative of a special kind of actin defect, since the rvs161Δ (48), rvs167Δ (48), and arp2Δ (49) mutants did not invade well in low or high concentrations of agar (not shown).
Nce102 appears to carry out a role in actin organization that is different from that of Bni1, since the details of their mutant phenotypes are distinct. The nce102Δ mutant cells can form normal-looking hyphae if given a higher dose of serum than is required for the wild type (Fig. 6). The bni1Δ mutant can initiate germ tube formation at doses similar to those for the wild type but does not maintain the highly polarized state and instead forms wider hyphae (43, 44). The sur7Δ mutant also has defects in actin localization and invasive growth, indicating that other components of MCC/eisosomes can influence actin organization (21). However, it is not clear how Sur7 influences actin, since MCC/eisosomes are distinct from sites of actin localization. In contrast, Nce102 was not restricted to MCC/eisosomes in log-phase cells (Fig. 4) and could therefore signal to actin from outside the MCC.
Role of Nce102 in PM organization.
Deletion of NCE102 caused about a 2-fold decrease in eisosome formation in C. albicans. Other aspects of PM organization seemed normal in the nce102Δ mutant, since we did not observe the extended invaginations of the cell wall or mislocalization of septin proteins that were seen in the sur7Δ cells. Thus, the effects of Nce102 appear relatively specific to actin organization. In contrast, an S. cerevisiae nce102Δ mutant displayed a stronger defect in that it also showed more diffuse localization of Sur7-GFP, which is one of the most stable MCC components, as well as altered localization of Pil1-GFP, Can1-GFP, and ergosterol (13, 27). Actin localization was not investigated in the Sc-nce102Δ mutant, but it is interesting that NCE102 expression is upregulated during invasive growth (50). Other studies revealed that an nce102Δ mutant of Schizosaccharomyces pombe produced fewer eisosomes and shorter versions of the long eisosomes typically formed in this species (51), and an nce102Δ mutant in Aspergillus fumigatus was defective in spore production (52). However, deletion of NCE102 from A. gossypii did not cause a significant defect in eisosome production (28), indicating that Nce102 function varies in different organisms.
MARVEL domain proteins.
MARVEL domain proteins comprise a subset of tetraspan proteins that carry out key functions in a wide range of cell types. Their mutation has been implicated in human diseases, such as schizophrenia and inflammation (29). One of their hallmarks is that MARVEL proteins are frequently identified at sites of membrane apposition. For example, synaptogyrin is found at sites of various stages of the synaptic vesicle cycle, including vesicle biogenesis, exocytosis, and endocytic recycling. Other MARVEL proteins, such as occludin, are found at tight junctions. A common aspect of many of the MARVEL proteins in animal cells is that they are present in cholesterol-rich raft regions and are therefore thought to be responsible for bringing specialized membrane domains into apposition. Interestingly, MCC/eisosome regions are thought to be enriched in ergosterol (53). If Nce102 is similarly involved at sites of membrane apposition, the most likely possibility is that it mediates an interaction across the PM furrows that were discovered at MCC/eisosomes in S. cerevisiae (11). This model could also explain why Nce102 influences the formation of the membrane furrows in S. cerevisiae (26, 27).
It was surprising that deletion of the other 7 MARVEL genes in C. albicans did not cause a readily detectable phenotype. Systematic gene deletion studies in S. cerevisiae also have not identified strong phenotypes for mutation of the two uncharacterized MARVEL genes, YNR061c and YDL281w. Interestingly, the MARVEL domain proteins in human cells that have been studied have not been reported to be essential for viability but do play important regulatory roles. For example, mice lacking occludin, the first integral membrane protein found at tight junctions, formed morphologically and functionally normal tight junctions, although they showed altered permeability (54, 55). Mice lacking the synaptic vesicle protein synaptogyrin did not show striking phenotypes (56), likely due to genetic redundancy, but mutations in the synaptogyrin 1 gene have been implicated in schizophrenia (30). Mutation of MAL in mice causes defects in protein sorting in the PM (57). Although the specific functions of these MARVEL proteins are not known, analysis of Nce102 in C. albicans suggests that some of the functions of the other MARVEL proteins could be mediated through effects on actin. In this regard, it is interesting that occludin promotes proper actin localization at tight junctions (55, 58). These similarities also suggest that MCC/eisosomes in C. albicans could be targeted for antifungal therapy in a manner analogous to the way in which drugs that alter tetraspan proteins in tight junctions and other microdomains in human cells are being identified (59, 60).
MATERIALS AND METHODS
Strains and media.
The C. albicans strains used in this study (Table 1) were grown in rich yeast extract-peptone-dextrose (YPD) medium with uridine (80 mg/ml) or in synthetic medium made with yeast nitrogen base (YNB) and lacking the appropriate nutrients.
TABLE 1 .
C. albicans strains used in this study
| Strain | Parent | Genotype | |
|---|---|---|---|
| BWP17 | Sc5314 | ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| DIC185 | BWP17 | ura3Δ::λimm434/URA3 his1::hisG/HIS1 arg4::hisG/ARG4 | |
| YHXW14 | YHXW13 | nce102∆::ARG4/nce102∆::HIS1 URA3/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD78-4-2-1 | YHXW13 | nce102∆::ARG4/nce102∆::HIS1 NCE102::URA3 ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD71-11-6-1 | YLD68-11-6 | orf19.3902∆::ARG4/orf19.3902Δ::HIS1 URA3/ ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD82-4-13-1 | YLD81-4-13 | orf19.3902-orf19.3908∆::ARG4/orf19.3902-orf19.3908Δ::HIS1 URA3/ ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD93-6-17-1 | YLD90-6-17 | orf19.4691∆::ARG4/orf19.4691Δ::HIS1 URA3 / ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD107-P | YLD104-P | nce102∆::ARG4/nce102∆::HIS1 orf19.3902-orf19.3908Δ/orf19.3902Δ-orf19.3908∆ URA3/ ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD125-P | YLD123-P |
nce102∆::ARG4/nce102∆::HIS1 orf19.4691Δ/orf19.4691Δ URA3/ ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/ arg4::hisG |
|
| YLD122-P | YLD118-P |
orf19.4691Δ::ARG4/orf19.4691Δ::HIS1 orf19.3902-orf19.3908Δ/ orf19.3902-orf19.3908Δ URA3/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG |
|
| YLD124-P | YLD120-P |
nce102∆::ARG4/nce102∆::HIS1 orf19.3902-orf19.3908Δ/orf19.3902Δ-orf19.3908Δ orf19.4691Δ/orf19.4691Δ URA3/ ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG |
|
| YLD91-1-9-1 | YLD89-1-9 | pkh1Δ::ARG4/pkh1Δ::HIS1 URA3/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD95-4-5-2 | YLD92-4-5 | pkh3Δ::ARG4/pkh3Δ::HIS1 URA3/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YLD146-6-54-1 | YLD135-6-54 | pkh1Δ::ARG4/pkh1Δ::HIS1 pkh3Δ URA3/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| YHXW15 | BWP17 | ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG NCE102-GFPγ::URA3 | |
| YLD76-1 | BWP17 | ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG LSP1-GFPγ::URA3 | |
| YLD84-1 | YHXW13 |
nce102∆::ARG4/nce102∆::HIS1 ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG LSP1-GFPγ::URA3 |
|
| YLD138-17 | YHXW15 |
ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG NCE102-GFPγ::URA3 SUR7-RFP::ARG4 |
|
| YLD168-4 | LLF018A |
ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG LSP-RFP::ARG4 NCE102-GFPγ::URA3 |
|
| YHXW-4 | BWP17 | ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG SUR7-GFPγ::URA3 | |
| YHXW-16 | YHXW-13 |
nce102∆::ARG4/nce102∆::HIS1 ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG SUR7-GFPγ::URA3 |
|
| YLD164-4 | YSM48-1 | bni1Δ::ARG4/bni1Δ::HIS1 URA3/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| SN180 | SN152 | cph1Δ::LEU2/cph1Δ::HIS1 ura3Δ::λimm434/ura3Δ::λimm434 his1::hisG/his1::hisG arg4::hisG/arg4::hisG | |
| DSY3332 | mkc1Δ::UAU/mkc1::URA3 his1::hisG/his1::hisG leu2/leu2 arg4/arg4 | ||
| CAGCO56 | msb2Δ::UAU/msb2::URA3 his1::hisG/his1::hisG leu2/leu2 arg4/arg4 | ||
Homozygous gene deletion mutant strains were constructed with strain BWP17 (61) by the sequential deletion of both copies of the targeted gene (C. albicans is diploid). Gene deletion cassettes were generated by PCR amplification of the ARG4 or HIS1 selectable marker gene, using primers that also included 80 bp of DNA sequence homologous to the upstream or downstream region of the targeted open reading frame. Cells that had undergone homologous recombination to delete the targeted gene were identified by PCR analysis using a combination of primers flanking sites of cassette integration and internal primers. A reconstituted strain, in which the nce102Δ mutation was complemented by the reintroduction of a wild-type copy of NCE102, was constructed by using PCR to amplify the entire open NCE102 reading frame plus ~1,000 bp upstream and 300 bp downstream and then cloning it between the SacI and SacII restriction sites in the plasmid pDDB57. The plasmid was linearized by restriction digestion in the promoter region and then transformed into the nce102Δ strain using URA3 for selection.
Homozygous double and triple deletion mutation strains were constructed by sequential deletion of both copies of the targeted gene using the SAT1 flipper method (62). Gene deletion cassettes containing a nourseothricin resistance marker CaSAT1, and a modified flippase gene, CaFLP, were generated by PCR amplification using primers that also included 80 bp of DNA sequence homologous to the upstream and downstream regions of the targeted open reading frame. The appropriate mutants were identified by PCR analysis using a combination of primers outside the sites of cassette integration and internal primers. Deletion strains used in all assays were transformed with a URA3-containing fragment to complement the remaining auxotrophy. The plasmid pBSK-URA3 was digested with the restriction enzymes Pst1 and Not1 to liberate the URA3-IRO1 sequence, which was then transformed into the mutant strains for integration by homologous recombination to restore URA3 at its native locus.
C. albicans strains expressing NCE102-GFP, SUR7-GFP, or LSP1-GFP were constructed by homologous recombination of GFP sequences into the 3′ end of the open reading frame of the designated gene. In brief, PCR primers containing ~70 bp of sequence homologous to the 3′ end of the open reading frame were used to amplify a cassette containing a version of enhanced GFP (C. albicans GFPγ) and a URA3 selectable marker (63). This cassette was transformed into either C. albicans wild-type strain BWP17 or the nce102Δ mutant, and the Ura+ colonies were screened to identify those carrying the GFP fusion protein. Cells carrying NCE102-GFP were also tagged with red fluorescent protein (RFP) sequences in the 3′ end of either the SUR7 or LSP1 open reading frame. Primers containing ~70 bp of sequence homologous to the 3′ end of the SUR7 or LSP1 open reading frame were used to amplify a cassette containing a cherry version of RFP (64) and an ARG4 selectable marker that was transformed into an NCE102-GFP fusion strain. To visualize actin, PCR primers were used to amplify a cassette containing the LIFEACT-GFP gene and URA3 that was optimized for C. albicans (49), which was then introduced into the genome by homologous recombination to place the fusion gene under control of the TDH3 promoter.
Hyphal growth assays.
Invasive growth of C. albicans strains in agar was assayed by spotting 2 µl of a 1 × 107-cells/ml dilution of an overnight YPD culture grown at 30°C onto the surface a semisolid medium containing 4% bovine calf serum (BCS) and the indicated concentrations of agar. Plates were incubated at 37°C for 7 days and then photographed. To assess invasive growth under embedding conditions, C. albicans strains were grown overnight in YPD at 30°C, diluted to 4 × 104 cells/ml in YPD, and incubated at 30 C for 4 to 5 h. Approximately 200 cells were then mixed with melted and cooled yeast extract-peptone-sucrose (YPS) medium containing 1.6% agar and incubated at 25°C for 3 or 5 days or mixed with melted and cooled 4% BCS containing 0.5, 1.5, 2, 4, or 6% agar and incubated for 5 days at 37°C. To assess hyphal growth in liquid, log-phase cells were diluted to 1 × 106 cells/ml in 0, 1, 3, 10, and 30% BCS and incubated for 1.75 h at 37°C. Microscopic images were then captured using an Olympus BH2 microscope equipped with a Zeiss AxioCam camera. To examine hyphal growth following exposure to the cAMP analog, N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate (db-cAMP) (Sigma Chemical Co.), log-phase cells grown in YNB medium containing 2% galactose at 37°C were diluted to 3 × 104 cells/ml in YNB medium containing 2% galactose, and 120 µl was added to wells of a 96-well polystyrene plate (Costar; Corning Inc., Corning, NY) with either 30 µl of sterile deionized H2O or 150 mM db-cAMP, for a final concentration of 30 mM. The plate was incubated at 37°C for 5 h and photographed using a Zeiss Axiovert 200 M microscope equipped with an AxioCam HRm camera.
Fluorescence microscopy.
To assess actin localization by phalloidin staining, log-phase cells grown in YPD at 30°C were initially fixed in 5% formaldehyde for 75 min at 30°C, followed by incubation at room temperature for 30 min in 0.1 M potassium phosphate buffer (pH 7.5) containing 0.1% Triton X-100. After the cells were washed two times in 0.1 M potassium phosphate buffer (pH 7.5), 5 units of rhodamine-phalloidin (Invitrogen, Carlsbad, CA) were added, and the cells were incubated at 4°C overnight, protected from light. The cells were then washed three times in 0.1 M potassium phosphate buffer (pH 7.5), resuspended in mounting medium containing 10 µg/ml phenylenediamine (Sigma Chemical Co.), and then examined by fluorescence microscopy. Strains carrying fluorescent fusion proteins were grown overnight in YPD and analyzed by microscopy using a Zeiss Axiovert 200 M microscope equipped with an AxioCam HRm camera and Zeiss AxioVision software for deconvoluting images.
Virulence assays.
C. albicans strains were grown overnight at 30°C in YPD medium with 80 µg/ml uridine, reinoculated into fresh medium, and incubated again overnight at 30°C. Cells were washed twice in phosphate-buffered saline (PBS), counted in a hemocytometer, and diluted to 1 × 107 cells/ml with PBS. Dilutions of cells were plated on solid YPD medium to confirm the quantitation. Eight BALB/c female mice per each C. albicans strain were inoculated with 1 × 106 cells in the lateral tail vein. The mice were monitored for 28 days, during which time a mouse was considered moribund if food and water could no longer be accessed. Humane euthanasia was then performed. All procedures were approved by the Stony Brook University IACUC Committee. Graphing and statistical analysis of survival after infection were carried out using a log rank test (Mantel-Haenszel test) with the software program Prism 4 (GraphPad Software, Inc., La Jolla, CA). Kidneys excised for fungal burden assessments were weighed, placed in 5 ml PBS, and homogenized for 30 s with a tissue homogenizer (Pro Scientific, Inc., Oxford, CT). Serial dilutions of the homogenate were plated on YPD medium plates and incubated at 30°C for 2 days to determine the number of CFU per gram of kidney tissue. Statistical analysis of the CFU data was carried out with Prism software using one-way analysis of variance with a nonparametric Kruskal-Wallis test and Dunn’s post-hoc test. Periodic acid-Schiff and hematoxylin-eosin staining for histological analysis was performed by a commercial laboratory (McClain Laboratories, Smithtown, NY). Kidney tissue homogenates were stained with 20 ng/ml calcofluor white for ~10 min, resuspended in 10% KOH for 5 min at room temperature, and then imaged by fluorescence microscopy.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI-47837, awarded to J.B.K., from the National Institute of Allergy and Infectious Diseases.
We thank the members of our labs for their helpful advice and comments on the manuscript. We also thank Martine Bassilana for the set of protein kinase plasmids, Malcolm Whiteway for a LIFEACT plasmid, and Patricio Mentaboada for assistance with experimental procedures.
Footnotes
Citation Douglas LM, Wang HX, Konopka JB. 2013. The MARVEL domain protein Nce102 regulates actin organization and invasive growth of Candida albicans. mBio 4(6):e00723-13. doi:10.1128/mBio.00723-13.
REFERENCES
- 1. Heitman J, Filler SG, Edwards JEJ, Mitchell AP. 2006. Molecular principles of fungal pathogenesis. ASM Press, Washington, DC [Google Scholar]
- 2. Odds FC. 1988. Candida and candidosis. Bailliere Tindall, Philadelphia, PA. [Google Scholar]
- 3. Pfaller MA, Diekema DJ. 2010. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 36:1–53 [DOI] [PubMed] [Google Scholar]
- 4. Sudbery PE. 2011. Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 9:737–748 [DOI] [PubMed] [Google Scholar]
- 5. Odds FC, Brown AJ, Gow NA. 2003. Antifungal agents: mechanisms of action. Trends Microbiol. 11:272–279 [DOI] [PubMed] [Google Scholar]
- 6. Malinsky J, Opekarová M, Tanner W. 2010. The lateral compartmentation of the yeast plasma membrane. Yeast 27:473–478 [DOI] [PubMed] [Google Scholar]
- 7. Olivera-Couto A, Aguilar PS. 2012. Eisosomes and plasma membrane organization. Mol. Genet. Genomics 287:607–620 [DOI] [PubMed] [Google Scholar]
- 8. Douglas LM, Wang HX, Li L, Konopka JB. 2011. Membrane compartment occupied by Can1 (MCC) and eisosome subdomains of the fungal plasma Membrane. Membranes (Basel) 1:394–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malínská K, Malínský J, Opekarová M, Tanner W. 2003. Visualization of protein compartmentation within the plasma membrane of living yeast cells. Mol. Biol. Cell 14:4427–4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malinska K, Malinsky J, Opekarova M, Tanner W. 2004. Distribution of Can1p into stable domains reflects lateral protein segregation within the plasma membrane of living S. cerevisiae cells. J. Cell Sci. 117:6031–6041 [DOI] [PubMed] [Google Scholar]
- 11. Strádalová V, Stahlschmidt W, Grossmann G, Blazíková M, Rachel R, Tanner W, Malinsky J. 2009. Furrow-like invaginations of the yeast plasma membrane correspond to membrane compartment of Can1. J. Cell Sci. 122:2887–2894 [DOI] [PubMed] [Google Scholar]
- 12. Young ME, Karpova TS, Brügger B, Moschenross DM, Wang GK, Schneiter R, Wieland FT, Cooper JA. 2002. The Sur7p family defines novel cortical domains in Saccharomyces cerevisiae, affects sphingolipid metabolism, and is involved in sporulation. Mol. Cell. Biol. 22:927–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grossmann G, Malinsky J, Stahlschmidt W, Loibl M, Weig-Meckl I, Frommer WB, Opekarová M, Tanner W. 2008. Plasma membrane microdomains regulate turnover of transport proteins in yeast. J. Cell Biol. 183:1075–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walther TC, Brickner JH, Aguilar PS, Bernales S, Pantoja C, Walter P. 2006. Eisosomes mark static sites of endocytosis. Nature 439:998–1003 [DOI] [PubMed] [Google Scholar]
- 15. Olivera-Couto A, Graña M, Harispe L, Aguilar PS. 2011. The eisosome core is composed of BAR domain proteins. Mol. Biol. Cell 22:2360–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ziółkowska NE, Karotki L, Rehman M, Huiskonen JT, Walther TC. 2011. Eisosome-driven plasma membrane organization is mediated by BAR domains. Nat. Struct. Mol. Biol. 18:854–856 [DOI] [PubMed] [Google Scholar]
- 17. Walther TC, Aguilar PS, Fröhlich F, Chu F, Moreira K, Burlingame AL, Walter P. 2007. Pkh-kinases control eisosome assembly and organization. EMBO J. 26:4946–4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo G, Gruhler A, Liu Y, Jensen ON, Dickson RC. 2008. The sphingolipid long-chain base-Pkh1/2-Ypk1/2 signaling pathway regulates eisosome assembly and turnover. J. Biol. Chem. 283:10433–10444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berchtold D, Walther TC. 2009. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol. Biol. Cell 20:1565–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spira F, Mueller NS, Beck G, von Olshausen P, Beig J, Wedlich-Söldner R. 2012. Patchwork organization of the yeast plasma membrane into numerous coexisting domains. Nat. Cell Biol. 14:640–648 [DOI] [PubMed] [Google Scholar]
- 21. Alvarez FJ, Douglas LM, Rosebrock A, Konopka JB. 2008. The Sur7 protein regulates plasma membrane organization and prevents intracellular cell wall growth in Candida albicans. Mol. Biol. Cell 19:5214–5225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang HX, Douglas LM, Aimanianda V, Latgé JP, Konopka JB. 2011. The Candida albicans Sur7 protein is needed for proper synthesis of the fibrillar component of the cell wall that confers strength. Eukaryot. Cell 10:72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Douglas LM, Wang HX, Keppler-Ross S, Dean N, Konopka JB. 2012. Sur7 promotes plasma membrane organization and is needed for resistance to stressful conditions and to the invasive growth and virulence of Candida albicans. mBio 3(1):e00254-11. 10.1128/mBio.00254-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vernay A, Schaub S, Guillas I, Bassilana M, Arkowitz RA. 2012. A steep phosphoinositide bis-phosphate gradient forms during fungal filamentous growth. J. Cell Biol. 198:711–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martin SW, Konopka JB. 2004. Lipid raft polarization contributes to hyphal growth in Candida albicans. Eukaryot. Cell 3:675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loibl M, Grossmann G, Stradalova V, Klingl A, Rachel R, Tanner W, Malinsky J, Opekarová M. 2010. C terminus of Nce102 determines the structure and function of microdomains in the Saccharomyces cerevisiae plasma membrane. Eukaryot. Cell 9:1184–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fröhlich F, Moreira K, Aguilar PS, Hubner NC, Mann M, Walter P, Walther TC. 2009. A genome-wide screen for genes affecting eisosomes reveals Nce102 function in sphingolipid signaling. J. Cell Biol. 185:1227–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seger S, Rischatsch R, Philippsen P. 2011. Formation and stability of eisosomes in the filamentous fungus Ashbya gossypii. J. Cell Sci. 124:1629–1634 [DOI] [PubMed] [Google Scholar]
- 29. Sánchez-Pulido L, Martín-Belmonte F, Valencia A, Alonso MA. 2002. MARVEL: a conserved domain involved in membrane apposition events. Trends Biochem. Sci. 27:599–601 [DOI] [PubMed] [Google Scholar]
- 30. Iatropoulos P, Gardella R, Valsecchi P, Magri C, Ratti C, Podavini D, Rossi G, Gennarelli M, Sacchetti E, Barlati S. 2009. Association study and mutational screening of SYNGR1 as a candidate susceptibility gene for schizophrenia. Psychiatr. Genet. 19:237–243 [DOI] [PubMed] [Google Scholar]
- 31. Leblanc MA, Penney LS, Gaston D, Shi Y, Aberg E, Nightingale M, Jiang H, Gillett RM, Fahiminiya S, Macgillivray C, Wood EP, Acott PD, Khan MN, Samuels ME, Majewski J, Orr A, McMaster CR, Bedard K. 2013. A novel rearrangement of occludin causes brain calcification and renal dysfunction. Genet. Hum. 132:1223–1234 [DOI] [PubMed] [Google Scholar]
- 32. Pérez JC, Kumamoto CA, Johnson AD. 2013. Candida albicans commensalism and pathogenicity are intertwined traits directed by a tightly knit transcriptional regulatory circuit. PLoS Biol. 11:e1001510. 10.1371/journal.pbio.1001510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warenda AJ, Kauffman S, Sherrill TP, Becker JM, Konopka JB. 2003. Candida albicans septin mutants are defective for invasive growth and virulence. Infect. Immun. 71:4045–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zucchi PC, Davis TR, Kumamoto CA. 2010. A candida albicans cell wall-linked protein promotes invasive filamentation into semi-solid medium. Mol. Microbiol. 76:733–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumamoto CA. 2005. A contact-activated kinase signals Candida albicans invasive growth and biofilm development. Proc. Natl. Acad. Sci. U. S. A. 102:5576–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Navarro-Garcia F, Alonso-Monge R, Rico H, Pla J, Sentandreu R, Nombela C. 1998. A role for the MAP kinase gene MKC1 in cell wall construction and morphological transitions in Candida albicans. Microbiology 144:411–424 [DOI] [PubMed] [Google Scholar]
- 37. Liu H, Köhler J, Fink GR. 1994. Suppression of hyphal formation in Candida albicans by mutation of a STE12 homolog. Science 266:1723–1726 [DOI] [PubMed] [Google Scholar]
- 38. Zhang X, Lester RL, Dickson RC. 2004. Pil1p and Lsp1p negatively regulate the 3-phosphoinositide-dependent protein kinase-like kinase Pkh1p and downstream signaling pathways Pkc1p and Ypk1p. J. Biol. Chem. 279:22030–22038 [DOI] [PubMed] [Google Scholar]
- 39. Casamayor A, Torrance PD, Kobayashi T, Thorner J, Alessi DR. 1999. Functional counterparts of mammalian protein kinases PDK1 and SGK in budding yeast. Curr. Biol. 9:186–197 [DOI] [PubMed] [Google Scholar]
- 40. Hope H, Schmauch C, Arkowitz RA, Bassilana M. 2010. The Candida albicans ELMO homologue functions together with Rac1 and Dck1, upstream of the MAP kinase Cek1, in invasive filamentous growth. Mol. Microbiol. 76:1572–1590 [DOI] [PubMed] [Google Scholar]
- 41. Zou H, Fang HM, Zhu Y, Wang Y. 2010. Candida albicans Cyr1, Cap1 and G-actin form a sensor/effector apparatus for activating cAMP synthesis in hyphal growth. Mol. Microbiol. 75:579–591 [DOI] [PubMed] [Google Scholar]
- 42. Wolyniak MJ, Sundstrom P. 2007. Role of actin cytoskeletal dynamics in activation of the cyclic AMP pathway and HWP1 gene expression in Candida albicans. Eukaryot. Cell 6:1824–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin R, Walther A, Wendland J. 2005. Ras1-induced hyphal development in Candida albicans requires the formin Bni1. Eukaryot. Cell 4:1712–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li CR, Wang YM, De Zheng X, Liang HY, Tang JC, Wang Y. 2005. The formin family protein CaBni1p has a role in cell polarity control during both yeast and hyphal growth in Candida albicans. J. Cell Sci. 118:2637–2648 [DOI] [PubMed] [Google Scholar]
- 45. Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5:605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lionakis MS, Lim JK, Lee CC, Murphy PM. 2011. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J. Innate Immun. 3:180–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hazan I, Liu H. 2002. Hyphal tip-associated localization of Cdc42 is F-actin dependent in Candida albicans. Eukaryot. Cell 1:856–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Douglas LM, Martin SW, Konopka JB. 2009. BAR domain proteins Rvs161 and Rvs167 contribute to Candida albicans endocytosis, morphogenesis, and virulence. Infect. Immun. 77:4150–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Epp E, Nazarova E, Regan H, Douglas LM, Konopka JB, Vogel J, Whiteway M. 2013. Clathrin- and Arp2/3-independent endocytosis in the fungal pathogen Candida albicans. mBio 4(5):e00476-13. 10.1128/mBio.00476-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foster HA, Cui M, Naveenathayalan A, Unden H, Schwanbeck R, Höfken T. 2013. The zinc cluster protein Sut1 contributes to filamentation in Saccharomyces cerevisiae. Eukaryot. Cell 12:244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kabeche R, Baldissard S, Hammond J, Howard L, Moseley JB. 2011. The filament-forming protein Pil1 assembles linear eisosomes in fission yeast. Mol. Biol. Cell 22:4059–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khalaj V, Azizi M, Enayati S, Khorasanizadeh D, Ardakani EM. 2012. NCE102 homologue in Aspergillus fumigatus is required for normal sporulation, not hyphal growth or pathogenesis. FEMS Microbiol. Lett. 329:138–145 [DOI] [PubMed] [Google Scholar]
- 53. Grossmann G, Opekarová M, Malinsky J, Weig-Meckl I, Tanner W. 2007. Membrane potential governs lateral segregation of plasma membrane proteins and lipids in yeast. EMBO J. 26:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. 2000. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 11:4131–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cummins PM. 2012. Occludin: one protein, many forms. Mol. Cell. Biol. 32:242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Janz R, Südhof TC, Hammer RE, Unni V, Siegelbaum SA, Bolshakov VY. 1999. Essential roles in synaptic plasticity for synaptogyrin I and synaptophysin I. Neuron 24:687–700 [DOI] [PubMed] [Google Scholar]
- 57. Zhou G, Liang FX, Romih R, Wang Z, Liao Y, Ghiso J, Luque-Garcia JL, Neubert TA, Kreibich G, Alonso MA, Schaeren-Wiemers N, Sun TT. 2012. MAL facilitates the incorporation of exocytic uroplakin-delivering vesicles into the apical membrane of urothelial umbrella cells. Mol. Biol. Cell 23:1354–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kuwabara H, Kokai Y, Kojima T, Takakuwa R, Mori M, Sawada N. 2001. Occludin regulates actin cytoskeleton in endothelial cells. Cell Struct. Funct. 26:109–116 [DOI] [PubMed] [Google Scholar]
- 59. Sala-Valdes M, Ailane N, Greco C, Rubinstein E, Boucheix C. 2012. Targeting tetraspanins in cancer. Expert Opin. Ther. Targets. 16:985–997 [DOI] [PubMed] [Google Scholar]
- 60. Suzuki H, Kondoh M, Takahashi A, Yagi K. 2012. Proof of concept for claudin-targeted drug development. Ann. N. Y. Acad. Sci. 1258:65–70 [DOI] [PubMed] [Google Scholar]
- 61. Wilson RB, Davis D, Mitchell AP. 1999. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J. Bacteriol. 181:1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reuss O, Vik A, Kolter R, Morschhäuser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127 [DOI] [PubMed] [Google Scholar]
- 63. Zhang C, Konopka JB. 2010. A photostable green fluorescent protein variant for analysis of protein localization in Candida albicans. Eukaryot. Cell 9:224–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Keppler-Ross S, Noffz C, Dean N. 2008. A new purple fluorescent color marker for genetic studies in Saccharomyces cerevisiae and Candida albicans. Genetics 179:705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]