

ABSTRACT
Tuberculosis, caused by Mycobacterium tuberculosis, remains a devastating human infectious disease, causing two million deaths annually. We previously demonstrated that M. tuberculosis induces an enzyme, heme oxygenase (HO1), that produces carbon monoxide (CO) gas and that M. tuberculosis adapts its transcriptome during CO exposure. We now demonstrate that M. tuberculosis carries a novel resistance gene to combat CO toxicity. We screened an M. tuberculosis transposon library for CO-susceptible mutants and found that disruption of Rv1829 (carbon monoxide resistance, Cor) leads to marked CO sensitivity. Heterologous expression of Cor in Escherichia coli rescued it from CO toxicity. Importantly, the virulence of the cor mutant is attenuated in a mouse model of tuberculosis. Thus, Cor is necessary and sufficient to protect bacteria from host-derived CO. Taken together, this represents the first report of a role for HO1-derived CO in controlling infection of an intracellular pathogen and the first identification of a CO resistance gene in a pathogenic organism.
IMPORTANCE
Macrophages produce a variety of antimicrobial molecules, including nitric oxide (NO), hydrogen peroxide (H2O2), and acid (H+), that serve to kill engulfed bacteria. In addition to these molecules, human and mouse macrophages also produce carbon monoxide (CO) gas by the heme oxygenase (HO1) enzyme. We observed that, in contrast to other bacteria, mycobacteria are resistant to CO, suggesting that this might be an evolutionary adaptation of mycobacteria for survival within macrophages. We screened a panel of ~2,500 M. tuberculosis mutants to determine which genes are required for survival of M. tuberculosis in the presence of CO. Within this panel, we identified one such gene, cor, that specifically confers CO resistance. Importantly, we found that the ability of M. tuberculosis cells carrying a mutated copy of this gene to cause tuberculosis in a mouse disease model is significantly attenuated. This indicates that CO resistance is essential for mycobacterial survival in vivo.
INTRODUCTION
Mycobacterium tuberculosis infects one-third of the world’s population and causes 8 million active cases of disease annually. Tissue macrophages are the primary cells within which much of the host-M. tuberculosis interaction occurs, and macrophages utilize multiple strategies to control M. tuberculosis infection, including elaboration of reactive oxygen and nitrogen intermediates, acidification of the phagosome, fusion of phagosomes with lysosomes, delivery of antimicrobial peptides to the phagolysosome, and autophagic engulfment of M. tuberculosis (1, 2). However, a complete understanding of the immune mechanisms that control M. tuberculosis infection is lacking. Likewise, how M. tuberculosis resists host immunity and persists indefinitely is only beginning to be elucidated.
Sustained in vivo and intracellular survival of M. tuberculosis, a hallmark of latent infection, has selected for bacteria that can resist host antimicrobial systems (3). These include oxidative radicals produced by the respiratory burst, NO toxicity from inducible nitric oxide synthase (NOS2), and phagosomal acidification (4). In response, M. tuberculosis has evolved strategies to interfere with host defenses such as excluding NOS2 from the phagosome (5, 6) and arresting phagosome maturation to prevent acidification (7). Furthermore, M. tuberculosis can intrinsically resist the toxicity mediated by host pathways such as hydrogen peroxide through catalase (8, 9), NO through DNA repair (10), protein degradation (11), and antioxidant defense (12), and acidification through adaptation (13), cell wall stabilization (14, 15), and nitrate respiration (16). Thus, in many cases, the presence of a host antimicrobial pathway has selected for mycobacterial genes that can prevent bacterial death.
Humans and mice produce CO by the enzyme heme oxygenase (HO1) (17, 18), which catalyzes the degradation of heme into biliverdin, iron, and CO in a reaction requiring O2 and NADPH (19, 20). HO1 is primarily expressed within alveolar, liver, and spleen macrophages and is induced by inflammatory mediators such as lipopolysaccharide, tumor necrosis factor, interleukin-1, and oxidative stress (21). The CO is exhaled, with the average, nonsmoking human exhaling approximately 2 ppm (22, 23), while patients with a variety of infectious and inflammatory conditions produce significantly more (21, 22, 24–26). No studies have been performed to date on the CO concentration in exhaled air from individuals with tuberculosis.
We (27) and others (28) have shown that M. tuberculosis infection of macrophages and mice induces HO1. HO1-derived CO then induces a set of ~50 genes known as the dormancy regulon via a two-component signal transduction system mediated by the sensor histidine kinases DosS and DosT, and this induction is diminished in HO1-deficient macrophages and with chemical inhibition of HO1 (27). Recently, it was shown that HO1-deficient mice are more susceptible to Mycobacterium avium infection (29, 30) and M. tuberculosis infection (30) than wild-type mice, although this enhanced susceptibility was attributed to either inappropriate granuloma formation (29, 30) or macrophage toxicity from heme accumulation (30) and not to an antimicrobial activity of CO. To that end, the identification of a specific CO resistance gene in M. tuberculosis, an organism that is highly evolved for human survival, would provide strong evidence that HO1-derived CO functions as an antibacterial mechanism in humans.
Although CO has been present since the beginning of life (31), little is known about how bacteria survive in its presence, as CO can be toxic to proteins containing iron and other transition metals. Weigel and Englund (32) showed that CO halts aerobic growth of Escherichia coli by rapidly inhibiting ATP production and preventing DNA replication. More recent work has confirmed that E. coli, Pseudomonas aeruginosa, and Staphylococcus aureus are susceptible to exogenous CO (33–35). In contrast, M. tuberculosis is resistant to high levels of CO (27). Because heme oxygenase is induced during an M. tuberculosis infection (27, 28), the CO produced is sensed by M. tuberculosis (27, 28), and M. tuberculosis is resistant to CO (27), we hypothesized that M. tuberculosis carries resistance genes for CO.
Here we identify a novel host-pathogen interaction centered on the effects of host-derived CO on mycobacterial pathogenesis. We found that M. tuberculosis carries a CO resistance gene that is both necessary and sufficient for CO resistance and that this gene is essential for long-term survival of M. tuberculosis in mice. This represents the first identification of a CO resistance gene in M. tuberculosis and highlights the critical importance of HO1 in controlling infection of an intracellular pathogen.
RESULTS
Identification of Rv1829 as a CO resistance gene.
Because HO1 is important for control of M. tuberculosis during mouse infection (29, 30), and because we previously showed that M. tuberculosis is resistant to exogenous CO (27), we hypothesized that M. tuberculosis carries CO resistance genes. We created a transposon library using the M. tuberculosis Erdman strain and the ΦMycomarT7 phage developed by Sassetti et al. (36) and screened for mutants that would not grow in the presence of CO. We screened ~2,500 individual mutants on plates and identified one mutant whose transposon insertion was mapped to Rv1829 that was unable to grow in the presence of CO both on plates (Fig. 1A and B) and in liquid culture (Fig. 1C). Because it conferred carbon monoxide resistance, we named the gene cor. Notably, cor mutant bacteria demonstrated normal growth under aerobic conditions (Fig. 1A, B, and C; CO). The gene organization of the region encompassing cor does not indicate that it is part of an operon (Fig. 2C). Nonetheless, to exclude the possibility that the transposon insertion resulted in a polar disruption of neighboring genes, we cloned cor under the control of its native promoter and integrated the construct into the attB site in the cor mutant. Heterologous expression of cor rescued the susceptibility of the mutant bacteria to grow in the presence of CO on plates (Fig. 1A) and in liquid culture (Fig. 1C). We confirmed that Cor was absent in the cor mutant and that its synthesis was restored in the complemented strain using an anti-Cor antibody (Fig. 1D). Thus, we conclude that the cor mutant phenotype is due specifically to disruption of cor and not to a polar effect on neighboring genes, including the neighboring SoxR-like transcription factors.
FIG 1 .

Identification of cor (Rv1829) as a CO resistance gene. (A) Serial dilutions of M. tuberculosis Erdman, cor mutant bacteria, or the cor mutant complemented with cor (cor-comp) were plated and exposed to ambient air or CO (0.2%) for 3 weeks. (B) Quantitation of CFU data from the experiment represented by panel A (one of three similar experiments shown). (C) Western blots of lysates of M. tuberculosis Erdman, the cor mutant, or the complemented strain were probed with anti-Cor antibody. WT, wild type. (D) M. tuberculosis Erdman, the cor mutant, and the complemented strain were grown in 7H9 liquid medium in the presence or absence of CO, and CFU were enumerated. *, P < 0.05 compared to M. tuberculosis Erdman (by Student’s t test).
FIG 2 .

Cor is a conserved, ancient protein. (A) Alignment of Rv1829 from M. tuberculosis to orthologues from Thermotoga maritima, M. leprae, M. smegmatis, Rhodococcus fascians, and Streptomyces species AA4. Sequence alignment of Rv1829 homologues shows conserved amino acids (in rainbow colors) by conservation, with invariant nonhydrophobic residues colored in red. This color scheme is identical in the modeled crystal structure in panel D. (B) Species-distribution taxonomic tree in “sunburst” format. The distribution and evolutionary conservation of all known homologues of Cor are shown. (C) Alignment of the Cor genomic region from M. tuberculosis with the orthologous genomic region from Rhodococcus sp. demonstrates conservation of multiple surrounding genes. (D) The Cor sequence was mapped to the representative DUF151 structure, which highlighted a conserved surface cleft formed at the interface (also relatively conserved) of two DUF151 monomers (shown in red). (E) Cor was purified from E. coli as a 6×His-tagged protein and then run on a denaturing SDS-PAGE gel (left panel) or a native gel (right panel) with appropriate molecular mass markers. The predicted molecular masses of cor are 18 kDa for the monomer and 36 kDa for the dimer. In the left panel, lanes are total lysate (lane 1), flowthrough (lane 2), final wash (lane 3), and imidazole elution (lane 4).
cor is a gene of unknown function in M. tuberculosis that encodes a 164-amino-acid protein (Fig. 2A). It is an evolutionarily ancient gene that is found in bacteria, archea, and plants and contains a DUF151 (domain of unknown function) domain (Fig. 2A). There are >500 sequences in the CDART database containing this domain, including other pathogenic mycobacteria (M. avium, M. kansasii, M. abscessus, and M. leprae) and other pathogens such as Rhodococcus sp. and corynebacteriae (Fig. 2B), and the genomic organization of the cor region is shared between mycobacteria and Rhodococcus sp. (Fig. 2C), suggesting shared functions. Bioinformatics and molecular modeling analysis using the crystal structure of the Cor homologue from Thermotoga maritima demonstrate that Cor likely dimerizes (Fig. 2D). We purified recombinant Cor in E. coli for biochemical experiments and found using native gel electrophoresis that Cor forms a dimer (Fig. 2E; compare the denaturing gel on the left to the native gel on the right). The structure of the Thermotoga maritima homologue is novel and therefore provides little insight with respect to function. However, the DUF151 domain containing wild rice Oryza minuta protein OmBBD was recently reported to exhibit nuclease activity (37). We tested recombinant Cor purified from E. coli as a 6×His–maltose-binding protein (MBP) fusion for nuclease activity and found that it lacked DNase or RNAse activity compared to His-MBP alone (see Fig. S1 in the supplemental material). Thus, cor appears to be a CO resistance gene of unknown function.
The cor mutant is not hypersusceptible to multiple stresses.
Although we isolated the cor mutant in a screen for CO sensitivity, cor may encode a general stress survival factor rather than providing isolated protection against CO toxicity. Therefore, we tested the ability of the mutant to survive when exposed to conditions expected to exist in vivo, namely, exposure to acid pH, NO, oxidative stress, and hypoxia. We found that the growth phenotype of the cor mutant when exposed to acid pH (Fig. 3A), NO (Fig. 3A), hydrogen peroxide via plumbagin treatment (Fig. 3B), or hypoxia (Fig. 3C and D) was indistinguishable from that of the wild type. Thus, we conclude that among the stresses tested, the mutant strain is increasingly susceptible to CO only.
FIG 3 .

The cor mutant is not hypersusceptible to acid pH, nitric oxide, hypoxia, or hydrogen peroxide. (A) M. tuberculosis Erdman, the cor mutant, and the cor mutant complemented with cor were diluted into 7H9-ADN to an OD600 of 0.05 at pH 5.5 or pH 5.5 plus sodium nitrite. Six days later, bacteria were diluted and plated and CFU enumerated after 3 weeks. (B) M. tuberculosis Erdman, the cor mutant, and the complemented strain (cor-comp) were grown to late log phase and resuspended to an OD600 of 0.1, and bacteria were plated to generate a dense lawn. Sterile paper discs placed on the plate were impregnated with 10 µl plumbagin, plates were incubated for 3 weeks, and the zone of inhibition was measured. (C and D) M. tuberculosis Erdman (black circles) and the cor mutant (red squares) were grown in 17-ml test tubes in triplicate, and gradual hypoxia was generated following the Wayne model. At each time point, tubes were sacrificed and OD600 (C) and CFU (D) were determined. Data represent the results of one of three similar experiments. Data represent means ± standard deviations (SD) for both panels.
The cor mutant develops a reducing environment.
On the basis of the crystal structure of the Thermatoga maritima homologue, we hypothesized that Cor might have enzymatic activity. Therefore, we tested if the cor mutant has a different small-molecule metabolite pool than wild-type bacteria when exposed to CO and asked if differences in metabolites might provide insight into Cor’s function. We extracted a small-molecule fraction from wild-type and cor mutant bacteria grown in the presence and absence of CO and profiled the mycobacterial metabolite pool. We found that for the roughly 150 metabolites surveyed, the NAD+ and mycothione levels were significantly reduced in the CO-treated mutant compared to the CO-treated wild type at the 10-day time point (Fig. 4A and B), indicating that the cor mutant has a dysregulated redox environment with accumulation of reducing equivalents (the full list of metabolites is available as Table S1 in the supplemental material). We also found that the cor mutant had significantly elevated levels of unsaturated (Fig. 4C to E) and saturated (Fig. 4F) long-chain fatty acids, likely indicative of increased anabolism of fatty acids. This result is consistent with a previous study of an M. tuberculosis WhiB3 mutant that develops a reducing environment and concomitant anabolism of a variety of lipids (38).
FIG 4 .

The cor mutant has a dysregulated intracellular redox environment, and its CO susceptibility can be rescued by the antioxidant α-tocopherol. (A and B) M. tuberculosis cultures were exposed to CO in vitro in quadruplicate and harvested after 0, 5, and 10 days. Small-molecule metabolites were extracted, and 145 known biochemicals were quantified using GC-MS and LC-MS. Normalized metabolite levels produced under the indicated treatment conditions, where the mean metabolite value for wild-type M. tuberculosis served as the denominator, are shown. Compared to wild-type bacteria, the cor mutant treated with CO had significantly reduced levels of mycothione (A) and NAD+ (B) but had increased levels of linoleate (C), linolenate (D), palmitoleate (E), and nonadecanoate (F). Differences between CO-treated M. tuberculosis Erdman and the mutant at 10 days were statistically significant. *, P < 0.05 (by Welch’s two-sample t test).
Expression of M. tuberculosis Cor in E. coli is sufficient to rescue CO toxicity.
To ask if Cor alone could confer CO resistance, we transformed E. coli with a vector containing cor under the control of a T7 promoter. Both E. coli transformed with the Cor expression vector but left uninduced and E. coli expressing an irrelevant protein, red fluorescent protein (RFP), were markedly susceptible to CO treatment (Fig. 5A and B), as has been previously reported (39). In contrast, when E. coli was induced to express Cor and exposed to CO, we observed significantly reduced CO toxicity such that Cor-expressing E. coli demonstrated a 3-to-4-log improvement in growth (Fig. 5A and B). Exogenous addition of recombinant Cor to CO-treated E. coli did not rescue E. coli from CO toxicity (data not shown). We confirmed expression of Cor with our anti-Cor polyclonal antibody (Fig. 5C). Thus, although the precise activity of Cor remains unknown, it is able to protect E. coli from CO toxicity.
FIG 5 .
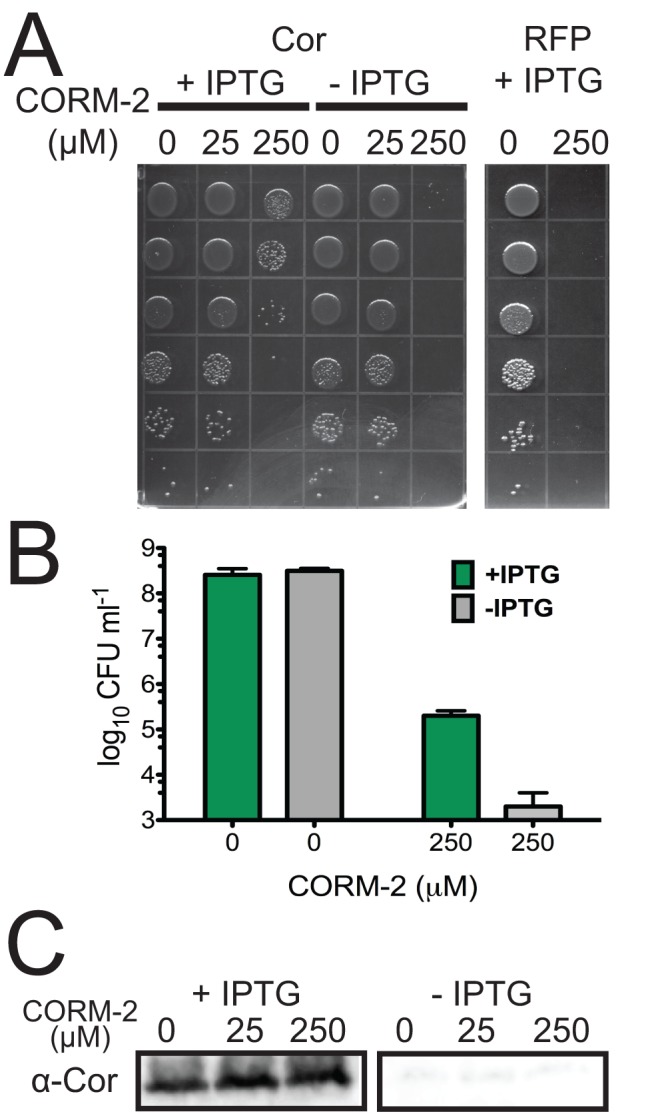
E. coli CO susceptibility is prevented by cor expression. (A) E. coli transformed with an IPTG-inducible vector expressing Cor or RFP was exposed to the CO donor corm-2 for 30 min with or without prior IPTG treatment, and serial dilutions were plated. (B) E. coli was grown as described above, and serial dilutions were plated to determine surviving CFU. (C) E. coli expressing Cor was lysed and Cor accumulation determined by Western blot analysis.
Virulence of the cor mutant is attenuated in mice.
To assess the role of Cor in vivo, we compared the outcomes of mouse infection with wild-type M. tuberculosis, the cor mutant, and the complemented strain. Mice infected with the cor mutant survived significantly longer than mice infected with the wild type or the complemented strain (Fig. 6A). Indeed, by 250 days postinfection, all the mice infected with the wild-type strain or the complemented strain had succumbed to infection, while more than 50% of the mutant mice were still alive. Likewise, the virulence of the cor mutant was attenuated in both mouse lung (Fig. 6B) and mouse liver (Fig. 6C) by an organ CFU assay, and this attenuation was most profound at later time points during the infection. Importantly, complementation restored full virulence, indicating that the attenuated phenotype of the cor mutant was due to disruption of cor.
FIG 6 .
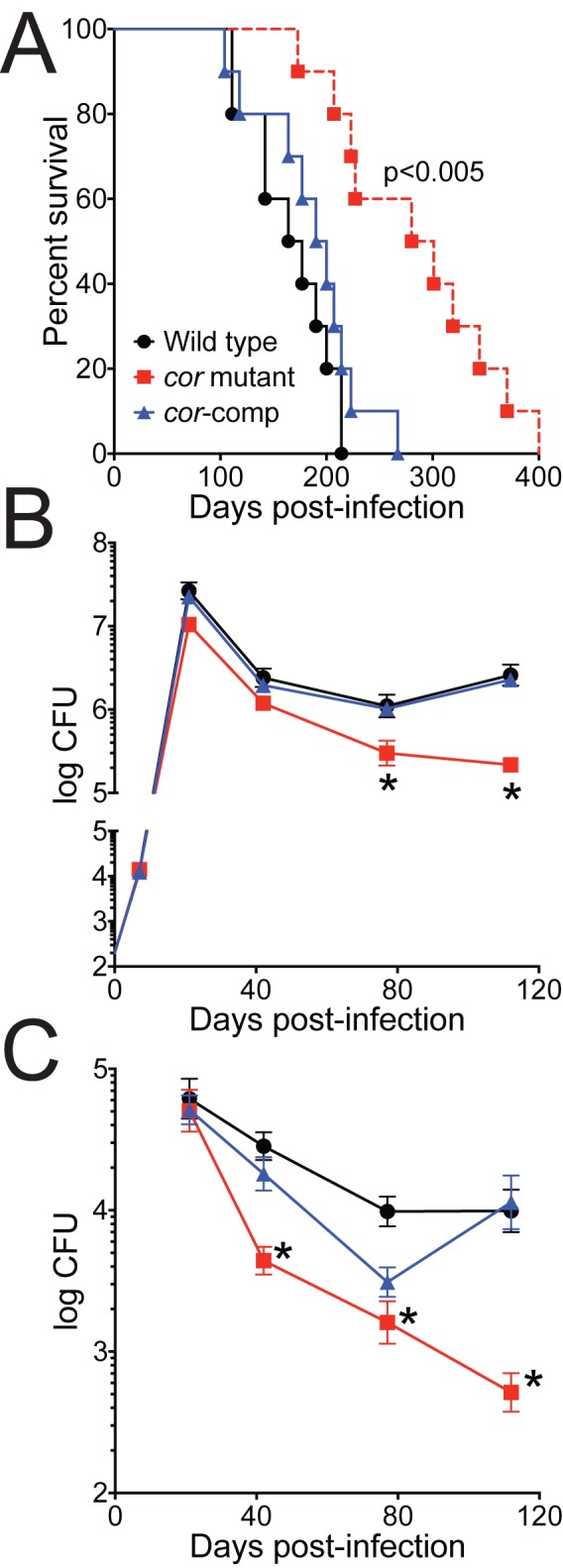
Virulence of the cor mutant is attenuated in mice. BALBc mice were infected with 102 CFU wild-type (WT) M. tuberculosis, the cor mutant, or the cor mutant complemented with cor via aerosol. (A) Ten mice per group were monitored over time for survival. By Kaplan-Meier analysis, differences between groups infected with the cor mutant and M. tuberculosis Erdman or the complemented strain are statistically significant. (B and C) Five mice per group were sacrificed, mouse lung (B) and mouse liver (C) were harvested and homogenized, and CFU were enumerated. *, P < 0.05 compared to WT (by the nonparametric Kruskal-Wallis test).
DISCUSSION
Our report identifies a novel host-pathogen relationship centered on the gas carbon monoxide. It was recently shown that HO1-deficient mice are more susceptible to M. tuberculosis infection (30), highlighting a role for HO1 in controlling M. tuberculosis. To combat CO in vivo, M. tuberculosis carries at least one CO resistance gene, cor. Although the precise biochemical mechanism of protection is unknown, the cor mutant appears to protect against accumulation of excess reducing equivalents. Further, cor can protect a heterologous bacterium, E. coli, from CO toxicity. Importantly, cor is important for M. tuberculosis survival after aerosol infection in mice.
Previous work demonstrated that some mycobacteria can utilize CO as a carbon source (40) by fixing CO (41). This raises the issue of how a gas such as CO could act as a potential metabolite, a signaling molecule, and a toxin. Notably, although the attenuated M. tuberculosis H37Ra strain can grow on CO as its sole carbon source (40), CO-dependent growth of virulent M. tuberculosis strains such as H37Rv, CDC 1551, and Erdman has yet to be demonstrated. Whether CO is toxic might also depend on the environment experienced by the bacteria. For example, we found that M. tuberculosis was more resistant to CO when it was exposed to CO at higher cell densities (data not shown). It is also possible that CO toxicity is concentration dependent, such that at low CO concentrations, some CO can be fixed by mycobacteria (41), but at higher CO concentrations, CO is toxic. Indeed, dose-dependent responses to host antimicrobial pathways are common for M. tuberculosis. For example, in vitro M. tuberculosis is susceptible only to high concentrations of H2O2 (42), but mutation of M. tuberculosis catalase leads to heightened susceptibility in vitro to H2O2 (43) and in vivo attenuation (9, 43). Likewise, whereas M. tuberculosis can tolerate (42, 44) and alter its transcription program in the presence of low, nontoxic NO concentrations (45), higher concentrations are toxic (42, 44), and a number of NO resistance genes have been identified that result in attenuation in vivo (10, 11, 46).
Since HO1-derived CO production during infection may stimulate innate immune activities and also be directly bactericidal, our data suggest that CO treatment might be useful for therapy against M. tuberculosis. CO-releasing molecules (CORM) are currently being synthesized to serve multiple therapeutic purposes (47), and the ruthenium complex known as tricarbonylchloro(glycinato)-ruthenium(II) (corm-3) is toxic to both Gram-negative and Gram-positive bacteria in vitro (33, 35, 39) and in vivo (35). Whether corm-3 or similar compounds could be effective treatments against M. tuberculosis in vivo is unknown.
Cor consists entirely of a DUF151 (domain of unknown function) domain, with homologues in a variety of organisms, including most mycobacteria, Bacteroides sp., Chlamydia sp., Streptomyces sp., and Rhodococcus sp. Although the DUF151 domain-containing protein OmBBD demonstrated DNase and RNAse activity in vitro (37), we failed to detect similar activity with recombinant Cor. OmBBD carries a C-terminal UvrB domain that is absent in the mycobacterial sequences. UvrB is one component of the UvrABC endonuclease system, and we propose that OmBBD’s observed nuclease activity may come not from the DUF151 domain but from an interaction of the C-terminal UvrB with the catalytic UvrC nuclease likely copurifying from E. coli.
Since the crystal structure of Cor’s homologue from Thermatoga maritima lacks an obvious CO ligand such as a heme, iron, or other transition metal (Fig. 2D) (48), how might Cor be mediating CO resistance? Some bacteria, including E. coli, Pseudomonas aeruginosa, and Staphylococcus aureus, are susceptible to exogenous CO (33, 35, 39), and this susceptibility has been attributed to the ability of CO to poison the bacterial respiratory chain (35) or to the ability of some CORM molecules to generate hydroxyl radicals (39). The respiratory chain of M. tuberculosis appears to not be as susceptible to CO toxicity, perhaps owing to the ability of M. tuberculosis to survive under microaerophilic or anaerobic conditions (49), and we did not identify mutants in the respiratory chain in our CO screen, though the results may have been limited by the sample size of the mutants tested.
Treatment of the cor mutant with CO led to marked depletion of NAD+ and mycothione, suggesting excessive reductive stress mediated by CO in the cor mutant. This depletion was also observed in wild-type M. tuberculosis 5 days after treatment with CO (Fig. 4A and B), but, whereas wild-type M. tuberculosis could recover from and adapt to exposure to CO, the cor mutant could not. We also observed marked increases in intracellular fatty acids in the CO-treated cor mutant which could be a result of either decreased catabolism of reduced fatty acids in the setting of an altered NAD+/NADH ratio or increased fatty acid anabolism if NADPH is also increased in the mutant owing to accumulation of reducing equivalents (38). Taken together, the data suggest that Cor functions to restore redox homeostasis in the setting of CO-mediated toxicity. Finally, the ability of Cor to protect E. coli from CO toxicity argues for a direct protective effect of Cor and experiments are ongoing to test this hypothesis.
In conclusion, we describe a novel host-pathogen interaction that directly impacts M. tuberculosis pathogenesis. HO1 is important for controlling mycobacterial growth in mice (29, 30), and mycobacteria express Cor to resist CO toxicity. Genetic disruption of cor attenuates mycobacterial virulence in a mouse model of chronic tuberculosis infection. These findings provide evidence that successful CO resistance is key to M. tuberculosis pathogenesis. The capacity of host-derived CO to restrict bacterial growth may reflect the presence of a general innate immune antimicrobial mechanism against other intracellular pathogens.
MATERIALS AND METHODS
Strains and media.
We grew M. tuberculosis Erdman and mutants in Middlebrook 7H9 medium or on Middlebrook 7H10 agar (Difco) plates containing 10% oleic acid-albumin-dextrose-catalase (Remel). Liquid medium contained 0.05% Tween 80. When needed, 7H10 plates were supplemented with α-tocopherol (Sigma) at a final concentration of 12.5 µg/ml.
Antibodies.
We prepared a rabbit-polyclonal antibody against Cor (Rv1829) by purifying 6×His-tagged cor from E. coli and immunizing two rabbits with recombinant Cor using incomplete Freund’s adjuvant. Loading control antibody against Mpt32 was from BEI Resources.
Screen.
We transduced M. tuberculosis Erdman with the ΦMycoMarT7 phage (36) and selected for kanamycin-resistant mutants. Individual mutants were arrayed in 96-well plates. For CO susceptibility assays, mutants were grown to the stationary phase and then spotted onto 7H10 plates with a 96-pin replicator. Plates were then exposed to 2% CO or air for 3 weeks, and growth was assessed by visualization. To identify transposon insertions, we isolated genomic DNA, digested with BamHI, religated the DNA to generate kanamycin-resistant plasmids, and transformed E. coli pir-116 cells (EPICENTRE Biotechnologies). We then isolated plasmids and sequenced the insertion sites (50).
Complementation.
We cloned wild-type cor under the control of its endogenous promoter into an integrating vector (pMV306 [51]) conferring hygromycin resistance. We transformed the cor mutant by electroporation and selected transformants with 50 µg ml−1 hygromycin.
Immunoblotting.
For immunoblotting, M. tuberculosis strains were grown to late log phase and bacteria lysed by bead beating. After boiling, protein content was determined and protein accumulation determined by Western blotting with rabbit anti-Cor and anti-Mpt32 antibodies.
Bioinformatic analysis of cor sequence.
DUF151 sequences were collected with a PSI-BLAST (52) search (E value cutoff of 0.01, 5 iterations) against the NR database (posted date, 17 April 2011; 2,015,132 sequences) using the Cor query sequence (gi|15608966). Identified sequences were aligned using PROMALS3D (53) and clustered using CLANS (54). Sequence groups that were remote from the Rv1829 cluster (including plant DUF151 sequences and others) were removed. To gain insight into DUF151 functional sites, residue conservations were calculated using AL2CO (55) on (i) a multiple-sequence alignment that included the close-sequence cluster (depicted in red and cyan in the CLANS map) and on (ii) a multiple-sequence alignment that included the extended sequence set (all sequences in the CLANS map).
Protein purification, gel electrophoresis, and nucleic acid degradation assay.
Recombinant His-MBP Cor and His-MBP were expressed in BL21 E. coli and were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Proteins were purified using cobalt-charged TALON resin (Clontech) under native, nondenaturing conditions. Denaturing gel electrophoresis was performed using a 4%-to-20% Tris-glycine (SDS-PAGE) gel with a PageRuler Plus prestained protein ladder. Native gel electrophoresis was performed using a NativePAGE Novex 4%-to-16% bis-Tris gel with a NativeMARK unstained protein standard. For the nucleic acid degradation assay, recombinant Cor or His-MBP alone at different concentrations (0.5, 2.5, and 5.0 µM) was incubated with either 4 µg of M. tuberculosis total RNA or 4 µg of M. smegmatis (mc2 155) genomic DNA in NEBuffer 3 for 1 h at 37°C. RNA samples were loaded with RNA sample loading buffer (Sigma-Aldrich) and were run on a 1.2% ethidium bromide-stained formaldehyde gel, whereas DNA samples were loaded with DNA loading dye (New England Biolabs) and were run on a 1% ethidium bromide-stained agarose gel.
Metabolomics.
M. tuberculosis Erdman and the cor mutant were grown in quadruplicate in roller bottles and exposed to air or 0.2% CO. At time zero (no treatment), day 5, and day 10, 1 × 109 bacteria were pelleted and resuspended in cold 100% methanol. Acetonitrile and water were then added for a final 40:40:20 ratio, the samples bead beated, and the supernatants collected and dried. The samples were then analyzed by gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-MS (LC-MS) and statistically significant differences determined by Welch’s two-sample test (Metabolon, Inc.).
Susceptibility to acid, nitric oxide, hypoxia, and plumbagin.
We grew M. tuberculosis Erdman and the cor mutant to the late log phase, washed cells in phosphate-buffered saline (PBS), resuspended the cells at an optical density at 600 nm (OD600) of 0.1 in 7H9 at either pH 6.6 or 5.0 in the absence or presence of NaNO2 (5 mM), and then measured OD600 daily. Alternatively, after washing the cells, we resuspended the bacteria in 7H9-ADN (7H9, 0.2% glycerol, 0.05% Tween 80, 0.5% bovine serum albumin, 0.2% dextrose, 0.085% NaCl) at pH 5.5 and exposed the bacteria to NaNO2 (3 mM) for 5 days. We determined CFU by plating serial dilutions of the suspensions on 7H10 agar plates. For growth under conditions of hypoxia, cells were grown in 17-ml test tubes in triplicate and gradual hypoxia was generated following the Wayne model (56). At each time point, tubes were sacrificed and CFU recorded. For testing of plumbagin via disc diffusion, M. tuberculosis strains (wild type, cor mutant, and cor complement) were grown to an OD600 of 0.6 to 0.8 and were back diluted to an OD600 of 0.1. Cultures were uniformly spread onto 7H10 plates supplemented with oleic acid, albumin fraction V, and dextrose, excluding catalase. Four discs (Oxoid/Thermo Scientific) were placed onto each plate, and 10 µl of plumbagin dissolved in ethanol was spotted onto the discs (0 mM, 20 mM, and 100 mM). Treatment of discs with plumbagin was performed in duplicate per strain used in the experiment. The zone of inhibition surrounding the discs was measured in millimeters.
CO treatment of E. coli.
All BL21(DE3) E. coli cultures were grown under aerobic conditions at 37°C with shaking at 250 rpm. The E. coli strain containing the pJ401 IPTG-inducible expression vector (DNA 2.0, Inc.) with the cor gene insert and the control E. coli strain (pJ401-RFP) were grown in LB medium plus kanamycin (50 µg/ml) to an OD600 of 0.35 and induced with IPTG (1 mM final concentration). The cultures were induced for 3 h with IPTG to allow maximal expression of protein (detected by Coomassie; data not shown). The cultures were then back-diluted to an OD600 of 0.1 in M9 minimal salts (BD Difco) media supplemented with MgSo4 and CaCl2. At an OD600 of 0.3, E. coli cultures were treated with tricarbonyldichlororuthenium (II) dimer (corm-2; Sigma-Aldrich) as previously described by Tavares et al. (39). Briefly, corm-2 was prepared as stock solutions dissolved in dimethyl sulfoxide (DMSO) and used at a final concentration of 0 µM (DMSO only), 25 µM, or 250 µM. E. coli bacteria were treated for 30 min at the indicated concentrations and were serially diluted and spotted onto square LB agar plates with kanamycin (50 µg/ml) and incubated overnight at 37° C. For bacterial enumeration, E. coli bacteria were spread onto plates and also incubated overnight at 37° C to determine CFU. Cor protein expression was determined by Western blot analysis using the anti-Cor antibody. Lysates from ~3 × 107 bacteria were loaded per well.
Mouse infections.
We infected BALBc mice (Jackson Laboratories) using a Madison aerosol exposure chamber to deliver ~200 bacilli per mouse. Prior to aerosolization, bacteria were washed repeatedly and sonicated to generate a single-cell suspension. At day zero, we plated total organ homogenates from both lungs (5 mice per group) to determine the initial inoculum per M. tuberculosis strain. At subsequent time points, we plated serial dilutions of organ homogenates from lung (left lung) or liver (left lobe) from 5 mice per group. Animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of University of California at San Francisco and University of Texas (UT) Southwestern. For organ CFU comparisons, statistically significant differences were determined by the nonparametric Kruskal-Wallis test. For survival experiments (10 mice per strain), mice were sacrificed when they had lost 15% of their maximal body weight as we had previously demonstrated that this degree of weight loss predicted imminent mouse death (57, 58). Mouse survival curves were compared by Kaplan-Meier analysis.
SUPPLEMENTAL MATERIAL
Nuclease activity of purified recombinant Cor. (A) Coomassie-stained SDS gel of purified His-MBP-Cor and His-MBP alone. Lanes: molecular weight marker (lane 1), whole-cell lysate (WCL) of uninduced BL21 expressing His-MBP and His-MBP-Cor (lanes 2 and 7, respectively), WCL of induced (1 mM IPTG) BL21 expressing His-MBP and His-MBP-Cor (lanes 3 and 8, respectively), purified His-MBP and His-MBP-Cor (lane 5 and 10, respectively). (B) M. tuberculosis total RNA incubated with increasing concentrations (0.5, 2.5, and 5.0 µM) of purified His-MBP or His-MBP-Cor or buffer alone. RNase A incubated with M. tuberculosis RNA was used as a positive control. Arrowheads indicate different RNA species. (C) M. smegmatis (mc2 155) genomic DNA was incubated with increasing concentrations (0.5, 2.5, and 5.0 µM) of purified His-MBP or His-MBP-Cor. The arrowhead indicates bands of copurifying RNA. Download
Heat map of statistically significant biochemicals profiled in this study.
ACKNOWLEDGMENTS
We thank C. Sassetti (University of Massachusetts) for the ΦMycomarT7 phage, Z. Zheng for help with immunohistochemistry and immunofluorescence, and members of the Cox and Shiloh laboratories for helpful suggestions.
Funding for this work was provided by NIH grants AI076632 and AI099439 (M.U.S.), AI081727 (J.S.C.), DK081668 (D.K.M.), and 5T32AI007520 (V.M.Z.). M.U.S. acknowledges support from the Disease Oriented Clinical Scholars Program at UT Southwestern.
Footnotes
Citation Zacharia VM, Manzanillo PS, Nair VR, Marciano DK, Kinch LN, Grishin NV, Cox JS, Shiloh MU. 2013. cor, a novel carbon monoxide resistance gene, is essential for Mycobacterium tuberculosis pathogenesis. mBio 4(6):e00721-13. doi:10.1128/mBio.00721-13
REFERENCES
- 1. Huynh KK, Joshi SA, Brown EJ. 2011. A delicate dance: host response to mycobacteria. Curr. Opin. Immunol. 23:464–472 [DOI] [PubMed] [Google Scholar]
- 2. Liu PT, Modlin RL. 2008. Human macrophage host defense against Mycobacterium tuberculosis. Curr. Opin. Immunol. 20:371–376 [DOI] [PubMed] [Google Scholar]
- 3. Nathan C, Shiloh MU. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U. S. A. 97:8841–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rohde K, Yates RM, Purdy GE, Russell DG. 2007. Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219:37–54 [DOI] [PubMed] [Google Scholar]
- 5. Davis AS, Vergne I, Master SS, Kyei GB, Chua J, Deretic V. 2007. Mechanism of inducible nitric oxide synthase exclusion from mycobacterial phagosomes. PLOS Pathog. 3:e186. 10.1371/journal.ppat.0030186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deghmane AE, Soualhine H, Bach H, Sendide K, Itoh S, Tam A, Noubir S, Talal A, Lo R, Toyoshima S, Av-Gay Y, Hmama Z. 2007. Lipoamide dehydrogenase mediates retention of coronin-1 on BCG vacuoles, leading to arrest in phagosome maturation. J. Cell Sci. 120:2796–2806 [DOI] [PubMed] [Google Scholar]
- 7. Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, Allen RD, Gluck SL, Heuser J, Russell DG. 1994. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263:678–681 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y, Heym B, Allen B, Young D, Cole S. 1992. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 358:591–593 [DOI] [PubMed] [Google Scholar]
- 9. Li Z, Kelley C, Collins F, Rouse D, Morris S. 1998. Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. J. Infect. Dis. 177:1030–1035 [DOI] [PubMed] [Google Scholar]
- 10. Darwin KH, Nathan CF. 2005. Role for nucleotide excision repair in virulence of Mycobacterium tuberculosis. Infect. Immun. 73:4581–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966 [DOI] [PubMed] [Google Scholar]
- 12. Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. 2002. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 295:1073–1077 [DOI] [PubMed] [Google Scholar]
- 13. Vandal OH, Pierini LM, Schnappinger D, Nathan CF, Ehrt S. 2008. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 14:849–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vandal OH, Roberts JA, Odaira T, Schnappinger D, Nathan CF, Ehrt S. 2009. Acid-susceptible mutants of Mycobacterium tuberculosis share hypersusceptibility to cell wall and oxidative stress and to the host environment. J. Bacteriol. 191:625–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abramovitch RB, Rohde KH, Hsu FF, Russell DG. 2011. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol. Microbiol. 80:678–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan MP, Sequeira P, Lin WW, Phong WY, Cliff P, Ng SH, Lee BH, Camacho L, Schnappinger D, Ehrt S, Dick T, Pethe K, Alonso S. 2010. Nitrate respiration protects hypoxic Mycobacterium tuberculosis against acid- and reactive nitrogen species stresses. PLoS One 5:e13356. 10.1371/journal.pone.0013356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sjostrand T. 1952. The formation of carbon monoxide by the decomposition of haemoglobin in vivo. Acta Physiol. Scand. 26:338–344 [DOI] [PubMed] [Google Scholar]
- 18. Tenhunen R, Marver HS, Schmid R. 1968. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. U. S. A. 61:748–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maines MD. 1988. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 2:2557–2568 [PubMed] [Google Scholar]
- 20. Maines MD. 2004. The heme oxygenase system: past, present, and future. Antioxid. Redox Signal. 6:797–801 [DOI] [PubMed] [Google Scholar]
- 21. Ryter SW, Morse D, Choi AM. 2004. Carbon monoxide: to boldly go where NO has gone before. Sci. STKE 2004:RE6. 10.1126/stke.2302004re6 [DOI] [PubMed] [Google Scholar]
- 22. Biernacki WA, Kharitonov SA, Barnes PJ. 2001. Exhaled carbon monoxide in patients with lower respiratory tract infection. Respir. Med. 95:1003–1005 [DOI] [PubMed] [Google Scholar]
- 23. Paredi P, Shah PL, Montuschi P, Sullivan P, Hodson ME, Kharitonov SA, Barnes PJ. 1999. Increased carbon monoxide in exhaled air of patients with cystic fibrosis. Thorax 54:917–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uasuf CG, Jatakanon A, James A, Kharitonov SA, Wilson NM, Barnes PJ. 1999. Exhaled carbon monoxide in childhood asthma. J. Pediatr. 135:569–574 [DOI] [PubMed] [Google Scholar]
- 25. Antuni JD, Kharitonov SA, Hughes D, Hodson ME, Barnes PJ. 2000. Increase in exhaled carbon monoxide during exacerbations of cystic fibrosis. Thorax 55:138–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paredi P, Kharitonov SA, Barnes PJ. 2003. Exhaled carbon monoxide in lung disease. Eur. Respir. J. 21:197–198. 10.1183/09031936.02.00071802 (Author reply.) [DOI] [PubMed] [Google Scholar]
- 27. Shiloh MU, Manzanillo P, Cox JS. 2008. Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe 3:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar A, Deshane JS, Crossman DK, Bolisetty S, Yan BS, Kramnik I, Agarwal A, Steyn AJ. 2008. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 283:18032–18039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Regev D, Surolia R, Karki S, Zolak J, Montes-Worboys A, Oliva O, Guroji P, Saini V, Steyn AJ, Agarwal A, Antony VB. 2012. Heme oxygenase-1 promotes granuloma development and protects against dissemination of mycobacteria. Lab. Invest. 92:1541–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silva-Gomes S, Appelberg R, Larsen R, Soares MP, Gomes MS. 2013. Heme catabolism by heme oxygenase-1 confers host resistance to mycobacterium infection. Infect. Immun. 81:2536–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bach FH. 2006. Carbon monoxide: from the origin of life to molecular medicine. Trends Mol. Med. 12:348–350 [DOI] [PubMed] [Google Scholar]
- 32. Weigel PH, Englund PT. 1975. Inhibition of DNA replication in Escherichia coli by cyanide and carbon monoxide. J. Biol. Chem. 250:8536–8542 [PubMed] [Google Scholar]
- 33. Nobre LS, Seixas JD, Romão CC, Saraiva LM. 2007. Antimicrobial action of carbon monoxide-releasing compounds. Antimicrob. Agents Chemother. 51:4303–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davidge KS, Sanguinetti G, Yee CH, Cox AG, McLeod CW, Monk CE, Mann BE, Motterlini R, Poole RK. 2009. Carbon monoxide-releasing antibacterial molecules target respiration and global transcriptional regulators. J. Biol. Chem. 284:4516–4524 [DOI] [PubMed] [Google Scholar]
- 35. Desmard M, Davidge KS, Bouvet O, Morin D, Roux D, Foresti R, Ricard JD, Denamur E, Poole RK, Montravers P, Motterlini R, Boczkowski J. 2008. A carbon monoxide-releasing molecule (corm-3) exerts bactericidal activity against Pseudomonas aeruginosa and improves survival in an animal model of bacteraemia. FASEB J. 23:1023–1031 [DOI] [PubMed] [Google Scholar]
- 36. Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84 [DOI] [PubMed] [Google Scholar]
- 37. You MK, Shin HY, Kim YJ, Ok SH, Cho SK, Jeung JU, Yoo SD, Kim JK, Shin JS. 2010. Novel bifunctional nucleases, OmBBD and AtBBD1, are involved in abscisic acid-mediated callose deposition in Arabidopsis. Plant Physiol. 152:1015–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJ. 2009. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 5:e1000545. 10.1371/journal.ppat.1000545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tavares AF, Teixeira M, Romão CC, Seixas JD, Nobre LS, Saraiva LM. 2011. Reactive oxygen species mediate bactericidal killing elicited by carbon monoxide-releasing molecules. J. Biol. Chem. 286:26708–26717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park SW, Hwang EH, Park H, Kim JA, Heo J, Lee KH, Song T, Kim E, Ro YT, Kim SW, Kim YM. 2003. Growth of mycobacteria on carbon monoxide and methanol. J. Bacteriol. 185:142–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. King GM. 2003. Uptake of carbon monoxide and hydrogen at environmentally relevant concentrations by mycobacteria. Appl. Environ. Microbiol. 69:7266–7272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan J, Xing Y, Magliozzo RS, Bloom BR. 1992. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 175:1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. 2004. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52:1291–1302 [DOI] [PubMed] [Google Scholar]
- 44. O’Brien L, Carmichael J, Lowrie DB, Andrew PW. 1994. Strains of mycobacterium tuberculosis differ in susceptibility to reactive nitrogen intermediates in vitro. Infect. Immun. 62:5187–5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK. 2003. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198:705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Venugopal A, Bryk R, Shi S, Rhee K, Rath P, Schnappinger D, Ehrt S, Nathan C. 2011. Virulence of mycobacterium tuberculosis depends on lipoamide dehydrogenase, a member of three multienzyme complexes. Cell Host Microbe 9:21–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Motterlini R, Otterbein LE. 2010. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 9:728–743 [DOI] [PubMed] [Google Scholar]
- 48. Boczkowski J, Poderoso JJ, Motterlini R. 2006. Co-metal interaction: vital signaling from a lethal gas. Trends Biochem. Sci. 31:614–621 [DOI] [PubMed] [Google Scholar]
- 49. Wayne LG. 1976. Dynamics of submerged growth of Mycobacterium tuberculosis under aerobic and microaerophilic conditions. Am. Rev. Respir. Dis. 114:807–811 [DOI] [PubMed] [Google Scholar]
- 50. Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 96:1645–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Jr, Bloom BR. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460 [DOI] [PubMed] [Google Scholar]
- 52. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pei J, Tang M, Grishin NV. 2008. PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucleic Acids Res. 36:W30–W34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Frickey T, Lupas A. 2004. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20:3702–3704 [DOI] [PubMed] [Google Scholar]
- 55. Pei J, Grishin NV. 2001. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17:700–712 [DOI] [PubMed] [Google Scholar]
- 56. Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64:2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ohol YM, Goetz DH, Chan K, Shiloh MU, Craik CS, Cox JS. 2010. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe 7:210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nuclease activity of purified recombinant Cor. (A) Coomassie-stained SDS gel of purified His-MBP-Cor and His-MBP alone. Lanes: molecular weight marker (lane 1), whole-cell lysate (WCL) of uninduced BL21 expressing His-MBP and His-MBP-Cor (lanes 2 and 7, respectively), WCL of induced (1 mM IPTG) BL21 expressing His-MBP and His-MBP-Cor (lanes 3 and 8, respectively), purified His-MBP and His-MBP-Cor (lane 5 and 10, respectively). (B) M. tuberculosis total RNA incubated with increasing concentrations (0.5, 2.5, and 5.0 µM) of purified His-MBP or His-MBP-Cor or buffer alone. RNase A incubated with M. tuberculosis RNA was used as a positive control. Arrowheads indicate different RNA species. (C) M. smegmatis (mc2 155) genomic DNA was incubated with increasing concentrations (0.5, 2.5, and 5.0 µM) of purified His-MBP or His-MBP-Cor. The arrowhead indicates bands of copurifying RNA. Download
Heat map of statistically significant biochemicals profiled in this study.