

ABSTRACT
Disulfide bond formation is required for the folding of many bacterial virulence factors. However, whereas the Escherichia coli disulfide bond-forming system is well characterized, not much is known on the pathways that oxidatively fold proteins in pathogenic bacteria. Here, we report the detailed unraveling of the pathway that introduces disulfide bonds in the periplasm of the human pathogen Pseudomonas aeruginosa. The genome of P. aeruginosa uniquely encodes two DsbA proteins (P. aeruginosa DsbA1 [PaDsbA1] and PaDsbA2) and two DsbB proteins (PaDsbB1 and PaDsbB2). We found that PaDsbA1, the primary donor of disulfide bonds to secreted proteins, is maintained oxidized in vivo by both PaDsbB1 and PaDsbB2. In vitro reconstitution of the pathway confirms that both PaDsbB1 and PaDsbB2 shuttle electrons from PaDsbA1 to membrane-bound quinones. Accordingly, deletion of both P. aeruginosa dsbB1 (PadsbB1) and PadsbB2 is required to prevent the folding of several P. aeruginosa virulence factors and to lead to a significant decrease in pathogenicity. Using a high-throughput proteomic approach, we also analyzed the impact of PadsbA1 deletion on the global periplasmic proteome of P. aeruginosa, which allowed us to identify more than 20 new potential substrates of this major oxidoreductase. Finally, we report the biochemical and structural characterization of PaDsbA2, a highly oxidizing oxidoreductase, which seems to be expressed under specific conditions. By fully dissecting the machinery that introduces disulfide bonds in P. aeruginosa, our work opens the way to the design of novel antibacterial molecules able to disarm this pathogen by preventing the proper assembly of its arsenal of virulence factors.
IMPORTANCE
The human pathogen Pseudomonas aeruginosa causes life-threatening infections in immunodepressed and cystic fibrosis patients. The emergence of P. aeruginosa strains resistant to all of the available antibacterial agents calls for the urgent development of new antibiotics active against this bacterium. The pathogenic power of P. aeruginosa is mediated by an arsenal of extracellular virulence factors, most of which are stabilized by disulfide bonds. Thus, targeting the machinery that introduces disulfide bonds appears to be a promising strategy to combat P. aeruginosa. Here, we unraveled the oxidative protein folding system of P. aeruginosa in full detail. The system uniquely consists of two membrane proteins that generate disulfide bonds de novo to deliver them to P. aeruginosa DsbA1 (PaDsbA1), a soluble oxidoreductase. PaDsbA1 in turn donates disulfide bonds to secreted proteins, including virulence factors. Disruption of the disulfide bond formation machinery dramatically decreases P. aeruginosa virulence, confirming that disulfide formation systems are valid targets for the design of antimicrobial drugs.
INTRODUCTION
The structures of many secreted proteins are stabilized by the formation of disulfide bonds. In Gram-negative bacteria, disulfide bond formation takes place in the periplasm, a viscous compartment that separates the outer membrane from the inner membrane (1–3). The first bacterial machinery catalyzing the formation of disulfide bonds has been discovered in Escherichia coli where it involves a soluble oxidoreductase, E. coli DsbA (EcDsbA) (4), and an inner membrane protein, EcDsbB (5). EcDsbA functions as the primary donor of disulfide bonds to proteins exported to the periplasm; it is a soluble monomeric protein that adopts a thioredoxin (Trx) fold and has a CXXC catalytic motif (4). The cysteine residues of the catalytic motif of EcDsbA are maintained in the oxidized state in vivo, which enables EcDsbA to react with proteins entering the periplasm to oxidize them (4). After transfer of the catalytic disulfide bond to target proteins, EcDsbA is released in the reduced state and reoxidized by EcDsbB (5). The membrane protein EcDsbB has four transmembrane segments and two small hydrophilic regions, both containing two cysteine residues that are exposed to the periplasm. These cysteine residues are required for EcDsbB activity: they shuttle the electrons away from EcDsbA and deliver them to bound quinone molecules. The electrons are then transferred to the respiratory chain, which finally results in the reduction of molecular oxygen (6). Under anaerobic conditions, EcDsbB transfers the electrons to menaquinone and then to other terminal electron acceptors such as fumarate and nitrate (6–8). More than 30 EcDsbA substrates have been identified so far using various proteomic techniques, but the number of proteins predicted to depend on EcDsbA for folding is much higher (9–12).
EcDsbA preferentially introduces disulfide bonds in a vectorial manner into proteins entering the periplasm (13), i.e., between cysteine residues that are consecutive in the amino acid sequence. Therefore, EcDsbA often incorrectly oxidizes proteins whose folding involves the formation of disulfide bonds between nonconsecutive cysteines. In E. coli, these nonnative disulfide bonds are corrected by an isomerization system, which involves EcDsbC and EcDsbD (14–18).
The E. coli disulfide bond formation pathway is often considered the paradigm of oxidative protein folding machinery in bacteria. However, genome analyses have revealed that the E. coli system cannot serve as a model for all bacteria (19, 20). For instance, many bacterial genomes encode a repertoire of thiol-disulfide oxidoreductases that is significantly expanded than the E. coli DsbA-DsbB system (19). As an illustrative example, the pathogenic bacterium Neisseria meningitidis has three DsbA proteins, which differ in terms of localization, redox properties, and surface characteristics (21–23). Furthermore, in certain bacteria, such as Mycobacterium tuberculosis, DsbA is not reoxidized by DsbB, but by a membrane protein homologous to the eukaryotic vitamin K epoxide reductase (VKOR) (20).
In this work, we characterize in detail the machinery that forms disulfide bonds in the periplasm of Pseudomonas aeruginosa, an opportunistic human pathogen that causes life-threatening infections in patients suffering from leukemia, AIDS, cancer, and cystic fibrosis (24). We show that the P. aeruginosa genome encodes multiple Dsb proteins, including two DsbA proteins (P. aeruginosa DsbA1 [PaDsbA1] and PaDsbA2) and two DsbB proteins (PaDsbB1 and PaDsbB2). PaDsbA1, which is the primary donor of disulfide bonds to proteins secreted to the envelope, is recycled by both PaDsbB1 and PaDsbB2. Accordingly, the simultaneous deletion of both P. aeruginosa dsbB (PadsbB) genes is required to impair the folding of several P. aeruginosa virulence factors, leading to a global decrease in pathogenicity. We also reconstituted the P. aeruginosa disulfide bond formation system in vitro and determined the kinetic parameters of the oxidation reaction of PaDsbA1 by both PaDsbB1 and PaDsbB2. Finally, we report the identification of 22 new potential substrates of PaDsbA1 and the biochemical and structural characterization of PaDsbA2, an oxidoreductase which seems to be expressed under specific conditions.
RESULTS
The P. aeruginosa genome encodes two DsbA proteins and two DsbB proteins.
We initiated our study by searching the genome of P. aeruginosa strain PA14 for homologs of EcDsbA and EcDsbB using the psi-BLAST algorithm (25). We found two proteins homologous to EcDsbA (PaDsbA1 [PA14_72450] and PaDsbA2 [PA14_59960] that have 27% and 14% sequence identity with EcDsbA, respectively) and two proteins homologous to EcDsbB (PaDsbB1 [PA14_07000] and PaDsbB2 [PA14_69400] that have 29% and 27% sequence identity with EcDsbB, respectively). The structural and biochemical properties of PaDsbA1 have been reported elsewhere (24, 26). Moreover, PaDsbA1 has been shown to be involved in the oxidative folding of various virulence factors, including elastase (26, 27), and PadsbA1 mutants have been described as less virulent than wild-type strains in several models, including Drosophila melanogaster (28), Caenorhabditis elegans (29), Arabidopsis thaliana, and mice (29, 30). However, nothing is known about PaDsbA2, PaDsbB1, and PaDsbB2. Interestingly, we noticed that PaDsbA2 belongs to a different subclass of DsbA proteins than EcDsbA and PaDsbA1 (see Fig. S1A in the supplemental material). The proteins from this subclass, mostly present in alphaproteobacteria, are characterized by the presence of an additional pair of conserved cysteine residues (Fig. S1B), as discussed further below. Thus, the machinery at work in P. aeruginosa differs from the E. coli system by the fact that multiple DsbA and DsbB proteins are encoded by the genome of this microorganism, raising the question of their respective role and contribution in oxidative protein folding.
PaDsbB1 and PaDsbB2 cooperate in the recycling of PaDsbA1.
It was important to first confirm the physiological importance and the functional roles of the various Dsb proteins identified by the bioinformatic analysis. First, we decided to determine the in vivo redox state of both PaDsbA1 and PaDsbA2 to confirm their involvement in a pathway that introduces disulfide bonds into substrates. In general, proteins that function in an oxidizing pathway accumulate in the oxidized state in vivo, whereas proteins that function as a reductase or isomerase involved in the correction of nonnative disulfide bonds, such as EcDsbC, accumulate in the reduced state (31). The in vivo redox state of PaDsbA1 and PaDsbA2 was determined using 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), a 490-Da reagent that covalently reacts with free thiol groups, leading to a mobility shift of the modified protein in SDS-polyacrylamide gels. As shown in Fig. 1, we found that PaDsbA1 is predominantly oxidized in vivo (Fig. 1, compare lane 1 to lane 6 showing the migration of reduced PaDsbA1 modified with AMS), which is consistent with PaDsbA1 playing a role as a thiol oxidase. PaDsbA2 was not detected, despite repeated attempts in different culture media, which suggests that this protein is expressed only under specific conditions that remain to be determined. For that reason, we were not able to further investigate the in vivo function of PaDsbA2. However, the results from the biochemical and structural characterization of this protein are presented below.
FIG 1 .

PaDsbA1 is maintained oxidized in vivo by both PaDsbB1 and PaDsbB2. Exponentially growing cells (in LB) were TCA precipitated, free cysteines were modified by AMS (+) (except for the sample shown in lane 7), and PaDsbA1 was detected by Western blotting using an anti-PaDsbA1 antibody. The strains used in this experiment are the wild-type P. aeruginosa PA14 strain (lanes 1, 6, and 7) and the PadsbA1 (lane 2), PadsbB1 (lane 3), PadsbB2 (lane 4), and PadsbB1B2 (lane 5) mutants. To prepare the sample shown in lane 6, wild-type cells were incubated with 10 mM dithiothreitol (DTT) (+) prior to TCA precipitation in order to reduce PaDsbA1. Abbreviations: red, reduced; ox, oxidized.
We then sought to determine the respective roles of PaDsbB1 and PaDsbB2 in the reoxidation of PaDsbA1 by monitoring the redox state of this oxidoreductase in PadsbB1 and PadsbB2 mutants. As shown in Fig. 1, deletion of either PadsbB1 or PadsbB2 does not have a significant impact on the redox state of PaDsbA1 (Fig. 1, lanes 3 and 4). However, when both PaDsbB proteins (PaDsbBs) are absent, PaDsbA1 accumulates mostly in the reduced form (Fig. 1, lane 5). These results indicate that both PaDsbB1 and PaDsbB2 control the redox state of PaDsbA1 and that both proteins are able to compensate for each other. It is noteworthy that expression of either PaDsbB protein in an E. coli dsbB (EcdsbB) mutant complements the motility defect of this mutant, indicating that both PaDsbB1 and PaDsbB2 are able to reoxidize EcDsbA (see Fig. S2 in the supplemental material).
In vitro reconstitution of the disulfide bond formation machinery of P. aeruginosa.
We decided to reconstitute the disulfide bond formation pathway of P. aeruginosa in vitro to determine the kinetic parameters of the reoxidation reaction of PaDsbA1 by PaDsbB1 and PaDsbB2. PaDsbB1 and PaDsbB2 were expressed in an EcdsbB mutant and partially purified. We then tested the ability of these enzymes to directly oxidize PaDsbA1 by using a fluorescence assay developed for DsbB activity (6, 32, 33). This assay is based on the 1.7-fold fluorescence decrease that accompanies PaDsbA1 oxidation (see Fig. S3 in the supplemental material). As shown in Fig. 2A, the fluorescence of reduced PaDsbA1 decreases slowly when the protein is incubated at 30°C, probably as a result of air oxidation. Addition of quinones that function as electron acceptors in the DsbB-catalyzed reaction (6, 34) has no impact. In contrast, addition of PaDsbB1 to the mixture leads to a rapid decrease in PaDsbA1 fluorescence (Fig. 2A). Similar results were obtained for PaDsbB2 (not shown), confirming that both PaDsbB1 and PaDsbB2 are able to reoxidize PaDsbA1. Importantly, we confirmed using AMS trapping experiments that the observed decrease in fluorescence is due to the oxidation of PaDsbA1 cysteines (not shown). We also measured the dependence of the initial velocities of fluorescence decrease on the concentration of PaDsbA1. As shown in Fig. 2, the obtained data could be fit to the Michaelis-Menten equation for both PaDsbB1 (Fig. 2B) and PaDsbB2 (Fig. 2C). We calculated a Km of 8.1 µM PaDsbA1 for PaDsbB1 and 9.0 µM for PaDsbB2. These values are similar to the value reported for the Km of EcDsbB for EcDsbA (33). Thus, PaDsbB1 and PaDsbB2 have similar affinities for PaDsbA1.
FIG 2 .

Both PaDsbB1 and PaDsbB2 recycle PaDsbA1 in vitro. (A) The reoxidation of PaDsbA1 by PaDsbB1 was monitored by measuring the decrease in fluorescence (excitation wavelength, 295 nm; emission wavelength, 330 nm) that accompanies PaDsbA1 oxidation. The reaction was performed in the presence of quinones (decylubiquinone). The various components were added sequentially to the reaction mixture as indicated. (B and C) We measured the initial velocities (v) of PaDsbA1 reoxidation by PaDsbB1 (B) and PaDsbB2 (C) to determine the kinetic parameters of the reaction. The plots show a fit of the data to the Michaelis-Menten equation. The experimental conditions are described in Materials and Methods.
Both PaDsbB1 and PaDsbB2 play a role in the oxidation of P. aeruginosa virulence factors.
From the data presented above, we can conclude that PaDsbB1 and PaDsbB2 control the redox state of PaDsbA1 both in vitro and in vivo. However, the respective importance of the two DsbB proteins for the pathogenicity of P. aeruginosa remains unknown, which prompted us to investigate their roles in the assembly of virulence factors.
We first tested the involvement of PaDsbB1 and PaDsbB2 in the assembly of type IV pili and flagellum, two structures involved in bacterial pathogenesis and whose formation requires PaDsbA1 (Fig. 3A) (26, 35). As shown in Fig. 3A, we found that the PadsbB1B2 double mutant is defective in both twitching and swimming motilities, which require functional type IV pili and flagellum, respectively, but that the single PadsbB mutants have a normal phenotype. The PadsbB1B2 mutant also exhibits a significant defect in the early steps of biofilm formation, which most likely results from defective assembly of the pili and flagellum (see Fig. S4A in the supplemental material) (26, 27). Moreover, the PadsbB1B2 double mutant, but not the single PadsbB mutants, exhibits a reduced elastase activity (Fig. S4B). The elastase LasB is an extracellular protease with two disulfide bonds in its native structure (36) whose formation has been reported to depend on PaDsbA1 (26, 27). It is important to note that the phenotypes described above could be complemented by trans expression of the corresponding genes (Fig. S4C), the double PadsbB1B2 mutant being complemented by expression of either individual PadsbB1 or PadsbB2 gene.
FIG 3 .
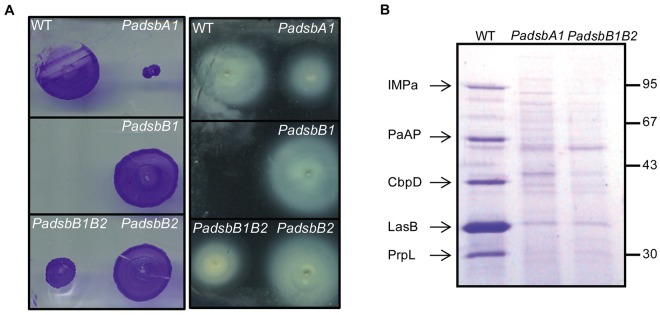
Deletion of both PadsbB1 and PadsbB2 impairs the oxidative folding of extracellular appendages and exoproteins. (A) Strains lacking PadsbA1 or PadsbB1 and PadsbB2 have twitching (left) and swimming (right) motility defects, but not the single PadsbB1 and PadsbB2 mutants. (B) In order to identify additional exoproteins affected by the impairment of the disulfide formation machinery, we analyzed the culture supernatants prepared from the wild type (WT) and the various Padsb mutants on a Coomassie brilliant blue-stained SDS-polyacrylamide gel (12%). The five proteins that are secreted by the wild type but not by the PadsbA1 and PadsbB1B2 mutants are indicated on the left. The molecular weight markers (in kilodaltons) are indicated on the right.
We also analyzed the protein contents of culture supernatants prepared from the wild type, the PadsbA1 mutant, and the PadsbB1B2 double mutant in order to find out whether the simultaneous deletion of PadsbB1 and PadsbB2 impacts the assembly of exoproteins other than elastase. As shown in Fig. 3B, we found four additional exoproteins, including the chitin-binding protein CbpD (PA14_53250), the recently characterized immunomodulating metalloprotease IMPa (PA14_07430) (37) and the proteases PaAP (PA14_26020) and PrpL (PA14_09900) that were absent in the supernatants of the PadsbB1B2 double mutant and the PadsbA1 mutant. These four proteins have not yet been reported to require disulfide bonds for folding. However, the fact that they all contain conserved cysteine residues in their sequence (4 for CbpD, 5 for IMPa, 2 for PaAP, and 6 for PrpL) strongly suggests that they are new substrates of the disulfide bond formation machinery of P. aeruginosa and that, like the elastase LasB, they need to oxidatively fold in the periplasm prior to secretion (27). In the Discussion, we explain why we think that the observed decrease in secretion levels results from an impairment of the folding process of these proteins and not from defective assembly of the type II secretion machinery.
Finally, we investigated the global roles of PaDsbB1 and PaDsbB2 in P. aeruginosa virulence by measuring the ability of the various PadsbB mutant strains to kill C. elegans using a previously established assay (38). Remarkably, we found that only the PadsbB1B2 mutant is significantly less virulent than the wild-type strain but that the single PadsbB mutants are not less virulent (see Fig. S4D in the supplemental material). Altogether, these data lead to the conclusion that both PaDsbB1 and PaDsbB2 participate in the folding of several virulence factors of P. aeruginosa and that both PadsbB genes need to be deleted to impact the global assembly of virulence factors, confirming that these genes have a redundant function.
New proteins that depend on the disulfide bond formation pathway for folding.
A few P. aeruginosa virulence factors have been shown to depend on PaDsbA1 for folding, including the protease LasB (27) and the pilus component PilA (35). However, most PaDsbA1 substrates remain to be identified. Thus, in order to fully grasp the role of the disulfide bond-forming system involving PaDsbA1, PaDsbB1, and PaDsbB2 in the folding of periplasmic proteins in P. aeruginosa and to identify new virulence factors depending on the disulfide bond formation machinery for assembly, we used a differential proteomic approach based on label-free two-dimensional liquid chromatography coupled to tandem mass spectrometry (2D-LC-MS/MS) to compare the periplasmic proteome of PadsbA1 and wild-type strains. The rationale behind this approach, which has proved to be useful in identifying the substrates of EcDsbA (12) and the E. coli periplasmic chaperone SurA (39, 40), is that proteins whose folding depends on the presence of a functional disulfide bond formation pathway will be less abundant in the PadsbA1 strain than in the wild type due to misfolding and protease digestion.
We prepared periplasmic extracts containing cell envelope proteins using a protocol adapted from the method of Hiniker and Bardwell (11) and optimized for P. aeruginosa in order to limit contamination by cytoplasmic proteins. Periplasmic proteins were digested with trypsin, and the peptides were analyzed by 2D-LC-MS/MS. The experiments were repeated on three biological replicates for both the PadsbA1 and wild-type strains. For quantification of protein abundance, we used the number of normalized spectral counts (SC) reported for each protein (see Tables S1 and S2 in the supplemental material), a value which is linearly correlated with protein abundance (12, 39–41).
An average of 499 ± 40 and 453 ± 73 proteins (with spectral count values of ≥5) were identified for the wild type and the PadsbA1 strain, respectively. The localization of all the identified proteins was predicted using PSORT (42). Each run allowed us to identify up to 112 envelope proteins (periplasmic, outer membrane, and extracellular proteins). The remaining proteins were soluble cytoplasmic proteins, inner membrane proteins, and proteins of unknown localization (according to PSORT) that contaminated the envelope fraction.
We then compared the relative abundance of the identified proteins in the wild-type and PadsbA1 strains to determine the global impact of the PadsbA1 deletion on the periplasmic proteome. Postulating that misfolding of PaDsbA1 substrates leads to their degradation, we selected proteins whose abundance was decreased by at least twofold (Table 1) in the PadsbA1 mutant. To test the statistical significance of the data, we used the unpaired Student’s t test and defined statistical significance as a P value of <0.05 (two-tailed two-sample equal variance test). We found 23 envelope proteins whose abundance was significantly decreased in the mutant (Table 1). These proteins include periplasmic binding proteins (PA14_03930, PA14_71030, and PA14_07870), putative periplasmic enzymes involved in peptidoglycan remodeling (PA14_24690 and PA14_25000), and potential extracellular virulence factors (PA14_26020 and PA14_39780). Importantly, to the exclusion of PbpG (PA14_53020) and ChiC (PA14_34870), all the less abundant proteins contain an even number of cysteine residues, which strongly suggests that they are PaDsbA1 substrates. Moreover, CbpD, IMPa, and PaAP, three proteins identified above as PaDsbA1 substrates (Fig. 3B), and PilA, a known PaDsbA1 substrate (35), were also found to be less abundant in the periplasm of the PadsbA1 mutant, which further validates our proteomic data.
TABLE 1 .
Proteins more than twofold less abundant in a PadsbA1 mutant than in a wild-type P. aeruginosa PA14 strain (P < 0.05)
| UniProtKB/TrEMBL accession no. |
Description of protein | No. of Cysa | Localizationb | Ratio (PadsbA1/WT)c |
|---|---|---|---|---|
| Q02I29 | d-Alanyl-d-alanine-endopeptidase PbpG (PA14_53020) | 1 | P | 0.31 |
| Q02UB6 | Polyamine transport protein SpuE (PA14_03930) | 2d | P | 0.42 |
| Q02DY4 | Putative glycine betaine/l-proline ABC transporter periplasmic component (PA14_71030) | 2 | P | 0.29 |
| Q02N84 | Putative periplasmic spermidine/putrescine-binding protein PotF (PA14_30570) | 2d | P | 0.21 |
| Q02TF9 | Putative binding protein component of ABC transporter (PA14_07870) | 2 | P | 0.11 |
| Q02QW4 | Cyclohexadienyl dehydratase PheC (PA14_19140) | 2d | P | 0.33 |
| Q02EI0 | Putative periplasmic monofunctional chorismate mutase (PA14_68480) | 2d | P | 0.00 |
| Q02PI4 | Putative soluble lytic transglycosylase Slt (PA14_25000) | 2 | P | 0.00 |
| Q02PL1 | Putative d-alanyl-d-alanine-carboxypeptidase DacB (PA14_24690) | 2 | P | 0.00 |
| Q02GT9 | Putative binding protein component of ABC dipeptide (PA14_58420) | 4d | P | 0.26 |
| Q02E45 | Putative binding protein component of ABC dipeptide transporter (PA14_70200) | 4d | P | 0.23 |
| Q02GU4 | Putative binding protein component of ABC transporter (PA14_58360) | 4d | P | 0.40 |
| Q02ER3 | Putative ABC transporter periplasmic substrate-binding protein (PA14_67400) | 4 | P | 0.08 |
| Q02S18 | Putative amino acid ABC transporter-binding protein YhdW (PA14_14100) | 4 | P | 0.00 |
| Q02TC1 | Putative phage-related protein tail component JF1 (PA14_08300) | 4 | OM | 0.38 |
| Q02DR8 | Putative uncharacterized protein (PA14_71840) | 8 | OM | 0.06 |
| Q02M97 | Chitinase ChiC (PA14_34870) | 1 | E | 0.47 |
| Q02GR5 | Type IV pilin structural subunit PilA (PA14_58730) | 2d | E | 0.32 |
| Q02L18 | Staphylolytic protease preproenzyme LasA (PA14_40290) | 4d | E | 0.18 |
| Q02PA2 | Putative aminopeptidase (PA14_26020) | 6 | E | 0.10 |
| Q02I11 | Chitin-binding protein CbpD (PA14_53250) | 8d | E | 0.22 |
| Q02TJ3 | Putative uncharacterized protein (PA14_07430) | 8 | E | 0.03 |
| Q02L61 | Putative halovibrin Hvn (PA14_39780) | 10 | E | 0.17 |
Number of cysteines present in the mature protein sequence.
The localizations of the proteins are shown as follows: P, periplasm; OM, outer membrane; and E, extracellular.
Ratio of the amount of protein in a PadsbA1 mutant to the amount of protein in the wild type (WT).
Homologous proteins from other bacteria that possess at least one conserved disulfide bond in their three-dimensional structure.
Biochemical and structural characterization of PaDsbA2.
As explained above, the genome of P. aeruginosa encodes a second DsbA, PaDsbA2, which belongs to a different subclass of DsbA proteins characterized by the presence of an additional pair of cysteine residues. We found that expression of PaDsbA2 from a plasmid does not complement the phenotype of a PadsbA1 mutant (not shown), suggesting that these two proteins have different roles in P. aeruginosa and different substrate specificities. However, as we failed to find the conditions under which PaDsbA2 is expressed, we could not investigate further the in vivo function of this protein.
In order to gain some insights into the role of PaDsbA2, we decided to characterize its biochemical, redox, and structural properties. First, we tested whether PaDsbA2 exhibits an oxidoreductase activity by evaluating its ability to catalyze the reduction of insulin by dithiothreitol (DTT). The reduction of insulin disulfides causes precipitation of the β-chain, which can be monitored by following the absorbance at 650 nm (43). As shown in Fig. 4A, PaDsbA2 is able to catalyze insulin reduction, although less efficiently than EcDsbA and PaDsbA1. Second, we determined whether PaDsbA2 can be reoxidized in vitro by PaDsbB1 and PaDsbB2, using purified components. Because PaDsbA2 does not exhibit a change in fluorescence, we followed its reoxidation using AMS trapping experiments. As shown in Fig. 5, we found that PaDsbA2 can be reoxidized by PaDsbB1 and PaDsbB2, confirming that it is a substrate for both P. aeruginosa DsbBs. We then determined the redox potential (Eo′) of the CXXC catalytic motif of PaDsbA2 by equilibrating the protein in various glutathione buffers. The redox potential of the CXXC motif was found to have a value of −67.5 ± 2.5 mV, which makes PaDsbA2 one of the most oxidizing oxidoreductases ever characterized (Fig. 4B). Such an oxidizing redox potential is also consistent with a function for PaDsbA2 as a thiol oxidase. Interestingly, mutation of the additional cysteine residues to serines decreases the redox potential of the CXXC motif to −118.2 ± 1.8 mV (see Fig. S5 in the supplemental material). As these cysteines form a disulfide bond in the structure of PaDsbA2 (see below), this indicates that the additional disulfide bond of PaDsbA2 controls the highly oxidizing redox potential of the catalytic site.
FIG 4 .

PaDsbA2 is an oxidoreductase with a highly oxidizing redox potential. (A) We tested the ability of PaDsbA2 to function as an oxidoreductase by measuring its ability to catalyze the reduction of insulin by DTT. Insulin (150 µM) was incubated with 10 µM of either PaDsbA1 (▼), PaDsbA2 (●), or EcDsbA (♦). The noncatalyzed reaction (DTT alone; ○) was also monitored. DTT (0.8 mM) was added at time zero, and the reduction of insulin was measured by monitoring the increase in absorbance at 650 nm. The data shown are averages of three independent experiments. (B) We determined the redox potential of PaDsbA2 by equilibrating the protein in redox buffers containing different GSH/GSSG ratios. The redox potential was calculated from the ratio (R) between the amounts of oxidized and reduced PaDsbA2 present at equilibrium and determined using AMS trapping experiments. The data shown are averages (± standard deviations [error bars]) of three independent experiments.
FIG 5 .

PaDsbA2 is reoxidized by both PaDsbB1 and PaDsbB2 in vitro. The reoxidation of PaDsbA2 by PaDsbB1 and PaDsbB2 was monitored over time by AMS trapping. The reaction was performed in the presence of quinones (decylubiquinone) when indicated. PaDsbA2 alone and PaDsbA2 with quinones (+) (top right panel) were used as negative controls. The experimental conditions are described in Materials and Methods. Abbreviations: red, reduced; ox, oxidized.
We also solved the structure of PaDsbA2 using single anomalous dispersion (SAD) (see Text S1 in the supplemental material for more information on the crystallization conditions and data analysis). The structure, which was refined to 1.3 Å with an R/Rfree ratio of 19.32%/21.96% (Text S1), consists of a thioredoxin fold in which a helical domain is inserted (Fig. 6A). This architecture resembles that of other DsbA proteins, confirming the identification of PaDsbA2 as a DsbA. In particular, the structure of PaDsbA2 superimposes onto the structures of PaDsbA1 (PDB accession no. 3H93, 14% sequence identity) and EcDsbA (PDB accession no. 1FVK, 14% sequence identity) with root mean square deviations (RMSDs) of 2.4 Å and 2.9 Å, respectively. The closest structural homolog of PaDsbA2 from a Dali search (44) is a 27-kDa outer membrane protein from Silibacter pomeroyi DSS-3, a bacterioplankton (PDB accession no. 3GYK, 24% sequence identity, RMSD 2.2 Å). Wolbachia pipientis alpha-DsbA1 (α-DsbA1) (PDB accession no. 3F4R), whose sequence presents 26% identity with PaDsbA2, has a lower Z score and an RMSD of 2.7 Å. A major difference in the structure of PaDsbA2 compared to its closest neighbors and other DsbAs is located in the helical domain where residues 131 to 151 form a coil instead of a helix. This coil links helix 4 to helix 5 that is disulfide bonded to helix 3. The first cysteine (Cys66) of the CXXC motif, which is reduced in the structure, is located at the beginning of helix 1. The S-γ of the catalytic cysteines are 3.6 Å apart, which is too long for a stable hydrogen bond, whereas the S-γ of other Dsb proteins are generally within hydrogen bond distance (45–47). On the other hand, the S-γ of Cys66 receives five hydrogen bonds: two from the N-α of Cys69 and Phe68, two from the O-α and O-γ of Thr183, a residue of the conserved cis-Pro loop of proteins with a thioredoxin fold, and one from a water molecule (Fig. 6B). These five hydrogen bonds to the S-γ of Cys66 contribute in stabilizing the thiolate state, which could explain the very oxidizing redox potential of PaDsbA2. As observed in the structure of α-DsbA1 from W. pipientis, a disulfide bond is formed between the additional cysteines of the helical domain, Cys111 and Cys157. This disulfide bond links helices 3 and 5 of the helical domain and is approximately 20 Å away from the CXXC catalytic motif. An important feature that varies between DsbA proteins is their surface electrostatics. Here, we see that the surface of PaDsbA2 is more basic than that of EcDsbA and, to a lesser extent, that of PaDsbA1 (Fig. 6C). Moreover, EcDsbA has a hydrophobic patch and a hydrophobic groove surrounding the CXXC active site. These regions, the hydrophobic patch and hydrophobic groove, which are involved in substrate and DsbB binding, respectively (45), were shown to be truncated in PaDsbA1 compared to EcDsbA (24). In the case of PaDsbA2, many amino acids contributing to the hydrophobic groove of EcDsbA are replaced by polar or charged residues, as in the structure of α-DsbA1 from W. pipientis (48). As the hydrophobic groove is important for the interaction between EcDsbA and EcDsbB (45), this may explain why EcDsbB does not reoxidize PaDsbA2 (not shown).
FIG 6 .

PaDsbA2 adopts a typical DsbA-like structure, with a thioredoxin (Trx) domain and an α-helical domain. (A) The structure of PaDsbA2 was solved using single anomalous dispersion and refined to 1.3 Å with an R/Rfree ratio of 19.32%/21.96% and consists of a thioredoxin fold (in gray) in which a helical domain is inserted (in blue). The catalytic cysteines (C66 and C69) and the additional cysteines (C111 and C157) are shown in red. (B) The first cysteine (Cys66) of the CXXC motif is located at the beginning of helix 1 and is 3.6 Å away from the second catalytic cysteine (Cys69). The S-γ of Cys66 receives five hydrogen bonds: two from the N-α of Cys69 and Phe68, two from the O-α and O-γ of Thr183, and one from a water molecule. These five hydrogen bonds to the S-γ of Cys66 help stabilize the thiolate state, which could explain the very oxidizing redox potential of PaDsbA2. (C) The relative electrostatic surface potentials of PaDsbA1, PaDsbA2, and EcDsbA are shown. The electrostatic potential is given from −3 to 3 kT/e with k the Boltzmann constant, T the temperature, and e the elementary charge (charge of a proton). The respective active site cysteines and cis-proline are annotated.
DISCUSSION
The number of infections with multidrug-resistant Gram-negative bacteria, including P. aeruginosa, has dramatically increased in recent years, calling for the urgent development of new classes of antimicrobial drugs. Molecules targeting bacterial virulence rather than bacterial growth appear to be a particularly promising therapeutic approach to combat bacterial infections. Indeed, as virulence factors are usually not required for bacterial survival, antivirulence molecules provide the advantage of decreasing pathogenicity without applying a strong selective pressure for the development of bacterial resistance. By highlighting the crucial role played by PaDsbA1, PaDsbB1, and PaDsbB2 in the assembly of the arsenal of virulence factors of P. aeruginosa, our study provides the fundamental knowledge required to develop antivirulence molecules able to disrupt disulfide bond formation in this microorganism. In particular, the central role played by PaDsbA1 in the disulfide bond formation machinery makes it a target of choice for antivirulence strategy. However, it will be important to identify drugs targeting PaDsbA1 without interfering with the cellular proteins from the host that also present a thioredoxin fold. In that respect, the available structure of PaDsbA1 (24) should facilitate the design of molecules interacting with the specific surface characteristics of this protein. An alternative approach is the development of drugs inhibiting recycling of PaDsbA1 by PaDsbB1 and PaDsbB2, which do not have eukaryotic homologs. However, our study shows that only molecules capable of inhibiting both PaDsbBs with high affinity will be useful due to the functional redundancy between these two proteins.
Our work also raises the question of why two seemingly redundant DsbB proteins are encoded by the P. aeruginosa genome. Indeed, in the absence of a selective advantage for keeping two redundant genes, one of them should have progressively become nonfunctional. However, this is not the case, as we show that PaDsbB1 and PaDsbB2 are both able to reoxidize PaDsbA1 and PaDsbA2. It has been proposed that redundant genes may provide a cumulative advantage (49), each contributing to the global quantity of a protein involved in a specific cellular process. Thus, a first hypothesis is that under certain growth conditions, the presence of a single dsbB gene is not sufficient to drive disulfide bond formation and that efficient oxidative folding requires the presence of both PadsbB1 and PadsbB2. Alternatively, possession of two redundant dsbB genes may allow a broader spectrum of regulation than a single one: the expression of PadsbB1 and PadsbB2 may be differently modulated according to the environmental conditions, which would de facto give rise to more individualized functions for these two genes. For instance, it has recently been shown that the respective expression levels of two redundant operons involved in phenazine production in P. aeruginosa are differently modulated in liquid cultures and biofilms (50). Thus, the presence of two redundant dsbB genes may allow P. aeruginosa to adapt to different culture and environmental conditions.
We also report the identification of several new potential PaDsbA1 substrates. First, we found several secreted proteins, including virulence factors that are not secreted by the PadsbA1 and PadsbB1B2 mutants. The affected proteins all possess an even number of cysteine residues that are conserved in the sequence, which strongly suggests that they contain disulfide bonds in the native state (20). This is further reinforced by the fact that for one of these proteins, the chitin-binding protein CpbD, disulfide bonds have been observed in the structure of a homolog from Serratia marcescens (51, 52). It is striking that the affected exoproteins depend on the Xcp type II machinery for secretion to the extracellular environment (53). This pathway, unlike the type I and III systems that facilitate a one-step secretion across both the inner and outer membrane, generally translocates proteins that are first transported to the periplasm and then fold in this compartment prior to secretion (54). The absence of the exoproteins in the supernatants prepared from the PadsbB1B2 and PadsbA1 mutants is therefore likely to result from the inability of these proteins to oxidatively fold in the periplasm of these strains. Alternatively, the decrease in secretion could result from a defective assembly of the type II secretion machinery in the PadsbA1 and PadsbB1B2 mutants. However, we think that this is unlikely for at least three reasons. First, the type II secretion apparatus should not be affected by impairment of the disulfide bond machinery, as none of the proteins involved in its assembly contains disulfide bonds (54). Second, the hemolytic phospholipase PlcH, a type II secreted substrate that folds to the active form in the cytoplasm, is efficiently secreted by the PadsbB1B2 and PadsbA1 mutants (not shown). Third, three of the exoproteins whose abundance was decreased in the supernatants prepared from the PadsbB1B2 and PadsbA1 mutants (CpbD, IMPa, and PaAP) were found to be less abundant in the periplasm of the PadsbA1 mutant (see the results of the 2D-LC-MS/MS analysis), which is more consistent with a defect in folding than in secretion.
Additional PaDsbA1 substrates were also identified using a high-throughput proteomic approach. Here also, almost all the proteins whose abundance is decreased in the PadsbA1 mutant contain an even number of conserved cysteine residues, strongly suggesting that the decreased abundance results from impaired oxidative folding. Furthermore, for several of these proteins (Table 1), disulfide bonds are observed in the structures of homologs from other bacteria. We cannot rule out the possibility that for some proteins, the decrease in abundance results from decreased synthesis. However, our previous work on the E. coli disulfide bond system showed that for almost all proteins whose abundance is decreased in the periplasm of an EcdsbA mutant, the lower protein abundance does not result from reduced transcription (12, 39).
We also report that P. aeruginosa encodes a second DsbA-like protein, which we named PaDsbA2. Although the in vivo function of PaDsbA2 remains unclear, we provide biochemical evidence that this protein is an oxidoreductase with a highly oxidizing redox potential. The PaDsbA2 protein belongs to the class of α-DsbA proteins, whose most representative members are found in alphaproteobacteria (48). Proteins from this class are characterized by the presence of four conserved cysteine residues, relatively longer sequences, and the presence of an invariant threonine residue preceding the cis-proline found in proteins with a thioredoxin (Trx) fold (48). The structure and biochemical properties of α-DsbA1 from Wolbachia pipientis, another α-DsbA, have recently been reported (48). This protein, which exhibits 26% sequence identity with PaDsbA2, is significantly more reducing (−163 mV) than PaDsbA2 (48). Importantly, α-DsbA1 also contains a second disulfide bond for which a regulatory role has been proposed (48). Although a single periplasmic DsbA is sufficient for oxidative folding in many bacterial species, the presence of additional DsbAs has been reported in some Gram-negative bacteria where they usually catalyze the oxidation of specific target proteins. In Legionella pneumophila for instance, a newly identified DsbA protein specifically catalyzes disulfide bond formation in proteins involved in the type IV secretion system (55). In Salmonella enterica serovar Typhimurium, DsbL, which is homologous to DsbA, specifically oxidizes a periplasmic arylsulfate sulfotransferase, an enzyme that detoxifies phenolic substances and antibiotics (56). Further research to explore the conditions under which PaDsbA2 is expressed and to identify the function of this protein remains to be done.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and protein purification.
The bacterial strains, plasmids, and growth conditions used in this study and the protein purification methods are described in Text S2 in the supplemental material.
Insulin reduction assay.
Insulin reduction assays were performed by using methods described previously (43). Briefly, insulin (150 µM) and the various DsbA proteins (10 µM) used as a catalyst were mixed in 100 mM KPi (potassium phosphate) (pH 7.0) and 1 mM EDTA. The reaction was started by adding 0.8 mM dithiothreitol (DTT) (final concentration), and the reduction of insulin by DTT was monitored spectrophotometrically at 650 nm.
Redox potential determination.
The fractions of reduced and oxidized PaDsbA2 were determined using 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) trapping by the method of Denoncin et al. (57). Briefly, PaDsbA2 and PaDsbA2CCSS (1 µM) were incubated overnight at room temperature in 50 mM KPi pH 7.0, 0.1 mM EDTA and various glutathione (GSH)/glutathione disulfide (GSSG) ratios. After incubation, proteins were precipitated with trichloroacetic acid (TCA) (10% final concentration). After 20-min incubation on ice, the samples were centrifuged (Eppendorf F45-24-11 rotor, 16,100 × g, 5 min, 4°C), and the pellets were washed with cold acetone. After a second centrifugation, pellets were dried and resuspended in a buffer containing 20 mM AMS, 0.1% SDS, 10 mM EDTA, and 50 mM Tris-HCl (pH 7.5). After 45-min incubation at 37°C, with 1,400 rpm shaking under light protection, samples were loaded onto 12% SDS-polyacrylamide gels under denaturing conditions. Fractions of reduced and oxidized protein were determined using ImageJ (http://rsbweb.nih.gov/). The redox potential was then calculated as described previously (58).
In vivo redox state determination.
The in vivo redox state of PaDsbA1 has been assessed using AMS trapping experiments as described by Denoncin et al. (57). Briefly, bacteria were cultured at 37°C with 130 rpm shaking in LB medium until they reached an optical density at 600 nm (OD600) of 0.5. The proteins were then TCA precipitated (10% cold TCA) and resuspended in 30 µl of 20 mM AMS, 0.1% SDS, 10 mM EDTA, and 50 mM Tris-HCl (pH 7.5). Samples were incubated for 45 min at 37°C, with 1,400 rpm shaking, protected from light. For a positive control, protein pellets were treated with 50 mM DTT in the presence of 200 mM Tris (pH 8) and 1% SDS. Samples were loaded onto 12% SDS-polyacrylamide gels under denaturing conditions. After electrophoresis, the proteins were transferred to a nitrocellulose membrane and probed with an anti-PaDsbA1 antibody (1/3,000) produced from a rabbit immunized with the purified protein (CER, Marloie, Belgium). Anti-rabbit IgG (Sigma) was used as the secondary antibody at a concentration of 1/5,000. Thermo Scientific Pierce enhanced chemiluminescence (ECL) Western blotting substrate and Fuji film (Fuji) were used to visualize the protein bands.
Reoxidation of PaDsbA1 and -A2 by PaDsbB1 and -B2.
The reoxidation of PaDsbA1 and PaDsbA2 by PaDsbB1 and PaDsbB2 was performed in the presence of 30 µM decylubiquinone (2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinone) in a buffer containing 50 mM NaPi (pH 6.0), 300 mM NaCl, and 0.1% N-dodecyl-β-d-maltoside (DDM). The reoxidation of PaDsbA1 was followed in a spectrofluorimeter using 295 nm as the excitation wavelength and 330 nm as the emission wavelength. The plot of substrate concentrations versus initial velocity was fitted, using Prism, to the Michaelis-Menten equation, and the Km values were determined. The reoxidation of PaDsbA2 was followed by AMS trapping as described by Denoncin et al. (57).
E. coli complementation assays.
Swarming motility was assayed as described previously (59). Strains IA32 and IA33 were inoculated on M63 minimal medium soft agar plates containing 15 mM (NH4)2SO4, 100 mM KH2PO4 (pH 7.0), 1 mM MgSO4, 0.4% glucose, 0.2% arabinose, 10 µg/ml thiamine, 5 µg/ml mix NAD-riboflavin, 0.4 µg/ml biotin, 174 µg/ml of a mix of amino acids (except cysteine), and 0.3% agar. After 16 h of incubation at 37°C, motility was analyzed. Strains IA34 and JFC234 were used as negative controls.
Analysis of culture supernatants of wild-type and mutant P. aeruginosa strains.
P. aeruginosa strains were grown at 37°C in TSB medium (Difco Laboratories). Cells and supernatants were separated by centrifugation (Beckman Coulter JA-10 rotor, 2,375 × g, 10 min, 4°C); proteins contained in the supernatant were TCA precipitated (10% final concentration) for 1 h at 4°C. Samples were subsequently centrifuged (Beckman Coulter JA-10 rotor, 16,000 × g, 30 min, 4°C). The pellets were then washed with 90% acetone and resuspended in SDS-PAGE sample buffer. The samples were normalized according to the OD600 of the initial bacterial culture (0.1 OD600 unit of cell culture per μl of sample buffer) and loaded onto a 10.5% SDS-polyacrylamide gel. CbpD and LasB were identified by Western blotting using specific antibodies (not shown), while the three other proteins were identified by mass spectrometry.
Plate assays.
The proteolytic activity of P. aeruginosa PA14 wild-type and mutant strains was tested by growing colonies on tryptic soy agar (TSA) plates containing 1.5% skim milk. Elastase activity was tested on TSA plates containing 1% elastin. Flagellar motility assay was performed by spotting bacteria with a toothpick at the surface of LB agar plates (0.3% agar). Bacteria were grown at 30°C, and halos corresponding to the spreading of bacteria from the point of inoculation were observed. Twitching motility assay was performed on LB agar plates by inoculating bacteria through the agar to the bottom of the plate. The plates were incubated overnight at 37°C. The halo at the bottom of the plate was visualized with crystal violet coloration.
Biofilm formation assay and quantification.
The biofilm formation assay was performed in 24-well polystyrene microtiter dishes as described by Vallet et al. (60). Phenotypes are visualized after 2, 4, 6, and 8 h of incubation at 30°C. Bacterial cells bound to the walls of the wells were stained with 1% crystal violet. For quantification, these cells were suspended in 400 µl of 95% ethanol and 600 µl of water, and the OD600 was measured.
Caenorhabditis elegans slow killing assay.
The slow killing assay was performed as previously described (38). Each assay consisted of four replicates. Adult stage C. elegans worms were selected from plates containing overnight growth of each bacterial strain, and on a daily basis worms were evaluated for viability. Worm survival was plotted using the Prism 5.00 computer program. Survival curves were considered significantly different from the control when P values were <0.05. Prism calculates survival fractions using the product limit (Kaplan-Meier) method and compares survival curves by two methods: the log rank test and the Gehan-Breslow-Wilcoxon test.
Preparation of periplasmic extracts and proteolytic digestion.
Periplasmic extracts of P. aeruginosa PA14 were prepared following a protocol adapted from the method of Hiniker and Bardwell (11). P. aeruginosa PA14 wild-type and PadsbA1 cells were grown aerobically in 100 ml M63 minimal medium containing 174 µg/ml of each amino acid (except cysteine), at 37°C with 130 rpm shaking, to an OD600 of 0.8. Cells were harvested by centrifugation (Beckman Coulter JA-10 rotor, 3,000 × g, 20 min, 4°C). Pellets were gently resuspended in 1 ml buffer containing 30 mM KPi (pH 8.0) and 20% sucrose. After 30 min of incubation at 4°C, suspensions were centrifuged (Eppendorf F45-24-11 rotor, 16,100 × g, 30 min, 4°C) in 2-ml Eppendorf tubes. Supernatants contain periplasmic proteins. Protein concentration was determined using the Bradford assay. Periplasmic proteins (100 µg) were then precipitated by adding TCA to a final concentration of 10% (wt/vol). Differential thiol labeling and protein digestion were performed as described previously (12). The reaction was stopped by adding TFA to a final concentration of 0.1% (vol/vol), and the sample was stored at −20°C.
Label-free differential 2D-LC-MS/MS.
Tryptic digests were first desalted and concentrated on HyperSep C18 cartridges (50 mg/ml; Thermo Scientific, San Jose, CA) according to the manufacturer’s instructions. Peptides were then loaded onto a hydrophilic interaction chromatography (HILIC) column TSKgel amide-80 (4.6 mm by 25 cm; Tosoh Bioscience, Stuttgart, Germany) equilibrated with solvent B (98% acetonitrile [ACN] [vol/vol], 0.1% [vol/vol] TFA in water) and connected to an Agilent 1100 high-performance liquid chromatography (HPLC) system. The peptides were separated using a 70-min elution gradient that consisted of 5 to 45% solvent A (2% ACN [vol/vol], 0.1 [vol/vol] TFA in water) at a flow rate of 500 µl/min. Absorbance was monitored at 214 nm to ensure that all samples contained similar amounts of material. Fractions were collected at 2-min intervals (20 in total) and dried using a Speedvac. Peptides were resuspended in 10 µl of solvent C (5% ACN [vol/vol], 0.01% [vol/vol] TFA in water) and analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) as described in Text S3 in the supplemental material.
Crystallization of wild-type PaDsbA2.
The conditions used to crystallize wild-type PaDsbA2, to collect data, to solve the structure, and to analyze the electrostatic surface are described in Text S1 in the supplemental material.
Protein database accession number.
The structure of PaDsbA2 has been deposited in the Protein Data Bank under accession number 4N30.
SUPPLEMENTAL MATERIAL
Additional information on analysis of the structure of PaDsbA2. Download
Bacterial strains, growth conditions, and protein purification. Download
Protein identification and quantification by 2D-LC-MS/MS. Download
Bioinformatic analysis of the DsbA proteins from P. aeruginosa. (A) Phylogenetic analysis of the DsbA family shows that DsbA proteins can be divided in two subgroups. PaDsbA1 and PaDsbA2 belong to subgroups 1 and 2, respectively. The tree was constructed using FigTree v1.3.1 (A. Rambaut) based on the amino acid sequences of 20 proteins homologous to PaDsbA1 or PaDsbA2. The purple bars represent sequences that align with PaDsbA1, and the blue bars represent sequences that align with PaDsbA2. Gammaproteobacteria are shown in black, alphaproteobacteria in pink, betaproteobacteria in indigo, and deltaproteobacteria in cyan. The scale bar, below the tree, indicates the number of substitutions per amino acid. (B) The sequences of PaDsbA1 and PaDsbA2 were aligned with the sequences of homologous proteins from Moraxella catarrhalis, Neisseria meningitidis, Escherichia coli, Methylobacterium populi, Wolbachia pipientis, and Polynucleobacter necessarius using MacVector (MacVector, Inc.). The cysteines of the catalytic CXXC motif are outlined in purple. The additional cysteines found in PaDsbA2 and other DsbA proteins from subgroup 2 are outlined in blue. Dark gray and light gray shading indicate identical amino acids and amino acids from the same group (nonpolar, polar, acidic, or basic), respectively. Download
PaDsbB1 and PaDsbB2 can both complement an EcdsbB deficiency. Complementation tests of the EcdsbB strain by PaDsbB1 and PaDsbB2 were performed on semisolid agar plates containing 0.2% arabinose. Restoration of the motility due to the correct folding of FlgI, a component of the flagellum, was observed when PaDsbB1 or PaDsbB2 is expressed in the EcdsbB mutant, indicating that both proteins are able to reoxidize EcDsbA. Download
Fluorescence emission spectra of oxidized and reduced PaDsbA1. Fluorescence emission spectra of oxidized PaDsbA1 (broken line) and reduced PaDsbA1 (solid line) were recorded at protein concentrations of 2 µM in 50 mM potassium phosphate (pH 7) and 0.1 mM EDTA at room temperature. At 340 nm, the intrinsic tryptophan fluorescence of PaDsbA1 increases about 1.7 upon reduction of the enzyme. Download
Deletion of both PadsbB1 and PadsbB2 impairs biofilm formation, prevents the oxidative folding of elastase, and decreases the global virulence of P. aeruginosa. (A) PadsbA1 and PadsbB1B2 mutants are defective in the early steps of biofilm formation. Biofilm formation was visualized in wells of a microtiter dish after crystal violet staining (purple circle that is visible only in the wells containing the wild-type [WT] strain). (B) Elastase, a protease with two disulfide bonds, is secreted by the Xcp type II secretion machinery in P. aeruginosa. The nonspecific and elastin-specific protease activity of elastase is tested on plates containing skim milk (left) and elastin (right), respectively. The halos observed around the colonies of the wild type and of the single PadsbB1 and PadsbB2 mutants correspond to milk or elastin degradation by elastase. The Paxcp mutant, used as a negative control, is unable to assemble the type II secretion machinery and therefore to secrete elastase. (C) Phenotypic complementation of Padsb mutants on skim milk plate. Wild-type secretion is restored when PadsbA1 (pJNPadsbA1) is expressed in trans in an P. aeruginosa PA14 PadsbA1 mutant and when PadsbB1 (pJNPadsbB1) or PadsbB2 (pJNPadsbB2) are expressed in trans in a PadsbB1B2 strain. The presence of the empty vector (pJN105) has no impact on elastase production by the P. aeruginosa PA14 wild-type strain. (D) Impairment of the disulfide bond formation machinery decreases the ability of P. aeruginosa to kill C. elegans. Worms were infected with the parental P. aeruginosa PA14 strain (●) or with the PadsbA1 (▪), PadsbB1 (▴), PadsbB2 (▾), or PadsbB1B2 (♦) mutant. The percentage of surviving nematodes is shown with respect to the number of days postinfection. The data shown are averages of four independent experiments. Download
Mutation of the additional disulfide decreases the redox potential of the CXXC motif of PaDsbA2. We determined the redox potential of PaDsbA2CCSS by equilibrating the protein in redox buffers containing different GSH/GSSG ratios. The redox potential was calculated from the ratio (R) between the amounts of oxidized and reduced PaDsbA2CCSS present at equilibrium and determined using AMS trapping experiments. The data shown are averages (± standard deviations) of three independent experiments. Download
Global analysis of the proteins identified by 2D-LC-MS/MS. Proteins in this table have an average spectral count value of ≥5 (before normalization) and were present in at least 3 of the 6 runs. # cys is the number of cysteines present in the unprocessed protein sequence (with the signal sequence).
Global analysis of the proteins more than twofold less abundant in a PadsbA1 strain. Proteins in this table were selected from Table S1.
ACKNOWLEDGMENTS
We thank Asma Boujtat, Khadija Wahni, and Gaetan Herinckx for technical assistance and all members of the J.-F.C. and R.V. research groups for fruitful discussions.
This work was supported by the Interuniversity Attraction Pole Program—Belgian Science Policy (J.-F.C) (network P7/44) and by grants from the FRS-FNRS awarded to J.-F.C, from the ANR to R.V., and from VIB and VUB to J.M. The X-ray generator was funded by the Hercules grant UABR/09/005. J.-F.C is Maitre de Recherches, P.L. is Chargée de Recherche, I.S.A. is Aspirant, and D.V. is Collaborateur Logistique of the Belgian FRS-FNRS. V.N. is a research fellow of the FRIA, and J.M. is a group leader of the VIB.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Citation Arts IS, Ball G, Leverrier P, Garvis S, Nicolaes V, Vertommen D, Ize B, Tamu Dufe V, Messens J, Voulhoux R, Collet J-F. 2013. Dissecting the machinery that introduces disulfide bonds in Pseudomonas aeruginosa. mBio 4(6):e00912-13. doi:10.1128/mBio.00912-13.
REFERENCES
- 1. Depuydt M, Messens J, Collet JF. 2011. How proteins form disulfide bonds. Antioxid. Redox Signal. 15:49–66 [DOI] [PubMed] [Google Scholar]
- 2. Holmgren A, Fagerstedt M. 1982. The in vivo distribution of oxidized and reduced thioredoxin in Escherichia coli. J. Biol. Chem. 257:6926–6930 [PubMed] [Google Scholar]
- 3. Messens J, Collet JF. 2006. Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 38:1050–1062 [DOI] [PubMed] [Google Scholar]
- 4. Bardwell JC, McGovern K, Beckwith J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589 [DOI] [PubMed] [Google Scholar]
- 5. Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J. 1993. A pathway for disulfide bond formation in vivo. Proc. Natl. Acad. Sci. U. S. A. 90:1038–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bader M, Muse W, Ballou DP, Gassner C, Bardwell JC. 1999. Oxidative protein folding is driven by the electron transport system. Cell 98:217–227 [DOI] [PubMed] [Google Scholar]
- 7. Tapley TL, Eichner T, Gleiter S, Ballou DP, Bardwell JC. 2007. Kinetic characterization of the disulfide bond-forming enzyme DsbB. J. Biol. Chem. 282:10263–10271 [DOI] [PubMed] [Google Scholar]
- 8. Takahashi YH, Inaba K, Ito K. 2004. Characterization of the menaquinone-dependent disulfide bond formation pathway of Escherichia coli. J. Biol. Chem. 279:47057–47065 [DOI] [PubMed] [Google Scholar]
- 9. Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. 2004. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303:534–537 [DOI] [PubMed] [Google Scholar]
- 10. Leichert LI, Jakob U. 2004. Protein thiol modifications visualized in vivo. PLoS Biol. 2:e333. 10.1371/journal.pbio.0020333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hiniker A, Bardwell JC. 2004. In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279:12967–12973 [DOI] [PubMed] [Google Scholar]
- 12. Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora JP, Messens J, Bardwell JC, Collet JF. 2008. The disulphide isomerase DsbC cooperates with the oxidase DsbA in a DsbD-independent manner. Mol. Microbiol. 67:336–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kadokura H, Beckwith J. 2009. Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138:1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rietsch A, Belin D, Martin N, Beckwith J. 1996. An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 93:13048–13053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shevchik VE, Condemine G, Robert-Baudouy J. 1994. Characterization of DsbC, a periplasmic protein of Erwinia chrysanthemi and Escherichia coli with disulfide isomerase activity. EMBO J. 13:2007–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stewart EJ, Katzen F, Beckwith J. 1999. Six conserved cysteines of the membrane protein DsbD are required for the transfer of electrons from the cytoplasm to the periplasm of Escherichia coli. EMBO J. 18:5963–5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Katzen F, Beckwith J. 2000. Transmembrane electron transfer by the membrane protein DsbD occurs via a disulfide bond cascade. Cell 103:769–779 [DOI] [PubMed] [Google Scholar]
- 18. Rietsch A, Bessette P, Georgiou G, Beckwith J. 1997. Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J. Bacteriol. 179:6602–6608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. 2009. DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7:215–225 [DOI] [PubMed] [Google Scholar]
- 20. Dutton RJ, Boyd D, Berkmen M, Beckwith J. 2008. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U. S. A. 105:11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tinsley CR, Voulhoux R, Beretti JL, Tommassen J, Nassif X. 2004. Three homologues, including two membrane-bound proteins, of the disulfide oxidoreductase DsbA in Neisseria meningitidis: effects on bacterial growth and biogenesis of functional type IV pili. J. Biol. Chem. 279:27078–27087 [DOI] [PubMed] [Google Scholar]
- 22. Lafaye C, Iwema T, Ferrer JL, Kroll JS, Griat M, Serre L. 2008. Preliminary crystallographic data of the three homologues of the thiol-disulfide oxidoreductase DsbA in Neisseria meningitidis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64:111–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lafaye C, Iwema T, Carpentier P, Jullian-Binard C, Kroll JS, Collet JF, Serre L. 2009. Biochemical and structural study of the homologues of the thiol-disulfide oxidoreductase DsbA in Neisseria meningitidis. J. Mol. Biol. 392:952–966 [DOI] [PubMed] [Google Scholar]
- 24. Shouldice SR, Heras B, Jarrott R, Sharma P, Scanlon MJ, Martin JL. 2010. Characterization of the DsbA oxidative folding catalyst from Pseudomonas aeruginosa reveals a highly oxidizing protein that binds small molecules. Antioxid. Redox Signal. 12:921–931 [DOI] [PubMed] [Google Scholar]
- 25. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Urban A, Leipelt M, Eggert T, Jaeger KE. 2001. DsbA and DsbC affect extracellular enzyme formation in Pseudomonas aeruginosa. J. Bacteriol. 183:587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Braun P, Ockhuijsen C, Eppens E, Koster M, Bitter W, Tommassen J. 2001. Maturation of Pseudomonas aeruginosa elastase. Formation of the disulfide bonds. J. Biol. Chem. 276:26030–26035 [DOI] [PubMed] [Google Scholar]
- 28. Kim SH, Park SY, Heo YJ, Cho YH. 2008. Drosophila melanogaster-based screening for multihost virulence factors of Pseudomonas aeruginosa PA14 and identification of a virulence-attenuating factor, HudA. Infect. Immun. 76:4152–4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan MW, Rahme LG, Sternberg JA, Tompkins RG, Ausubel FM. 1999. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc. Natl. Acad. Sci. U. S. A. 96:2408–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rahme LG, Tan MW, Le L, Wong SM, Tompkins RG, Calderwood SB, Ausubel FM. 1997. Use of model plant hosts to identify Pseudomonas aeruginosa virulence factors. Proc. Natl. Acad. Sci. U. S. A. 94:13245–13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Joly JC, Swartz JR. 1997. In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductases DsbA and DsbC. Biochemistry 36:10067–10072 [DOI] [PubMed] [Google Scholar]
- 32. Hennecke J, Sillen A, Huber-Wunderlich M, Engelborghs Y, Glockshuber R. 1997. Quenching of tryptophan fluorescence by the active-site disulfide bridge in the DsbA protein from Escherichia coli. Biochemistry 36:6391–6400 [DOI] [PubMed] [Google Scholar]
- 33. Bader M, Muse W, Zander T, Bardwell J. 1998. Reconstitution of a protein disulfide catalytic system. J. Biol. Chem. 273:10302–10307 [DOI] [PubMed] [Google Scholar]
- 34. Bader MW, Xie T, Yu CA, Bardwell JC. 2000. Disulfide bonds are generated by quinone reduction. J. Biol. Chem. 275:26082–26088 [DOI] [PubMed] [Google Scholar]
- 35. Ha UH, Wang Y, Jin S. 2003. DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect. Immun. 71:1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thayer MM, Flaherty KM, McKay DB. 1991. Three-dimensional structure of the elastase of Pseudomonas aeruginosa at 1.5-A resolution. J. Biol. Chem. 266:2864–2871 [DOI] [PubMed] [Google Scholar]
- 37. Bardoel BW, Hartsink D, Vughs MM, de Haas CJ, van Strijp JA, van Kessel KP. 2012. Identification of an immunomodulating metalloprotease of Pseudomonas aeruginosa (IMPa). Cell. Microbiol. 14:902–913 [DOI] [PubMed] [Google Scholar]
- 38. Garvis S, Munder A, Ball G, de Bentzmann S, Wiehlmann L, Ewbank JJ, Tümmler B, Filloux A. 2009. Caenorhabditis elegans semi-automated liquid screen reveals a specialized role for the chemotaxis gene cheB2 in Pseudomonas aeruginosa virulence. PLoS Pathog. 5:e1000540. 10.1371/journal.ppat.1000540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet JF. 2009. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Denoncin K, Schwalm J, Vertommen D, Silhavy TJ, Collet JF. 2012. Dissecting the Escherichia coli periplasmic chaperone network using differential proteomics. Proteomics 12:1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu H, Sadygov RG, Yates JR., III 2004. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76:4193–4201 [DOI] [PubMed] [Google Scholar]
- 42. Nakai K, Horton P. 1999. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 24:34–36 [DOI] [PubMed] [Google Scholar]
- 43. Holmgren A. 1979. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254:9627–9632 [PubMed] [Google Scholar]
- 44. Holm L, Rosenström P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38:W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martin JL, Bardwell JC, Kuriyan J. 1993. Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 365:464–468 [DOI] [PubMed] [Google Scholar]
- 46. McCarthy AA, Haebel PW, Torronen A, Rybin V, Baker EN, Metcalf P. 2000. Crystal structure of the protein disulfide bond isomerase, DsbC Escherichia coli. Nat. Struct. Biol. 7:196–199 [DOI] [PubMed] [Google Scholar]
- 47. Heras B, Edeling MA, Schirra HJ, Raina S, Martin JL. 2004. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc. Natl. Acad. Sci. U. S. A. 101:8876–8881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kurz M, Iturbe-Ormaetxe I, Jarrott R, Shouldice SR, Wouters MA, Frei P, Glockshuber R, O’Neill SL, Heras B, Martin JL. 2009. Structural and functional characterization of the oxidoreductase alpha-DsbA1 from Wolbachia pipientis. Antioxid. Redox Signal. 11:1485–1500 [DOI] [PubMed] [Google Scholar]
- 49. Thomas JH. 1993. Thinking about genetic redundancy. Trends Genet. 9:395–399 [DOI] [PubMed] [Google Scholar]
- 50. Recinos DA, Sekedat MD, Hernandez A, Cohen TS, Sakhtah H, Prince AS, Price-Whelan A, Dietrich LE. 2012. Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 109:19420–19425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vaaje-Kolstad G, Houston DR, Riemen AH, Eijsink VG, van Aalten DM. 2005. Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J. Biol. Chem. 280:11313–11319 [DOI] [PubMed] [Google Scholar]
- 52. Aachmann FL, Sørlie M, Skjak-Braek G, Eijsink VG, Vaaje-Kolstad G. 2012. NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U. S. A. 109:18779–18784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bleves S, Viarre V, Salacha R, Michel GP, Filloux A, Voulhoux R. 2010. Protein secretion systems in Pseudomonas aeruginosa: a wealth of pathogenic weapons. Int. J. Med. Microbiol. 300:534–543 [DOI] [PubMed] [Google Scholar]
- 54. Filloux A. 2004. The underlying mechanisms of type II protein secretion. Biochim. Biophys. Acta 1694:163–179 [DOI] [PubMed] [Google Scholar]
- 55. Jameson-Lee M, Garduño RA, Hoffman PS. 2011. DsbA2 (27 kDa Com1-like protein) of Legionella pneumophila catalyses extracytoplasmic disulphide-bond formation in proteins including the Dot/Icm type IV secretion system. Mol. Microbiol. 80:835–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lin D, Kim B, Slauch JM. 2009. DsbL and DsbI contribute to periplasmic disulfide bond formation in Salmonella enterica serovar. Typhimurium. Microbiology 155:4014–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Denoncin K, Nicolaes V, Cho SH, Leverrier P, Collet JF. 2013. Protein disulfide bond formation in the periplasm: determination of the in vivo redox state of cysteine residues. Methods Mol. Biol. 966:325–336 [DOI] [PubMed] [Google Scholar]
- 58. Wunderlich M, Glockshuber R. 1993. Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci. 2:717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wolfe AJ, Berg HC. 1989. Migration of bacteria in semisolid agar. Proc. Natl. Acad. Sci. U. S. A. 86:6973–6977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. 2001. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc. Natl. Acad. Sci. U. S. A. 98:6911–6916 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional information on analysis of the structure of PaDsbA2. Download
Bacterial strains, growth conditions, and protein purification. Download
Protein identification and quantification by 2D-LC-MS/MS. Download
Bioinformatic analysis of the DsbA proteins from P. aeruginosa. (A) Phylogenetic analysis of the DsbA family shows that DsbA proteins can be divided in two subgroups. PaDsbA1 and PaDsbA2 belong to subgroups 1 and 2, respectively. The tree was constructed using FigTree v1.3.1 (A. Rambaut) based on the amino acid sequences of 20 proteins homologous to PaDsbA1 or PaDsbA2. The purple bars represent sequences that align with PaDsbA1, and the blue bars represent sequences that align with PaDsbA2. Gammaproteobacteria are shown in black, alphaproteobacteria in pink, betaproteobacteria in indigo, and deltaproteobacteria in cyan. The scale bar, below the tree, indicates the number of substitutions per amino acid. (B) The sequences of PaDsbA1 and PaDsbA2 were aligned with the sequences of homologous proteins from Moraxella catarrhalis, Neisseria meningitidis, Escherichia coli, Methylobacterium populi, Wolbachia pipientis, and Polynucleobacter necessarius using MacVector (MacVector, Inc.). The cysteines of the catalytic CXXC motif are outlined in purple. The additional cysteines found in PaDsbA2 and other DsbA proteins from subgroup 2 are outlined in blue. Dark gray and light gray shading indicate identical amino acids and amino acids from the same group (nonpolar, polar, acidic, or basic), respectively. Download
PaDsbB1 and PaDsbB2 can both complement an EcdsbB deficiency. Complementation tests of the EcdsbB strain by PaDsbB1 and PaDsbB2 were performed on semisolid agar plates containing 0.2% arabinose. Restoration of the motility due to the correct folding of FlgI, a component of the flagellum, was observed when PaDsbB1 or PaDsbB2 is expressed in the EcdsbB mutant, indicating that both proteins are able to reoxidize EcDsbA. Download
Fluorescence emission spectra of oxidized and reduced PaDsbA1. Fluorescence emission spectra of oxidized PaDsbA1 (broken line) and reduced PaDsbA1 (solid line) were recorded at protein concentrations of 2 µM in 50 mM potassium phosphate (pH 7) and 0.1 mM EDTA at room temperature. At 340 nm, the intrinsic tryptophan fluorescence of PaDsbA1 increases about 1.7 upon reduction of the enzyme. Download
Deletion of both PadsbB1 and PadsbB2 impairs biofilm formation, prevents the oxidative folding of elastase, and decreases the global virulence of P. aeruginosa. (A) PadsbA1 and PadsbB1B2 mutants are defective in the early steps of biofilm formation. Biofilm formation was visualized in wells of a microtiter dish after crystal violet staining (purple circle that is visible only in the wells containing the wild-type [WT] strain). (B) Elastase, a protease with two disulfide bonds, is secreted by the Xcp type II secretion machinery in P. aeruginosa. The nonspecific and elastin-specific protease activity of elastase is tested on plates containing skim milk (left) and elastin (right), respectively. The halos observed around the colonies of the wild type and of the single PadsbB1 and PadsbB2 mutants correspond to milk or elastin degradation by elastase. The Paxcp mutant, used as a negative control, is unable to assemble the type II secretion machinery and therefore to secrete elastase. (C) Phenotypic complementation of Padsb mutants on skim milk plate. Wild-type secretion is restored when PadsbA1 (pJNPadsbA1) is expressed in trans in an P. aeruginosa PA14 PadsbA1 mutant and when PadsbB1 (pJNPadsbB1) or PadsbB2 (pJNPadsbB2) are expressed in trans in a PadsbB1B2 strain. The presence of the empty vector (pJN105) has no impact on elastase production by the P. aeruginosa PA14 wild-type strain. (D) Impairment of the disulfide bond formation machinery decreases the ability of P. aeruginosa to kill C. elegans. Worms were infected with the parental P. aeruginosa PA14 strain (●) or with the PadsbA1 (▪), PadsbB1 (▴), PadsbB2 (▾), or PadsbB1B2 (♦) mutant. The percentage of surviving nematodes is shown with respect to the number of days postinfection. The data shown are averages of four independent experiments. Download
Mutation of the additional disulfide decreases the redox potential of the CXXC motif of PaDsbA2. We determined the redox potential of PaDsbA2CCSS by equilibrating the protein in redox buffers containing different GSH/GSSG ratios. The redox potential was calculated from the ratio (R) between the amounts of oxidized and reduced PaDsbA2CCSS present at equilibrium and determined using AMS trapping experiments. The data shown are averages (± standard deviations) of three independent experiments. Download
Global analysis of the proteins identified by 2D-LC-MS/MS. Proteins in this table have an average spectral count value of ≥5 (before normalization) and were present in at least 3 of the 6 runs. # cys is the number of cysteines present in the unprocessed protein sequence (with the signal sequence).
Global analysis of the proteins more than twofold less abundant in a PadsbA1 strain. Proteins in this table were selected from Table S1.