

ABSTRACT
The Escherichia coli sequence type 131 (ST131) clone is notorious for extraintestinal infections, fluoroquinolone resistance, and extended-spectrum beta-lactamase (ESBL) production, attributable to a CTX-M-15-encoding mobile element. Here, we applied pulsed-field gel electrophoresis (PFGE) and whole-genome sequencing to reconstruct the evolutionary history of the ST131 clone. PFGE-based cluster analyses suggested that both fluoroquinolone resistance and ESBL production had been acquired by multiple ST131 sublineages through independent genetic events. In contrast, the more robust whole-genome-sequence-based phylogenomic analysis revealed that fluoroquinolone resistance was confined almost entirely to a single, rapidly expanding ST131 subclone, designated H30-R. Strikingly, 91% of the CTX-M-15-producing isolates also belonged to a single, well-defined clade nested within H30-R, which was named H30-Rx due to its more extensive resistance. Despite its tight clonal relationship with H30Rx, the CTX-M-15 mobile element was inserted variably in plasmid and chromosomal locations within the H30-Rx genome. Screening of a large collection of recent clinical E. coli isolates both confirmed the global clonal expansion of H30-Rx and revealed its disproportionate association with sepsis (relative risk, 7.5; P < 0.001). Together, these results suggest that the high prevalence of CTX-M-15 production among ST131 isolates is due primarily to the expansion of a single, highly virulent subclone, H30-Rx.
IMPORTANCE
We applied an advanced genomic approach to study the recent evolutionary history of one of the most important Escherichia coli strains in circulation today. This strain, called sequence type 131 (ST131), causes multidrug-resistant bladder, kidney, and bloodstream infections around the world. The rising prevalence of antibiotic resistance in E. coli is making these infections more difficult to treat and is leading to increased mortality. Past studies suggested that many different ST131 strains gained resistance to extended-spectrum cephalosporins independently. In contrast, our research indicates that most extended-spectrum-cephalosporin-resistant ST131 strains belong to a single highly pathogenic subclone, called H30-Rx. The clonal nature of H30-Rx may provide opportunities for vaccine or transmission prevention-based control strategies, which could gain importance as H30-Rx and other extraintestinal pathogenic E. coli subclones become resistant to our best antibiotics.
INTRODUCTION
Horizontal gene transfer is one of the most powerful forces in bacterial evolution. The transformative potential of this process is perhaps best exemplified by the acquisition of antimicrobial resistance determinants: in a single genetic event, an antimicrobial-susceptible bacterium can acquire a complex suite of resistance determinants and become resistant to multiple antimicrobials. Thus, frequent horizontal transfer between different strains can potentially drive the spread of antibiotic resistance within the bacterial population, without any change in the distribution of strains. However, when virulent bacterial clones acquire such elements, they can emerge rapidly within the population through clonal expansion and thereby gain local or even global predominance (1–3). Quantifying the relative contribution of horizontal (gene transfer) and vertical (clonal expansion) mechanisms to the emergence of multidrug-resistant bacterial pathogens will provide important insights into the evolution of these pathogens and inform novel intervention strategies.
In 2008, a previously unrecognized Escherichia coli clonal group, sequence type 131 (ST131), was identified in nine countries, spanning three continents (4, 5). Today, ST131 is the dominant extraintestinal pathogenic E. coli (ExPEC) strain worldwide, but retrospective analyses suggest that the pandemic emergence of ST131 took place over a period of fewer than 10 years (6, 7). ST131 is part of the virulent phylogenetic group B2 and has been reported to cause a wide range of infections, including meningitis, osteomyelitis, myositis, epididymo-orchitis, and peritonitis (6, 8–10). However, ST131 is most commonly associated with urinary tract infection (UTI) and is a major etiologic agent of bladder infections, kidney infections, and urosepsis in the United States and internationally (11–14). Population genetics analysis of ST131 isolates indicated that the recent epidemic spread of this group is driven by descendants of a single strain, named subclone H30, that differ from the members of other, less prevalent ST131 subclones by carriage of fimH30, an allele of the gene encoding the mannose-specific type 1 fimbrial adhesin, FimH (15).
Over the last decade, the emergence of multidrug-resistant ExPEC strains has made UTI treatment more problematic, leading to discordant antimicrobial therapy and increased morbidity and mortality (16–19). This increase in multidrug-resistant UTIs has in large part been due to the rapid rise in prevalence of ExPEC strains—particularly from ST131—harboring determinants for extended-spectrum β-lactamases (ESBLs) and resistance to trimethoprim-sulfamethoxazole and fluoroquinolones (FQ) (16, 20–27).
The CTX-M-15 β-lactamase is the dominant ESBL in ST131 and is increasingly found in isolates causing both UTI and bacteremia (13, 28–31). The CTX-M gene phylogeny suggests that these enzymes arose through mobilization of chromosomal β-lactamase (bla) genes from the gut commensal organism Kluyvera (32). A number of CTX-M enzymes have risen to prevalence since the 1990s, with a new CTX-M type frequently appearing in multiple distant countries simultaneously, suggesting independent transfer events (33). This, together with the substantial diversity in transferable resistance elements in ST131, has led some to conclude that horizontal transfer must be the dominant mechanism whereby ESBLs have gained prominence among strains of the ST131 clone (7, 34, 35). However, other evidence suggests that clonal expansion contributes significantly to the spread of antimicrobial resistance within E. coli (36–39).
Until recently, our knowledge of the epidemiology and dispersal of bacterial strains, including of ST131 origin, has been based largely on multilocus sequence typing (MLST), which has limited resolution at the subclone level, and on pulsed-field gel electrophoresis (PFGE) analysis, which is highly vulnerable to distortions from horizontal gene transfer events and subjective interpretation. In the current study, we used whole-genome single-nucleotide polymorphism (SNP) analysis to reconstruct the ST131 phylogeny and then overlaid resistance determinants and phenotypic susceptibility on this phylogeny to elucidate the evolutionary history of fluoroquinolone resistance and ESBL production within this prominent pathogen.
RESULTS
PFGE analyses.
A collection of 524 ST131 isolates cultured from humans and animals between 1967 and 2011 was analyzed using PFGE, which yielded a complex dendrogram (Fig. 1A). Within the PFGE-based dendrogram, the isolates that were fluoroquinolone resistant and/or blaCTX-M-15 positive, although exhibiting some clustering, were intermingled extensively with those that were fluoroquinolone susceptible and/or blaCTX-M-15 negative. As such, the PFGE analysis supported previous reports suggesting frequent horizontal acquisition of fluoroquinolone resistance determinants and blaCTX-M-15 among different ST131 subclones.
FIG 1 .
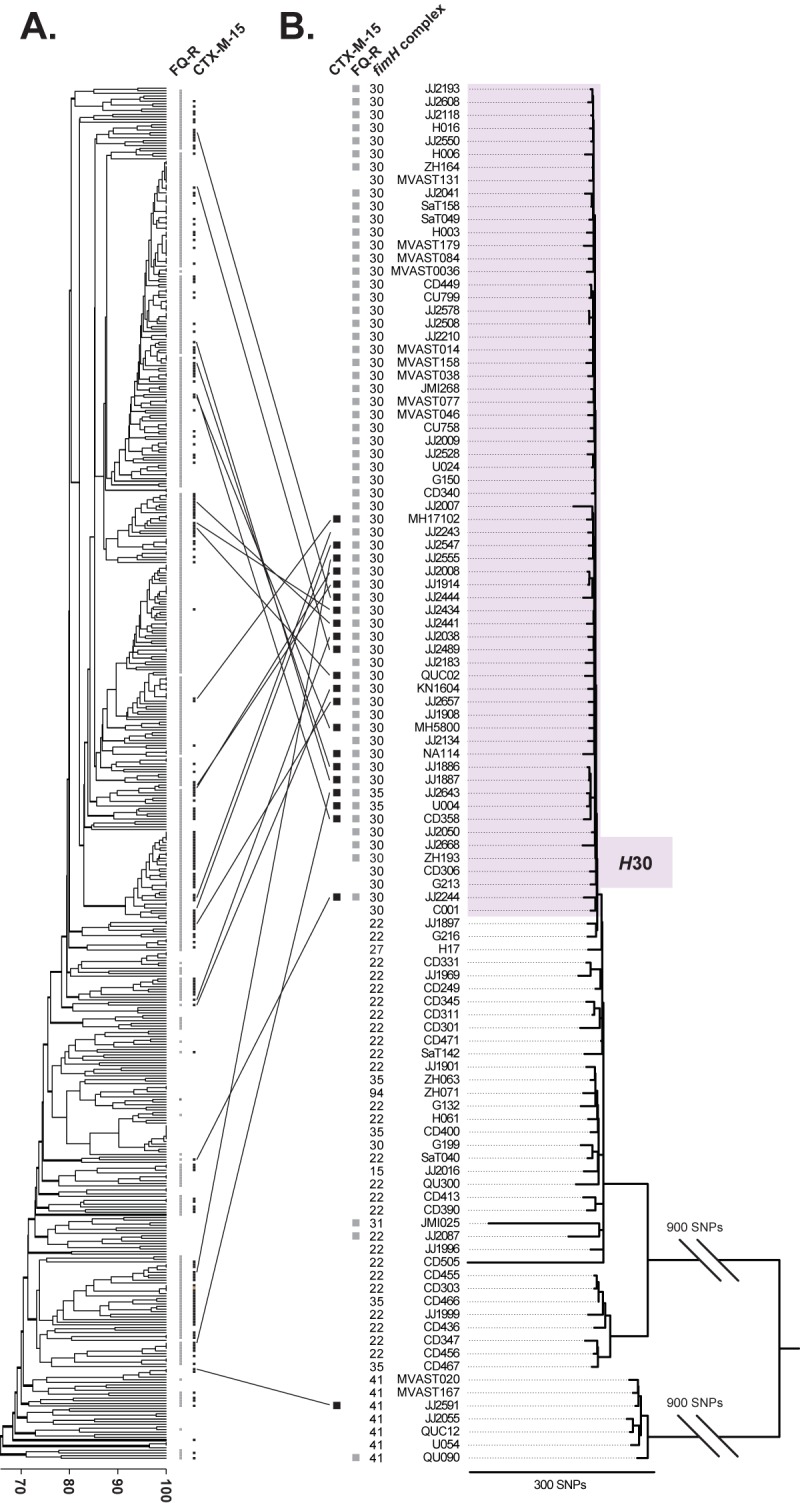
PFGE dendrogram and whole-genome SNP-based phylogeny of E. coli ST131. (A) PFGE-based dendrogram of E. coli ST131 isolates (n = 524), as inferred within BioNumerics according to the unweighted pair group method based on Dice similarity coefficients. (B) Whole-genome SNP-based phylogeny of selected ST131 isolates (n = 105) and the NA114 reference genome. SNPs were identified from genomic regions equivalent to approximately 44.7% of the reference genome that was shared among all isolates and sequenced at ≥10× coverage. Analysis of these shared genomic regions revealed 2,531 parsimony-informative and 4,000 total SNPs from the core genome (excluding horizontally acquired regions) that were used to construct the phylogeny presented here. Homoplasy index (HI) = 0.012. The purple block highlights the H30 subclone.
Whole-genome SNP-based phylogenetic reconstruction of ST131.
From the total collection that underwent PFGE analysis, 105 ST131 isolates were systematically selected for genome sequencing according to prespecified criteria that emphasized diversity of genetic backgrounds according to PFGE. The 105 isolates, which derived from five countries and 23 states and provinces in Canada and the United States, included 22 CTX-M-15-producing isolates, which were widely distributed across the PFGE dendrogram (Fig. 1A).
Genomic comparisons identified SNP loci that were present in all isolates and, therefore, informative for phylogenetic reconstruction. The first phylogenetic tree included non-ST131 strain AA86 (group B2; ST1876) (40) as an outgroup, to root the tree and to identify the basal clones within the ST131 phylogeny (see Fig. S1 in the supplemental material). Next, strain AA86 was excluded, and a new SNP matrix and phylogenetic tree were generated (see Fig. S2 in the supplemental material). Since (distant) strain AA86 lacks some of the genomic regions found within the ST131 clone, exclusion of AA86 increased the number of shared genomic regions in the sequence alignment and, therefore, increased the number of informative SNPs with which to resolve the ST131 phylogeny.
The homoplasy index (HI) for these two initial trees (see Fig. S1 and S2) was exceedingly high (>0.33), indicating substantial recombination. Phylogenetic reconstructions that include genomic regions acquired by horizontal gene transfer will not accurately represent the evolutionary history of clonal organisms. However, such phylogenies can be used to identify the regions acquired horizontally. This was accomplished here by mapping to the reference genome the HI values for individual SNPs, which revealed four large recombinant regions representing nearly 31% of the genome.
Exclusion of SNPs from the four horizontally acquired regions resulted in trees with minimal homoplasy (homoplasy index [HI] = 0.012) (see Fig. S3 in the supplemental material), suggestive of highly accurate phylogenies (41). Figure 1B shows the resultant whole-genome SNP phylogeny for the 105 ST131 isolates, plus the strain NA114 reference ST131 genome (42).
Whole-genome-based clustering of resistant subclones.
The whole-genome SNP-based phylogeny showed distinct clustering of strains carrying specific fimH alleles (Fig. 1B), as well as gyrA and parC alleles and O type (see Dataset S1 and Table S1 in the supplemental material). In particular, strains carrying the fimH30 allele clustered as a single low-diversity clade, designated H30, which included 58 (95%) of the 61 fluoroquinolone-resistant isolates. Moreover, nearly all of the CTX-M-15-producing isolates, despite appearing to have diverse genetic backgrounds according to the PFGE-based dendrogram (Fig. 1A), collapsed into a distinct subclade within the H30 clade (Fig. 1B).
To further resolve the evolutionary history of the H30 subclone, genomic sequences from the 64 H30 isolates and their three nearest neighbors were analyzed separately from the rest of the isolates (Fig. 2). Aligning these sequences to the finished NA114 reference genome increased the number of shared nucleotides and revealed additional informative SNPs that were used to generate the high-resolution and highly accurate (HI = 0.000) phylogenetic tree shown in Fig. 2. This tree suggested that acquisition of the fimH30 allele preceded the acquisition of fluoroquinolone resistance by a single ancestor within the H30 subclone, which was followed by a large clonal expansion of fluoroquinolone-resistant H30 strains. To distinguish the clonally related fluoroquinolone-resistant H30 isolates from the ancestral fluoroquinolone-susceptible H30 isolates, the resistant subclone within H30 was designated H30-R.
FIG 2 .

High-resolution phylogenetic analysis of the emergence of fluoroquinolone resistance and CTX-M-15 production. Approximately 51.8% of the reference genome was shared among all isolates and sequenced at ≥10× coverage. Analysis of these shared genomic regions revealed 72 parsimony-informative SNPs and 771 total SNPs from the core genome (excluding horizontally acquired regions) that were used to construct the phylogeny presented here. Homoplasy index (HI) = 0.000. The colored blocks highlight the three nested ST131 subclones, H30 (purple), H30-R (blue), H30-Rx (yellow).
In this high-resolution phylogeny, 20 (91%) of the 22 ST131 isolates that carried blaCTX-M-15—including isolates from Australia, South Korea, Portugal, Canada, and the United States—formed a distinct, single-ancestor subclone within H30-R. Because of its more extensive resistance characteristics, this blaCTX-M-15-associated subclone was designated H30-Rx (Fig. 2). Three canonical SNPs distinguished H30-Rx from the rest of H30-R with 100% fidelity.
Genomic location of the CTX-M-15 element.
Multiple previous studies have reported that blaCTX-M-15 is positioned on a conjugative IncFII-type plasmid as part of a Tn3-like ISEcp1-blaCTX-M-15-orf477 mobile element. Here, we performed an in silico analysis to characterize the structure and genomic location of the CTX-M-15 mobile element among the 22 blaCTX-M-15-positive ST131 isolates. In each instance, blaCTX-M-15 was part of a typical Tn3-like ISEcp1-blaCTX-M-15-orf477 transposable element. While no SNPs were identified among the different CTX-M-15 elements, the regions flanking blaCTX-M-15 were frequently degraded by insertion/deletion (not shown). The Illumina short-read sequences were sufficient to reliably identify the element’s insertion site for all but three of the 22 CTX-M-15-positive isolates sequenced in the current study (Table 1). The insertion site varied among the isolates: 13 carried a single copy on an IncFII-type plasmid, four carried a single copy on the chromosome, and two carried one copy on the chromosome and another copy on an IncFII-type plasmid (Table 1). Moreover, among the strains with a chromosomally located element, five distinct chromosomal insertion sites were identified; only two strains, JJ1886 and JJ1887, which are the nearest neighbors in the SNP phylogeny (Fig. 2), carried the element in the same chromosomal location (Table 1).
TABLE 1 .
CTX-M-15 element locations among the 22 Escherichia coli ST131 isolates
| Isolate name |
Subclone | Genomic locationa | Chromosomal insertion siteb |
|---|---|---|---|
| JJ2444 | H30-Rx | Chromosome | 2,037,134 |
| JJ2038 | H30-Rx | Chromosome | 2,127,735 |
| JJ1886 | H30-Rx | Chromosome | 1,473,842 |
| JJ1887 | H30-Rx | Chromosome | 1,473,842 |
| JJ2434 | H30-Rx | Plasmid and chromosome | 4,493,369 |
| MH5800 | H30-Rx | Plasmid and chromosome | 4,191,808 |
| JJ2547 | H30-Rx | Plasmid | NA |
| JJ2555 | H30-Rx | Plasmid | NA |
| JJ2008 | H30-Rx | Plasmid | NA |
| JJ1914 | H30-Rx | Plasmid | NA |
| JJ2441 | H30-Rx | Plasmid | NA |
| JJ2489 | H30-Rx | Plasmid | NA |
| JJ2657 | H30-Rx | Plasmid | NA |
| JJ2643 | H30-Rx | Plasmid | NA |
| U004 | H30-Rx | Plasmid | NA |
| CD358 | H30-Rx | Plasmid | NA |
| NA1114 | H30-Rx | Plasmidc | NA |
| MH17102 | H30-Rx | Undetermined | NA |
| QUC02 | H30-Rx | Undetermined | NA |
| KN1604 | H30-Rx | Undetermined | NA |
| JJ2244 | H30, non-H30-Rx | Plasmid | NA |
| JJ2591 | Non-H30 | Plasmid | NA |
Marked as undetermined if the in silico analyses provided equivocal results.
Chromosomal location based on the JJ1886 closed genome. NA, not applicable.
Reported previously.
Association of ESBL production and blaCTX-M-15 with the H30-Rx ST131 subclone.
Because most of the isolates in the phylogenetic trees were of historic (i.e., pre-2009) origin, we assessed the generalizability of the observed association of the H30-Rx subclone with ESBL production and blaCTX-M-15 by analyzing more recent clinical isolates, i.e., from 2011 to 2013. For this, a total of 261 ST131 isolates from Seattle, WA, Minneapolis, MN, and Münster, Germany, were assessed for fimH allele type, H30-Rx subclone membership, ESBL production, and possession of blaCTX-M-15 (Table 2).
TABLE 2 .
Association of ST131 subclones with resistance traits among 261 recent clinical isolates of Escherichia coli ST131 from the United States and Germany
| Resistance trait | Prevalence of resistance trait, no. (%) |
||||
|---|---|---|---|---|---|
| Total ST131 strains (n = 261) |
ST131 subclone(s) |
||||
| Non-H30 (n = 87) |
H30 (n = 174) |
||||
| Non-H30-R (n = 11) |
H30-R (n = 165) |
||||
| Non-H30-Rx (n = 119) |
H30-Rx (n = 44) |
||||
| FQ resistant | 163 (62) | 0 (0) | 0 (0) | 119 (100) | 44 (100)a |
| ESBL | 45 (17) | 3 (3) | 1 (9) | 7 (6) | 34 (77)b |
| blaCTX-M-15 | 39 (15) | 3 (7) | 1 (9) | 2 (2) | 33 (75)c |
For the fluoroquinolone (FQ)-resistant fraction, H30-Rx compared to other ST131 isolates, P < 0.001 (Fisher’s exact test [FET]).
For prevalence of extended-spectrum β-lactamase (ESBL) production, H30-Rx compared to other ST131 isolates, P < 0.001 (FET).
For prevalence of blaCTX-M-15, H30-Rx compared to other ST131 isolates, P < 0.001 (FET).
Among the 261 recent ST131 isolates, 174 (67%) belonged to the H30 subclone, whereas the remaining 87 (33%) carried one of several other ST131-associated fimH alleles, as described recently (15). Among the 174 H30 isolates, the 163 (94%) that were fluoroquinolone resistant were defined as H30-R. Detection of H30-Rx-specific SNPs showed that H30-Rx comprised 44 (27%) of 163 H30-R strains (Table 2).
Among the 44 H30-Rx isolates, 34 (77%) were ESBL producing, and 33 of these carried blaCTX-M-15. This was very similar to the 74% carriage of blaCTX-M-15 observed among the genome-sequenced historic H30-Rx isolates (Fig. 2) but significantly higher than the low prevalence of either ESBL production or blaCTX-M-15 carriage observed among the recent non-H30 ST131 isolates (3% for each trait), H30 but not H30-R isolates (9% for each trait), and H30-R but not H30-Rx isolates (6% and 2% for the two traits, respectively) (Table 2). Thus, blaCTX-M-15 accounted for nearly all ESBL-producing isolates within the H30-Rx subclone; conversely, within ST131 overall, the H30-Rx subclone accounted for the vast majority of ESBL production and, especially, blaCTX-M-15 carriage. Moreover, this tight association between H30-Rx and blaCTX-M-15 held true across the different laboratories that supplied the recent clinical isolates (data not shown).
Demographic, geographic, and clinical prevalence of H30-Rx.
We also assessed the relative prevalence of the H30-R and H30-Rx subclones within the total ST131 population in relation to patient population and locale by comparing urine isolates from Group Health Cooperative in Seattle, WA, which serves an almost exclusively outpatient population, with urine isolates from hospital laboratories in the United States and Germany that serve mixed inpatient and outpatient populations. The relative prevalence of H30-Rx was highest among the German Hospital isolates (where it exceeded the prevalence even of other H30-R isolates), intermediate among United States-based hospital isolates, and lowest among the Group Health outpatient isolates (Table 3).
TABLE 3 .
Prevalence of ST131 subclones in relation to source population among 261 recent clinical isolates of Escherichia coli ST131 from the United States and Germany
| Source population | Total no. of ST131 isolates | ST131 subclones, no. (%) |
|||
|---|---|---|---|---|---|
| Non-H30 (n = 87) |
H30 (n = 174) |
||||
| Non-H30-R (n = 11) |
H30-R (n = 165) |
||||
| Non-H30-Rx (n = 119) |
H30-Rx (n = 44) |
||||
| United States ambulatory | 86 | 35 (41) | 3 (4) | 42 (49) | 6 (7)a,b |
| United States hospital | 120 | 32 (27) | 4 (3) | 64 (53) | 20 (17)a,c |
| German hospital | 55 | 20 (36) | 4 (7) | 13 (24) | 18 (33)b,c |
For prevalence of H30-Rx, United States ambulatory compared to United States hospital isolates, P = 0.054 (FET).
For prevalence of H30-Rx, United States ambulatory compared to German hospital isolates, P < 0.001 (FET).
For prevalence of H30-Rx, United States hospital compared to German hospital isolates, P = 0.03 (FET).
Data regarding presence/absence of clinically diagnosed sepsis were available for 162 of the recent United States ST131 clinical isolates, among which 12 source patients (7%) overall were diagnosed with sepsis (Table 4), a value similar to the 5.2% overall prevalence of diagnosed sepsis among the 1,133 extraintestinal clinical isolates from which the 162 ST131 strains were derived (P = 0.26) (15). However, sepsis was diagnosed in 28% of the patients with an H30-Rx isolate (Table 4), a significantly greater proportion than among patients with a non-H30-Rx, H30-R isolate (6%; P = 0.02), a non-H30, ST131 isolate (4%; P = 0.01), any non-H30-Rx, ST131 isolate (5%; P = 0.005), or a non-ST131 isolate (5.6%; P = 0.003). For H30-Rx isolates compared to other ST131 isolates, the relative risk of associated sepsis was 7.5 (95% confidence interval, 2.3 to 23.8).
TABLE 4 .
Association of ST131 subclones with clinical sepsis among 162 recent clinical isolates of Escherichia coli ST131 from the United States
| Clinical presentation | No. (%) of isolates with associated clinical presentation |
||||
|---|---|---|---|---|---|
| Total ST131 isolates (n = 162) |
ST131 subclone(s) |
||||
| Non-H30 (n = 56) |
H30 (n = 174) |
||||
| Non-H30-R (n = 6) |
H30-R (n = 165) |
||||
| Non-H30-Rx (n = 82) |
H30-Rx (n = 18) |
||||
| No sepsis | 150 (92.6) | 54 (96) | 6 (100) | 77 (94) | 13 (72) |
| Sepsis | 12 (7.4) | 2 (4)a | 0 (0)b | 5 (6)c | 5 (28)a,b,c,d |
For prevalence of sepsis, H30-Rx compared to non-H30, P = 0.008 (FET).
For prevalence of sepsis, H30-Rx compared to non-H30-R (H30), P = 0.28 (FET).
For prevalence of sepsis, H30-Rx compared to non-H30-Rx (H30-R), P = 0.016 (FET).
d For prevalence of sepsis, H30-Rx compared to all other ST131 (7/144, 5%), P = 0.005 (FET).
DISCUSSION
The results of this study provide compelling evidence that clonal expansion is the dominant mechanism for the proliferation of both CTX-M-15 production and fluoroquinolone resistance in E. coli ST131. Past studies have shown that the determinants for both of these traits can be acquired through horizontal gene transfer or, in the case of fluoroquinolone resistance, independent mutation (15). However, the whole-genome SNP-based phylogenies presented here show that almost all of the fluoroquinolone-resistant ST131 isolates belong to a distinct subclone, H30-R, which was derived from a single common ancestor carrying the fimH30 allele (i.e., part of the H30 subclone). Likewise, 91% of the CTX-M-15-producing isolates form another distinct subclone, H30-Rx, which was derived from a single common ancestor within the H30-R subclone. These nested subclones form a Russian-doll-like configuration, within which each subsequent lineage is more extensively resistant than the former.
The nearly exclusive confinement of the CTX-M-15 element to the H30-Rx subclone was in striking contrast to this element’s promiscuity within the ST131 genome. We identified H30-Rx isolates with a copy of the element on the chromosome, on an IncFII-type plasmid, and, in some instances, on both the chromosome and a plasmid. Moreover, among those isolates with a chromosomal CTX-M-15 element, the element was inserted in five different locations. Notably, the only two isolates with identical chromosomal insertion sites were recovered from epidemiologically linked adult siblings who both suffered from UTIs of varying severity and were suspected of sharing the same ST131 strain (43), as supported here by the close proximity of these isolates in the high-resolution H30 phylogenetic tree (Fig. 2). Among the 22 CTX-M-15-producing strains, the CTX-M-15 elements exhibited no SNPs and, therefore, no phylogenetic signal that could be compared with the host strain phylogeny.
One cannot exclude the possibility that chromosomal and even some plasmid-borne CTX-M-15 elements were acquired horizontally by H30-RX on multiple occasions. However, three lines of evidence suggest that within H30-Rx, the chromosomally encoded CTX-M-15 is the result of repeated intragenomic mobilization of the plasmid-located Tn3-like ISEcp1-blaCTX-M-15-orf477 element, rather than independent horizontal acquisition events. First, the sequence identity of the CTX-M-15 elements suggests that they represent copies of the same recently derived genetic element. Second, the plasmid location is the most common among H30-Rx isolates. Third, some strains with chromosomally encoded CTX-M-15 (e.g., JJ1886 and JJ1887) also maintain an IncFII-type plasmid, albeit missing the CTX-M-15 element, consistent with the CTX-M-15 element having moved from the plasmid to the chromosome.
The exact evolutionary history of CTX-M-15 acquisition in H30-Rx also remains to be elucidated. It is possible that H30-Rx was founded by an ancestor that acquired CTX-M-15 on an incF-type plasmid, which expanded and differentiated along with the subclone. Under this model, the CTX-M-15-negative H30-Rx isolates would represent independent gene loss events. An alternative explanation is that the H30-Rx subclone was founded by a CTX-M-15-negative ancestor and partially expanded before becoming extensively cephalosporin resistant later, through horizontal acquisition of the CTX-M-15 element. Indeed, this subclone may be under differential third-generation cephalosporin selection due to its enhanced virulence, which might result in its being exposed to aggressive antimicrobial therapy more commonly than other E. coli strains. Further investigation, including total chromosome and plasmid closure, could clarify the most probable mechanism for the association between H30-Rx and CTX-M-15. Interestingly, however, the blaCTX-M-15-positive, FQ-resistant JJ2244 strain, which occupies a phylogenetic outgroup position relative to both H30-R and H30-Rx (see Fig. 2), likely represents an independently emerged multidrug-resistant clonal lineage within the H30 subclone. Indeed, this strain not only lacks all canonical H30-Rx SNPs but also has a distinct, recombinant FQ resistance-conferring gyrA-parC allele combination (i.e., 1AB/4A), compared with that present in H30-R (i.e., 1AB/1aAB).
The results of this whole-genome SNP-based analysis depicted a considerably different evolutionary history for ST131 from that derived from PFGE analysis. Our use of an iterative approach to identify and exclude SNPs from recombinant regions elucidated an evolutionary path marked by clonal expansions rather than frequent lateral gene acquisitions. This underscores one of the major advantages of a whole-genome SNP-based approach relative to PFGE. Although PFGE likewise uses signatures from throughout the genome, it is highly vulnerable to phylogenetic distortions from horizontal gene transfer, which can lead to false assumptions about the evolutionary history of an organism (44), and from subjective interpretation of banding patterns and the (often invalid) presumption that similarly migrating bands represent the same chromosomal region.
The biological basis for the proliferation of H30-R and H30-Rx remains unclear. It is possible that antimicrobial resistance, the seemingly obvious explanation, is not the sole selective characteristic leading to the successive proliferation of H30-R and H30-Rx from within the H30 subclone. This is suggested by the fact that certain non-H30 ST131 isolates were identified that possessed the same phenotypic resistance traits without having reached a comparable level of success as H30-Rx. Increased virulence, as suggested by the significant association between H30-Rx and sepsis, could be a factor contributing to the success of this important subclone. Further investigations, including detailed comparative genomic, epidemiological, and functional studies, are needed to determine the basis for the success of H30-R and the strong association of H30-Rx with sepsis.
Regardless of mechanisms, the association of H30-Rx with sepsis, its broad multidrug resistance profile, and its rapid expansion and geographic dispersal warrant attention from the public health and clinical communities. Although continued accumulation of antibiotic resistance determinants may limit therapeutic options in the future, the clonal nature of H30-Rx may facilitate effective control strategies involving vaccines or transmission prevention.
MATERIALS AND METHODS
Isolates and patients.
The molecular epidemiological analyses used a large collection (n = 1,908) of recent, consecutive, single-patient E. coli isolates from 6 clinical microbiology laboratories in the United States and Germany. The United States isolates (n = 1,518) were recovered in 2010 and 2011 from 5 locations, including Group Health Cooperative, Harborview Medical Center, Seattle Children’s Hospital, and University of Washington Medical Center (all in Seattle, WA) and the Veterans Affairs Medical Center in Minneapolis, MN, as described previously (15). The German isolates (n = 390) were recovered in 2012 at the University Hospital in Münster, Germany. All isolates underwent fumC-fimH (CH) clonotyping (45) to identify ST131 and its constituent CH clonotypes (i.e., fimH-specific subclones, including H30) and were assessed for ESBL production by disk diffusion as specified by the Clinical and Laboratory Standards Institute. Medical record data regarding presence of clinically diagnosed sepsis at the time of sample collection or during the subsequent 30 days were available for 1,133 (75%) of the 2010-2011 United States isolates. Each center’s institutional review board approved the study protocol.
PFGE analysis.
The 524 historical and recent ST131 isolates were subjected to standardized XbaI PFGE analysis, as described previously (46). The dendrogram was inferred within BioNumerics (Applied Maths) according to the unweighted pair group method based on Dice similarity coefficients.
Strain selection.
Selection of ST131 isolates for genome sequencing was done in successive phases. First, to sample the breadth of phylogenetic diversity within the ST (to the extent that this is reflected in PFGE profiles), 20 isolates were selected to represent widely distributed clusters within a PFGE profile dendrogram based on a published collection of 524 historical and recent ST131 isolates from diverse locales, years of isolation, and hosts (Fig. 1A). In selecting the representative isolate(s) for a given PFGE cluster, priority was given to (i) most recent year of isolation, (ii) human host, and (iii) fluoroquinolone resistance. Next, 28 additional isolates were selected from these same PFGE clusters based on (i) proximity in the dendrogram to the initially selected (index) isolate and (ii) differences from the index isolate with respect to host and/or fluoroquinolone phenotype. Subsequently, an additional 60 isolates were selected from both this initial collection and a large collection of recent human clinical ST131 isolates from Seattle, WA, and Minneapolis, MN, that had undergone sequence analysis of gyrA, parC, and fimH (to define subclones within ST131) and PFGE analysis. Here, selection criteria included (i) distinctive gyrA, parC, and/or fimH alleles, or combinations thereof, (ii) outliers with respect to fluoroquinolone phenotype, in comparison with other isolates sharing the same PFGE type or gyrA-parC-fimH allele combination, and (iii) distinctive host species, clinical presentations (e.g., published case report isolates), specimen types (e.g., food or environmental), or dates of isolation (e.g., oldest known and oldest published ST131 isolates). Of the 108 total selected isolates, four isolates were subsequently excluded due to questionable authenticity, leaving 104 isolates for genome sequencing.
Genome sequencing.
DNA samples were prepared for multiplexed, paired-end sequencing on an Illumina Genome Analyzer IIx (Illumina, Inc., San Diego, CA). For each isolate, 1 to 5 µg DNA in 200 µl was sheared in a 96-well plate with the SonicMAN (part no. SCM1000-3; Matrical BioScience, Spokane, WA) to a size range of 200 to 1,000 bp, with the majority of material at ca. 600 bp, using the following parameters: prechill, 0°C for 75 s; cycles, 20; sonication, 10 s; power, 100%; lid chill, 0°C for 75 s; plate chill, 0°C for 10 s; postchill, 0°C for 75 s. The sheared DNA was purified using the QIAquick PCR purification kit (catalogue no. 28106; Qiagen, Valencia, CA). The enzymatic processing (end repair, phosphorylation, A tailing, and adaptor ligation) of the DNA followed the guidelines described in the Illumina protocol (“Preparing Samples for Multiplexed Paired-End Sequencing,” catalogue no. PE-930-1002, part no. 1005361). The enzymes for processing were obtained from New England Biolabs (catalogue no. E6000L; New England Biolabs, Ipswich, MA), and the oligonucleotides and adaptors were obtained from Illumina (catalogue no. PE-400-1001).
After ligation of the adaptors, the DNA was run on a 2% agarose gel for 2 h, after which a gel slice containing 500- to 600-bp fragments of each DNA sample was isolated and purified using the QIAquick gel extraction kit (catalogue no. 28706; Qiagen, Valencia, CA). Individual libraries were quantified by quantitative PCR on an ABI 7900HT (part no. 4329001; Life Technologies Corporation, Carlsbad, CA) in triplicate at two concentrations, 1:1,000 and 1:2,000, using the Kapa library quantification kit (part no. KK4832 or KK4835; Kapa Biosystems, Woburn, MA). Based on the individual library concentrations, equimolar pools of no more than 12 indexed E. coli libraries were prepared at a concentration of at least 1 nM using 10 mM Tris-HCl (pH 8.0) and 0.05% Tween 20. To ensure accurate loading onto the flow cell, the same quantification method was used to quantify the final pools. The pooled paired-end libraries were sequenced on an Illumina Genome Analyzer IIx to a read length of at least 76 bp.
The genomes were sequenced at an average depth of 60.93× (standard deviation [SD] = 31.66, using the 4,971,461-base NA114 chromosome as a reference). An average of 4,654,457.54 bases (SD = 385,629.23) were sequenced at ≥10× coverage.
Identification of SNPs.
Illumina whole-genome sequence data sets were aligned against the chromosome of a published ST131 reference genome (strain NA114; GenBank accession no. CP002797) (42) using the short-read alignment component of the Burrows-Wheeler Aligner. Each alignment was analyzed for SNPs using SolSNP, a Java-based DNA variant-calling tool for next-generation sequencing alignment data. SolSNP uses a modified Kolmogorov-Smirnov statistic and data filtering to call variants on high-coverage, aligned genomes (http://sourceforge.net/projects/solsnp/). To avoid false calls due to sequencing errors, SNP loci were excluded if they did not meet a minimum coverage of 10× and if the variant was present in <90% of the base calls for that position. SNP calls were combined for all of the sequenced genomes such that, for the locus to be included in the final SNP matrix, it had to be present in all of the genomes. SNPs falling in the duplicated regions on the reference genome were discarded.
Phylogenetic analysis.
Phylogenetic trees were generated using the maximum-parsimony method in PAUP version 4.0b10. Using prior knowledge about near neighbors, a published E. coli strain belonging to the phylogenetic group B2 genome (strain AA86; GenBank accession no. AFET00000000) was selected as an outgroup to root the ST131 whole-genome sequence tree (45). ST131 isolates in the clade nearest to this bifurcation point were used to root subsequent trees.
The homoplasy index (HI) was calculated in PAUP using the formula HI = 1 − CI, where CI is the consistency index. CI serves to measure the relative amount of homoplasy in a cladogram, as assessed by the level of difficulty in fitting SNP alleles to a given tree. The CI is calculated using the formula CI = m/s, where m is the total number of expected character changes and s is the actual number of changes that occur in the tree.
Detection of H30-Rx-specific SNPs.
Two SNPs that differentiate the CTX-M-15-associated subclone (H30-Rx) within the H30-R subclone from the rest of the H30 subclone were interrogated using Sanger sequencing. SNP-200 was detected as a C-to-T transition at position 299 of the 460-bp PCR product generated using forward primer 5′ GACACCATGCGTTTTGCTTC 3′ and reverse primer 5′ TCGTACCGGCAACAATTGAC 3′. SNP-264 was detected as a G-to-A transition at position 287 of the 462-bp PCR product generated using forward primer 5′ GTGGCGATTTCACGCTGTTA 3′ and reverse primer 5′ TATCCAGCACGTTCCAGGTG 3′. Isolates that tested positive for both SNPs were regarded as members of the H30-Rx subclone.
PCR-based detection of blaCTX-M-15.
The CTX-M-15-encoding gene blaCTX-M-15 was detected by PCR using SNP-specific forward primer 5′ ATAAAACCGGCAGCGGTGG 3′ and universal reverse primer 5′ GAATTTTGACGATCGGGG 3′ (47). PCR conditions were 10 min of denaturation at 95°C, 33 cycles of 30 s at 94°C, and 30 s at 67°C, followed by 7 min at 72°C elongation. The blaCTX-M-15-specific 483-bp PCR product was detected by agarose gel electrophoresis
Location of CTX-M-15-encoding mobile element.
Illumina short reads were aligned to the closed chromosome of JJ1886 (CP006784, CP006785, CP006786, CP006787, CP006788, and CP006789) (54) and the pEC_L8 closed plasmid sequence using BWA-MEM (48) version 0.7.5a-r405 with the default settings for paired reads. Alignments were analyzed using the IGV tool (49, 50). The location of the conserved CTX-M-15 element (ISEcp1-blaCTX-M-15-orf477) was then inferred based on the sequence alignments. Close attention was paid to the boundaries of the element, since any unmapped or incorrectly mapped pairs or pairs with insertions or deletions provided clues as to the genomic location and possible rearrangements. Putative duplications were noted if the depth of coverage around the element was substantially higher than the flanking regions. Illumina short reads were also assembled with the Mira assembler (51). The contigs were aligned with Mauve (52) against JJ1886 and pEC_L8 to identify any chromosomal rearrangement in the contigs carrying the CTX-M-15 element.
fimH and gyrA-parC allele assignments.
Sequenced isolates were assembled using VelvetOptimiser (version 2.2.2) and Velvet (53). All fimH, gyrA, and parC sequences were compared to an in-house sequence library using nucleotide-nucleotide BLAST (version 2.2.25+). Sequence similarity matches were determined using thresholds of 100% nucleotide identity and 100% coverage of the query sequence length. Allele designations were assigned based on an in-house nomenclature for the gyrA-parC combination and fimH.
Statistical methods.
Comparisons of proportions were tested using Fisher’s exact test or a chi-square test (two tailed), with P values of <0.05 as the criterion for significance.
Accession number.
All Illumina sequences were deposited into the NCBI SRA (http://www.ncbi.nlm.nih.gov/sra), study accession number SRP027327.
SUPPLEMENTAL MATERIAL
Whole-genome-SNP-based phylogeny of E. coli ST131, including strain AA86 as an outgroup. Phylogeny is based on 4,780 parsimony-informative and 15,978 total SNPs from throughout the genome. Homoplasy index (HI) = 0.351. Download
Whole-genome-SNP-based phylogeny of E. coli ST131, excluding strain AA86. Phylogeny is based on 4,770 parsimony-informative and 8,007 total SNPs from throughout the genome. HI = 0.328. Download
Whole-genome-SNP-based phylogeny of E. coli ST131 after removing SNPs from the recombinant regions and including strain AA86 as an outgroup. Phylogeny is based on 2,518 parsimony-informative and 9,533 total SNPs from throughout the genome. HI = 0.044. Download
Consensus characteristics of the major subclones within the all-ST131 phylogeny.
Isolate metadata. Download
Genetic content of the four recombinant regions excluded for phylogenetic analyses. Download
ACKNOWLEDGMENTS
This material is based upon work supported by Office of Research and Development, Medical Research Service, Department of Veterans Affairs, Merit Review grant 1I01 CX000192 01 (J.R.J.), the TGen Foundation (L.B.P.), NIH grant RC4 AI092828 (E.V.S.), and USAMRMC grant W81XWH-10-1-0753 (L.B.P.).
We thank Tania Contente-Cuomo, Richard Lester, Michael Bork, Cindy M. Liu, and Laura Davis from TGen for their contributions to this project and the staff of the Clinical Microbiology Laboratories of Harborview Medical Center, Seattle, WA, University of Washington Medical Center, Seattle, WA, and Universitätsklinikum, Münster, Germany, for their excellent help in collecting isolates and associated clinical data. Providers of isolates (institutional affiliations) also include Javier Adachi (University of Texas M. D. Anderson Cancer Center, Houston, TX), Robert L. Bergsbaken (Health Partners and Regions Medical Center, St. Paul, MN), Susan Butler-Wu (Department of Laboratory Medicine, Seattle Children’s Hospital, Seattle, WA), Mariana Castanheira (JMI Laboratories, North Liberty, IA), Tim Cleary (University of Miami, Miami, FL), Brad T. Cookson (Department of Microbiology and Department of Laboratory Medicine, University of Washington School of Medicine, Seattle, WA), Michael Cooperstock (University of Missouri School of Medicine, Columbia, MO), Chitrita DebRoy (Pennsylvania State University, University Park, PA), Paul Edelstein (Hospital of the University of Pennsylvania, Philadelphia, PA), Peter T. Ender (St. Luke’s University Hospital and Health Network, Bethlehem, PA), Ferric C. Fang (Department of Microbiology and Department of Laboratory Medicine, University of Washington School of Medicine, Seattle, WA), Glen Hansen (University of Minnesota and Hennepin County Medical Center, Minneapolis, MN), John Holter (Minneapolis VA Health Care System, Minneapolis, MN), Thomas M. Hooton (University of Miami, Miami, FL), Marie-Hélène Nicolas-Chanoine (Hôpital Beaujon, Clichy, France), Rob Owens (Cubist Pharmaceuticals, Falmouth, ME), Johann Pitout (Calgary Laboratory Services and University of Calgary, Calgary, AB, Canada), JoAnn Platell (University of Queensland, Brisbane, QLD, Australia), Xuan Qin (Department of Laboratory Medicine, Seattle Children’s Hospital, Seattle, WA), John Quinn (Atra Zeneca, Waltham, MA), James Rice (Scripps Clinic, La Jolla, CA), Ari Robicsek (NorthShore University HealthSystem, Evanston, IL), Daniel Sahm (Eurofins-Medinet, Chantilly, VA), Patricia Stogsdill (Maine Medical Center, Portland, ME, and Tufts University School of Medicine, Boston, MA), Carl Urban (NY Hospital Queens, Flushing, NY, and NY University School of Medicine, New York, NY), Karen Vigil (University of Texas Health Sciences Center at Houston, Houston, TX), Scott J. Weissman (Seattle Children’s Research Institute, Seattle, WA), and George Zhanel (University of Manitoba, Winnipeg, MB, Canada).
Footnotes
Citation Price LB, Johnson JR, Aziz M, Clabots C, Johnston B, Tchesnokova V, Nordstrom L, Billig M, Chattopadhyay S, Stegger M, Andersen PS, Pearson T, Riddell K, Rogers P, Scholes D, Kahl B, Keim P, Sokurenko EV. 2013. The epidemic of extended-spectrum-β-lactamase-producing Escherichia coli ST131 is driven by a single highly pathogenic subclone, H30-Rx. mBio 4(6):e00377-13. doi:10.1128/mBio.00377-13.
REFERENCES
- 1. Freitas AR, Novais C, Ruiz-Garbajosa P, Coque TM, Peixe L. 2009. Clonal expansion within clonal complex 2 and spread of vancomycin-resistant plasmids among different genetic lineages of Enterococcus faecalis from Portugal. J. Antimicrob. Chemother. 63:1104–1111 [DOI] [PubMed] [Google Scholar]
- 2. Samuelsen O, Toleman MA, Sundsfjord A, Rydberg J, Leegaard TM, Walder M, Lia A, Ranheim TE, Rajendra Y, Hermansen NO, Walsh TR, Giske CG. 2010. Molecular epidemiology of metallo-beta-lactamase-producing Pseudomonas aeruginosa isolates from Norway and Sweden shows import of international clones and local clonal expansion. Antimicrob. Agents Chemother. 54:346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shu JC, Chia JH, Kuo AJ, Su LH, Wu TL. 2010. A 7-year surveillance for ESBL-producing Escherichia coli and Klebsiella pneumoniae at a university hospital in Taiwan: the increase of CTX-M-15 in the ICU. Epidemiol. Infect. 138:253–263 [DOI] [PubMed] [Google Scholar]
- 4. Coque TM, Novais A, Carattoli A, Poirel L, Pitout J, Peixe L, Baquero F, Cantón R, Nordmann P. 2008. Dissemination of clonally related Escherichia coli strains expressing extended-spectrum beta-lactamase CTX-M-15. Emerg. Infect. Dis. 14:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nicolas-Chanoine MH, Blanco J, Leflon-Guibout V, Demarty R, Alonso MP, Caniça MM, Park YJ, Lavigne JP, Pitout J, Johnson JR. 2008. Intercontinental emergence of Escherichia coli clone O25:H4-ST131 producing CTX-M-15. J. Antimicrob. Chemother. 61:273–281 [DOI] [PubMed] [Google Scholar]
- 6. Johnson JR, Johnston B, Clabots C, Kuskowski MA, Castanheira M. 2010. Escherichia coli sequence type ST131 as the major cause of serious multidrug-resistant E. coli infections in the United States. Clin. Infect. Dis. 51:286–294 [DOI] [PubMed] [Google Scholar]
- 7. Rogers BA, Sidjabat HE, Paterson DL. 2011. Escherichia coli O25b–ST131: a pandemic, multiresistant, community-associated strain. J. Antimicrob. Chemother. 66:1–14 [DOI] [PubMed] [Google Scholar]
- 8. Assimacopoulos A, Johnston B, Clabots C, Johnson JR. 2012. Post-prostate biopsy infection with Escherichia coli ST131 leading to epididymo-orchitis and meningitis caused by Gram-negative bacilli. J. Clin. Microbiol. 50:4157–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bert F, Johnson JR, Ouattara B, Leflon-Guibout V, Johnston B, Marcon E, Valla D, Moreau R, Nicolas-Chanoine MH. 2010. Genetic diversity and virulence profiles of Escherichia coli isolates causing spontaneous bacterial peritonitis and bacteremia in patients with cirrhosis. J. Clin. Microbiol. 48:2709–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vigil KJ, Johnson JR, Johnston BD, Kontoyiannis DP, Mulanovich VE, Raad II, Dupont HL, Adachi JA. 2010. Escherichia coli pyomyositis: an emerging infectious disease among patients with hematologic malignancies. Clin. Infect. Dis. 50:374–380 [DOI] [PubMed] [Google Scholar]
- 11. Courpon-Claudinon A, Lefort A, Panhard X, Clermont O, Dornic Q, Fantin B, Mentré F, Wolff M, Denamur E, Branger C, COLIBAFI Group 2011. Bacteraemia caused by third-generation cephalosporin-resistant Escherichia coli in France: prevalence, molecular epidemiology and clinical features. Clin. Microbiol. Infect. 17:557–565 [DOI] [PubMed] [Google Scholar]
- 12. Karfunkel D, Carmeli Y, Chmelnitsky I, Kotlovsky T, Navon-Venezia S. 2013. The emergence and dissemination of CTX-M-producing Escherichia coli sequence type 131 causing community-onset bacteremia in Israel. Eur. J. Clin. Microbiol. Infect. Dis. 32:513–521 [DOI] [PubMed] [Google Scholar]
- 13. Peirano G, Schreckenberger PC, Pitout JD. 2011. Characteristics of NDM-1-producing Escherichia coli isolates that belong to the successful and virulent clone ST131. Antimicrob. Agents Chemother. 55:2986–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vincent C, Boerlin P, Daignault D, Dozois CM, Dutil L, Galanakis C, Reid-Smith RJ, Tellier PP, Tellis PA, Ziebell K, Manges AR. 2010. Food reservoir for Escherichia coli causing urinary tract infections. Emerg. Infect. Dis. 16:88–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott D, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye A, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV. 2013. Abrupt emergence of a single dominant multi-drug-resistant strain of Escherichia coli. J. Infect. Dis. 207:191–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cullen IM, Manecksha RP, McCullagh E, Ahmad S, O’Kelly F, Flynn RJ, McDermott T, Murphy P, Grainger R, Fennell JP, Thornhill JA. 2012. The changing pattern of antimicrobial resistance within 42,033 Escherichia coli isolates from nosocomial, community and urology patient-specific urinary tract infections, Dublin, 1999–2009. BJU Int. 109:1198–1206 [DOI] [PubMed] [Google Scholar]
- 17. Foxman B. 2012. Extended spectrum beta lactamase (ESBL) producing Escherichia coli in the United States: time to re-think empirical treatment for suspected E. coli infections? Clin. Infect. Dis. 56:649–651 [DOI] [PubMed] [Google Scholar]
- 18. Johnson JR, Menard ME, Lauderdale TL, Kosmidis C, Gordon D, Collignon P, Maslow JN, Andrasević AT, Kuskowski MA, Trans-Global Initiative for Antimicrobial Resistance Analysis Investigators 2011. Global distribution and epidemiologic associations of Escherichia coli clonal group A, 1998–2007. Emerg. Infect. Dis. 17:2001–2009 [DOI] [PubMed] [Google Scholar]
- 19. Lu PL, Liu YC, Toh HS, Lee YL, Liu YM, Ho CM, Huang CC, Liu CE, Ko WC, Wang JH, Tang HJ, Yu KW, Chen YS, Chuang YC, Xu Y, Ni Y, Chen YH, Hsueh PR. 2012. Epidemiology and antimicrobial susceptibility profiles of Gram-negative bacteria causing urinary tract infections in the Asia-Pacific region: 2009–2010 results from the Study for Monitoring Antimicrobial Resistance Trends (SMART). Int. J. Antimicrob. Agents 40(Suppl):S37–S43 [DOI] [PubMed] [Google Scholar]
- 20. Christiansen N, Nielsen L, Jakobsen L, Stegger M, Hansen LH, Frimodt-Møller N. 2011. Fluoroquinolone resistance mechanisms in urinary tract pathogenic Escherichia coli isolated during rapidly increasing fluoroquinolone consumption in a low-use country. Microb. Drug Resist. 17:395–406 [DOI] [PubMed] [Google Scholar]
- 21. Johnson JR, Johnston B, Clabots C, Kuskowski MA, Pendyala S, Debroy C, Nowicki B, Rice J. 2010. Escherichia coli sequence type ST131 as an emerging fluoroquinolone-resistant uropathogen among renal transplant recipients. Antimicrob. Agents Chemother. 54:546–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson TJ, Logue CM, Johnson JR, Kuskowski MA, Sherwood JS, Barnes HJ, DebRoy C, Wannemuehler YM, Obata-Yasuoka M, Spanjaard L, Nolan LK. 2012. Associations between multidrug resistance, plasmid content, and virulence potential among extraintestinal pathogenic and commensal Escherichia coli from humans and poultry. Foodborne Pathog. Dis. 9:37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Longhi C, Conte MP, Marazzato M, Iebba V, Totino V, Santangelo F, Gallinelli C, Pallecchi L, Riccobono E, Schippa S, Comanducci A. 2012. Plasmid-mediated fluoroquinolone resistance determinants in Escherichia coli from community uncomplicated urinary tract infection in an area of high prevalence of quinolone resistance. Eur. J. Clin. Microbiol. Infect. Dis. 31:1917–1921 [DOI] [PubMed] [Google Scholar]
- 24. Pitout JD. 2012. Extraintestinal pathogenic Escherichia coli: a combination of virulence with antibiotic resistance. Front. Microbiol. 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tiruvury H, Johnson JR, Mariano N, Grenner L, Colon-Urban R, Erritouni M, Wehbeh W, Segal-Maurer S, Rahal JJ, Johnston B, Urban C. 2012. Identification of CTX-M beta-lactamases among Escherichia coli from the community in New York City. Diagn. Microbiol. Infect. Dis. 72:248–252 [DOI] [PubMed] [Google Scholar]
- 26. van der Donk CF, van de Bovenkamp JH, De Brauwer EI, De Mol P, Feldhoff KH, Kalka-Moll WM, Nys S, Thoelen I, Trienekens TA, Stobberingh EE. 2012. Antimicrobial resistance and spread of multi drug resistant Escherichia coli isolates collected from nine urology services in the Euregion Meuse-Rhine. PLoS One 7:e47707. 10.1371/journal.pone.0047707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang H, Dzink-Fox JL, Chen M, Levy SB. 2001. Genetic characterization of highly fluoroquinolone-resistant clinical Escherichia coli strains from China: role of acrR mutations. Antimicrob. Agents Chemother. 45:1515–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cantón R, Coque TM. 2006. The CTX-M beta-lactamase pandemic. Curr. Opin. Microbiol. 9:466–475 [DOI] [PubMed] [Google Scholar]
- 29. Doi Y, Park YS, Rivera JI, Adams-Haduch JM, Hingwe A, Sordillo EM, Lewis JS, II, Howard WJ, Johnson LE, Polsky B, Jorgensen JH, Richter SS, Shutt KA, Paterson DL. 2012. Community-associated extended-spectrum beta-lactamase-producing Escherichia coli infection in the United States. Clin. Infect. Dis. 56:641–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peirano G, Pitout JD. 2010. Molecular epidemiology of Escherichia coli producing CTX-M beta-lactamases: the worldwide emergence of clone ST131 (O25:H4). Int. J. Antimicrob. Agents 35:316–321 [DOI] [PubMed] [Google Scholar]
- 31. Peirano G, Richardson D, Nigrin J, McGeer A, Loo V, Toye B, Alfa M, Pienaar C, Kibsey P, Pitout JD. 2010. High prevalence of ST131 isolates producing CTX-M-15 and CTX-M-14 among extended-spectrum-beta-lactamase-producing Escherichia coli isolates from Canada. Antimicrob. Agents Chemother. 54:1327–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cantón R, Novais A, Valverde A, Machado E, Peixe L, Baquero F, Coque TM. 2008. Prevalence and spread of extended-spectrum beta-lactamase-producing Enterobacteriaceae in Europe. Clin. Microbiol. Infect. 14(Suppl 1):144–153 [DOI] [PubMed] [Google Scholar]
- 33. Canton R, Gonzalez-Alba JM, Galan JC. 2012. CTX-M enzymes: origin and diffusion. Front. Microbiol. 3:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dimude JU, Amyes SG. 2013. Molecular diversity associated with the dissemination of CTX-M-15 beta-lactamase gene in blood culture isolates of Escherichia coli from Edinburgh. Scand. J. Infect. Dis. 45:32–37 [DOI] [PubMed] [Google Scholar]
- 35. Lee MY, Choi HJ, Choi JY, Song M, Song Y, Kim SW, Chang HH, Jung SI, Kim YS, Ki HK, Son JS, Kwon KT, Heo ST, Yeom JS, Shin SY, Chung DR, Peck KR, Song JH, Ko KS. 2010. Dissemination of ST131 and ST393 community-onset, ciprofloxacin-resistant Escherichia coli clones causing urinary tract infections in Korea. J. Infect. 60:146–153 [DOI] [PubMed] [Google Scholar]
- 36. Clark G, Paszkiewicz K, Hale J, Weston V, Constantinidou C, Penn C, Achtman M, McNally A. 2012. Genomic analysis uncovers a phenotypically diverse but genetically homogeneous Escherichia coli ST131 clone circulating in unrelated urinary tract infections. J. Antimicrob. Chemother. 67:868–877 [DOI] [PubMed] [Google Scholar]
- 37. Doumith M, Dhanji H, Ellington MJ, Hawkey P, Woodford N. 2012. Characterization of plasmids encoding extended-spectrum beta-lactamases and their addiction systems circulating among Escherichia coli clinical isolates in the UK. J. Antimicrob. Chemother. 67:878–885 [DOI] [PubMed] [Google Scholar]
- 38. Grude N, Strand L, Mykland H, Nowrouzian FL, Nyhus J, Jenkins A, Kristiansen BE. 2008. Fluoroquinolone-resistant uropathogenic Escherichia coli in Norway: evidence of clonal spread. Clin. Microbiol. Infect. 14:498–500 [DOI] [PubMed] [Google Scholar]
- 39. Novais A, Pires J, Ferreira H, Costa L, Montenegro C, Vuotto C, Donelli G, Coque TM, Peixe L. 2012. Characterization of globally spread Escherichia coli ST131 isolates (1991–2010). Antimicrob. Agents Chemother. 56:3973–3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yi H, Cho YJ, Hur HG, Chun J. 2011. Genome sequence of Escherichia coli AA86, isolated from cow feces. J. Bacteriol. 193:3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pearson T, Okinaka RT, Foster JT, Keim P. 2009. Phylogenetic understanding of clonal populations in an era of whole genome sequencing. Infect. Genet. Evol. 9:1010–1019 [DOI] [PubMed] [Google Scholar]
- 42. Avasthi TS, Kumar N, Baddam R, Hussain A, Nandanwar N, Jadhav S, Ahmed N. 2011. Genome of multidrug-resistant uropathogenic Escherichia coli strain NA114 from India. J. Bacteriol. 193:4272–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Owens RC, Jr, Johnson JR, Stogsdill P, Yarmus L, Lolans K, Quinn J. 2011. Community transmission in the United States of a CTX-M-15-producing sequence type ST131 Escherichia coli strain resulting in death. J. Clin. Microbiol. 49:3406–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hao W, Allen VG, Jamieson FB, Low DE, Alexander DC. 2012. Phylogenetic incongruence in E. coli O104: understanding the evolutionary relationships of emerging pathogens in the face of homologous recombination. PLoS One 7:e33971. 10.1371/journal.pone.0033971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Weissman SJ, Johnson JR, Tchesnokova V, Billig M, Dykhuizen D, Riddell K, Rogers P, Qin X, Butler-Wu S, Cookson BT, Fang FC, Scholes D, Chattopadhyay S, Sokurenko E. 2012. High-resolution two-locus clonal typing of extraintestinal pathogenic Escherichia coli. Appl. Environ. Microbiol. 78:1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johnson JR, Nicolas-Chanoine MH, DebRoy C, Castanheira M, Robicsek A, Hansen G, Weissman S, Urban C, Platell J, Trott D, Zhanel G, Clabots C, Johnston BD, Kuskowski MA, MASTER Investigators 2012. Comparison of Escherichia coli ST131 pulsotypes, by epidemiologic traits, 1967–2009. Emerg. Infect. Dis. 18:598–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnson JR, Urban C, Weissman SJ, Jorgensen JH, Lewis JS, II, Hansen G, Edelstein PH, Robicsek A, Cleary T, Adachi J, Paterson D, Quinn J, Hanson ND, Johnston BD, Clabots C, Kuskowski MA, AMERECUS Investigators 2012. Molecular epidemiological analysis of Escherichia coli sequence type ST131 (O25: H4) and blaCTX-M-15 among extended-spectrum-beta-lactamase-producing E. coli from the United States, 2000–2009. Antimicrob. Agents Chemother. 56:2364–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14:178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chevreux B, Wetter T, Suhai S. 1999. Genome sequence assembly using trace signals and additional sequence information, p 45–56 Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB)
- 52. Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Andersen PS, Stegger M, Aziz M, Contente-Cuomo T, Gibbons HS, Keim P, Sokurenko EV, Johnson JR, Price LB. Complete genome of the epidemic and highly virulent CTX-M-15-producing H30-Rx subclone of Escherichia coli ST131. Genome Announc., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Whole-genome-SNP-based phylogeny of E. coli ST131, including strain AA86 as an outgroup. Phylogeny is based on 4,780 parsimony-informative and 15,978 total SNPs from throughout the genome. Homoplasy index (HI) = 0.351. Download
Whole-genome-SNP-based phylogeny of E. coli ST131, excluding strain AA86. Phylogeny is based on 4,770 parsimony-informative and 8,007 total SNPs from throughout the genome. HI = 0.328. Download
Whole-genome-SNP-based phylogeny of E. coli ST131 after removing SNPs from the recombinant regions and including strain AA86 as an outgroup. Phylogeny is based on 2,518 parsimony-informative and 9,533 total SNPs from throughout the genome. HI = 0.044. Download
Consensus characteristics of the major subclones within the all-ST131 phylogeny.
Isolate metadata. Download
Genetic content of the four recombinant regions excluded for phylogenetic analyses. Download