

ABSTRACT
Candida albicans invades endothelial cells by binding to N-cadherin and other cell surface receptors. This binding induces rearrangement of endothelial cell actin microfilaments, which results in the formation of pseudopods that surround the organism and pull it into the endothelial cell. Here, we investigated the role of endothelial cell septin 7 (SEPT7) in the endocytosis of C. albicans hyphae. Using confocal microscopy, we determined that SEPT7 accumulated with N-cadherin and actin microfilaments around C. albicans as it was endocytosed by endothelial cells. Affinity purification studies indicated that a complex containing N-cadherin and SEPT7 was recruited by C. albicans and that formation of this complex around C. albicans was mediated by the fungal Als3 and Ssa1 invasins. Knockdown of N-cadherin by small interfering RNA (siRNA) reduced recruitment of SEPT7 to C. albicans, suggesting that N-cadherin functions as a link between SEPT7 and the fungus. Also, depolymerization of actin microfilaments with cytochalasin D decreased the association between SEPT7 and N-cadherin and inhibited recruitment of both SEPT7 and N-cadherin to C. albicans, indicating the necessity of an intact cytoskeleton in the functional interaction between SEPT7 and N-cadherin. Importantly, knockdown of SEPT7 decreased accumulation of N-cadherin around C. albicans in intact endothelial cells and reduced binding of N-cadherin to this organism, as revealed by the affinity purification assay. Furthermore, SEPT7 knockdown significantly inhibited the endocytosis of C. albicans. Therefore, in response to C. albicans infection, SEPT7 forms a complex with endothelial cell N-cadherin, is required for normal accumulation of N-cadherin around C. albicans hyphae, and is necessary for maximal endocytosis of the organism.
IMPORTANCE
During hematogenously disseminated infection, Candida albicans invades the endothelial cell lining of the blood vessels to invade the deep tissues. C. albicans can invade endothelial cells by inducing its own endocytosis, which is triggered when the C. albicans Als3 and Ssa1 invasins bind to N-cadherin on the endothelial cell surface. How this binding induces endocytosis is incompletely understood. Septins are intracellular GTP-binding proteins that influence the function and localization of cell surface proteins. We found that C. albicans Als3 and Ssa1 bind to a complex containing N-cadherin and septin 7, which in turn interacts with endothelial cell microfilaments, thereby inducing endocytosis of the organism. The key role of septin 7 in governing receptor-mediated endocytosis is likely relevant to host cell invasion by other microbial pathogens, in addition to C. albicans.
INTRODUCTION
Hematogenously disseminated candidiasis is a severe fungal infection that occurs in hospitalized patients and is associated with a mortality of almost 40%, even with currently available therapy. About 50% of these life-threatening infections are caused by Candida albicans; the remaining cases are caused by other species of Candida (1, 2).
In susceptible hosts, hematogenously disseminated candidiasis is initiated when C. albicans enters the bloodstream, either by traversing the wall of the digestive tract or via an intravenous catheter. To escape from the bloodstream and proliferate in the deep tissues, the blood borne organisms must invade the endothelial cells that line the blood vessels (3). One mechanism by which C. albicans invades endothelial cells is by inducing its own endocytosis. This organism expresses the Als3 and Ssa1 invasins, which bind to N-cadherin and other receptors on the endothelial cell surface (4–7). Normally, N-cadherin on one endothelial cell binds to N-cadherin on other host cells to allow cross-communication among cells. However, when C. albicans binds to this receptor, it triggers rearrangement of actin microfilaments by a clathrin-dependent mechanism (8). This results in the formation of endothelial cell pseudopods, which surround the organism and pull it into the endothelial cell (4, 5). Because C. albicans hyphae are relatively long compared to the size of the endothelial cell, they are not endocytosed all at once (9). Instead, endothelial cell pseudopods form around part of the hypha, usually starting at the distal end, and progressively pull the organism into the cell. N-cadherin, actin, and components of the clathrin-related endocytic pathway accumulate only around the portion of the organism that is in the process of being endocytosed (8).
Host cell invasion is a critical step in the initiation of disseminated candidiasis. Thus, strategies to block this process can potentially lead to new approaches to treat this infection. Developing such therapeutic approaches requires a comprehensive understanding of the mechanisms by which C. albicans invades endothelial cells. Although some of the fundamental components of C. albicans uptake are already known, the underlying mechanisms by which N-cadherin localizes to the correct regions on the cell surface, subsequently signals actin rearrangement, and induces pseudopod formation are incompletely understood.
One potential link between N-cadherin and actin microfilaments is the septin family of proteins, which consists of 30- to 65-kDa intracellular GTP-binding proteins. Originally identified in yeast, septins are present in most eukaryotic cells, except for plants. In mammalian cells, these proteins form hetero-oligomeric filaments that associate with actin microfilaments, microtubules, and other elements of the cytoskeletal network. Septins contribute to protein recruitment, cytokinesis, and vesicle fusion (10, 11). Importantly, septins also play a key role in anchoring cell surface proteins to specific regions of the cell membrane (12, 13). For example, SEPT2 is required for the normal expression and function of Met on the surface of epithelial cells. Because Met is a receptor for Listeria monocytogenes, small interfering RNA (siRNA) knockdown of SEPT2 significantly inhibits epithelial cell internalization of this organism (14, 15).
Although SEPT7 is expressed by virtually all cell types and is a key component of most types of septin filaments (11), its role in governing the interactions of host cells with microbial pathogens has not been studied previously. Herein, we investigated the role of SEPT7 in the endocytosis of C. albicans by endothelial cells. We found that in response to C. albicans infection, SEPT7 forms a complex with endothelial cell N-cadherin, is required for N-cadherin to accumulate around C. albicans hyphae, and is necessary for maximal endocytosis of C. albicans.
RESULTS
SEPT7 colocalizes with N-cadherin around C. albicans hyphae.
For C. albicans to induce its own endocytosis, the fungus must first bind to endothelial cell N-cadherin. Downregulation of this protein inhibits endothelial cell endocytosis of C. albicans (4, 5). We hypothesized that septins play a role in the endocytosis of C. albicans either by interacting with N-cadherin or by stabilizing this protein on the cell surface. Because SEPT7 is a key component of most septin filaments and is present in virtually all types of host cells (16), we selected it for in-depth study.
We first used indirect immunofluorescence and confocal microscopy to determine if N-cadherin and SEPT7 colocalized around C. albicans hyphae in infected endothelial cells. A time course study was performed in which, after 30, 60, and 90 min of infection, the infected endothelial cells were fixed and stained for both N-cadherin and SEPT7. Next, we selected 15 microscopic fields at random and counted the number of organisms around which there was accumulation of N-cadherin alone, SEPT7 alone, or both N-cadherin and SEPT7. In uninfected cells, both N-cadherin and SEPT7 were present on or near the apical surface of the endothelial cells, but there was limited colocalization (see Fig. S1 in the supplemental material). After 30 min of infection, almost no N-cadherin had accumulated around any organisms, and SEPT7 had accumulated around only a few organisms (Fig. 1). However, after 60 and 90 min of infection, progressively more N-cadherin and SEPT7 accumulated around the organisms. Virtually all organisms that were surrounded by N-cadherin were also surrounded by SEPT7. However, approximately 43% of the organisms were surrounded by SEPT7 but not N-cadherin. In addition, we observed that N-cadherin and SEPT7 frequently accumulated around only part of an organism, usually a portion of a hypha, and almost never around the yeast. In fact, N-cadherin and SEPT7 generally accumulated around the portion of the organism that was adjacent to the apical surface of the endothelial cells; these proteins no longer surrounded portions of organisms that were completely inside the endothelial cell. These results are consistent with our previous data showing that N-cadherin and components of the endocytic pathway accumulate only around the portion of the hypha that is in the process of being endocytosed (9). Collectively, these data suggest that SEPT7 and N-cadherin may interact during the endocytosis of C. albicans. The finding that some organisms were surrounded by SEPT7, but not N-cadherin, also indicates that SEPT7 likely interacts with another endothelial cell receptor for C. albicans, other than N-cadherin.
FIG 1 .
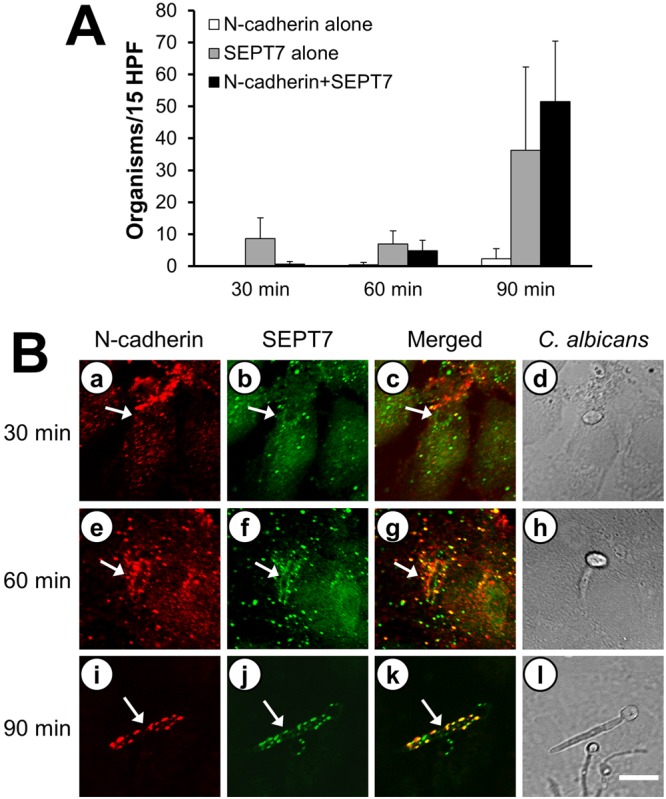
Colocalization of N-cadherin and SEPT7 during the endocytosis of C. albicans. Endothelial cells were infected with wild-type C. albicans for the indicated times and then fixed and stained for N-cadherin and SEPT7. (A) The number of organisms in 15 high-power fields (HPF) that were surrounded by N-cadherin alone, SEPT7 alone, or N-cadherin plus SEPT7 were counted at the indicated times after infection. Results are the means ± standard deviations (SD) from three experiments, each performed in triplicate. (B) Confocal microscopic images of N-cadherin (a, e, i) and SEPT7 (b, f, j) accumulating around the same organism. The merged images are shown in panels c, g, and k, and images of the corresponding microscopic fields viewed by differential interference contrast are shown in panels d, h, and l. Arrows indicate the N-cadherin and SEPT7 that accumulated around the hyphae. Scale bar = 5 µm.
SEPT7 accumulation precedes actin accumulation around C. albicans cells.
The endothelial cell pseudopods that are formed around C. albicans cells contain actin microfilaments, which are required for the endocytosis of this organism (17, 18). Therefore, we investigated whether SEPT7 and actin microfilaments colocalized around C. albicans cells as they adhered to and were endocytosed by endothelial cells. The endocytosed organisms were identified by a network of phalloidin-labeled actin microfilaments that formed around them (18). When yeast-phase organisms were added to endothelial cells, these organisms rapidly adhered and began to germinate within 30 min. At this time point, there was scant accumulation of SEPT7 underneath a few of the organisms and no accumulation of actin (Fig. 2). After 60 min, some C. albicans hyphae were surrounded by SEPT7. A few of these organisms were also surrounded by a small amount of actin, indicating early pseudopod formation during the initiation of endocytosis. By 90 min, the hyphae were surrounded by both SEPT7 and actin. However, approximately equal numbers of organisms were surrounded by SEPT7 alone, actin alone, and SEPT7 plus actin. Collectively, these observations indicate that SEPT7 accumulation around C. albicans precedes the accumulation of actin and subsequent induction of endocytosis. The finding that some organisms were surrounded by actin alone suggests that, once pseudopods have formed, SEPT7 is no longer present at the site of endocytosis.
FIG 2 .
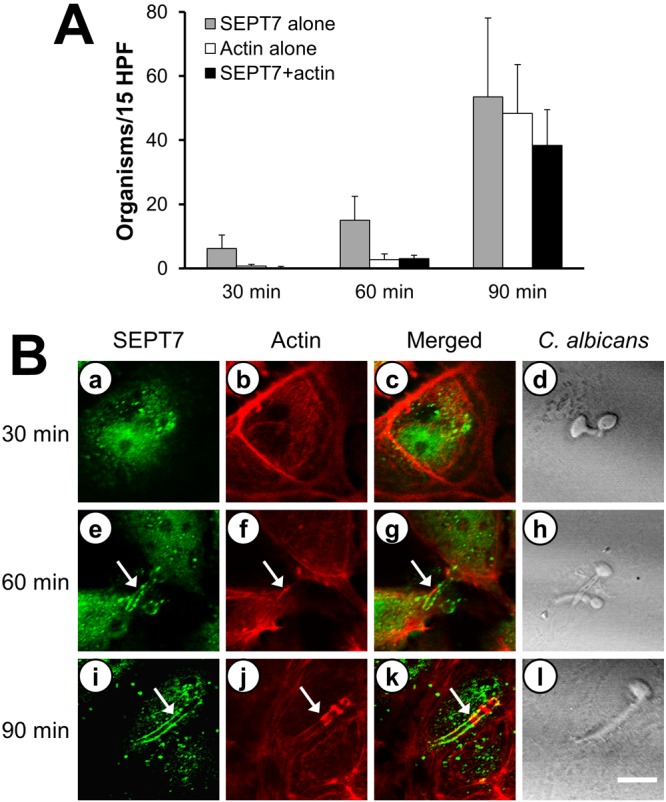
SEPT7 and actin colocalize during the endocytosis of C. albicans. Endothelial cells were infected with C. albicans for the indicated times and then fixed and stained for SEPT7 and actin. (A) The numbers of organisms in 15 HPF that were surrounded by SEPT7 alone, actin alone, or SEPT7 plus actin were counted at the indicated times after infection. Results are the means ± SD from three experiments, each performed in triplicate. (B) Confocal microscopic images of SEPT7 (a, e, i) and actin (b, f, j) accumulation. The merged images are shown in panels c, g, and k, and images of the corresponding microscopic fields viewed by differential interference contrast are shown in panels d, h, and l. Arrows indicate the SEPT7 and actin that accumulated around the hyphae. Scale bar = 5 µm.
N-cadherin and SEPT7 are part of a complex that is recruited by C. albicans.
The colocalization of N-cadherin and SEPT7 in the indirect immunofluorescence experiments suggested that these two proteins either form a complex with each other or are constituents of the same complex. To investigate this possibility, we infected endothelial cells with live C. albicans for various times and then lysed the endothelial cells with octyl-glucopyranoside, leaving the fungi intact. The hyphae and the endothelial cell proteins that were attached to them (either directly or as part of a protein complex) were collected by centrifugation and then rinsed extensively. Next, the bound endothelial cell proteins were eluted from the hyphae and separated by SDS-PAGE, after which the amount of N-cadherin and SEPT7 in these eluted proteins was determined by immunoblotting. In time course studies, we observed that there was increased binding of N-cadherin to C. albicans hyphae after 60 min of infection, and even more N-cadherin had bound to the hyphae after 90 min (Fig. 3A and B). C. albicans hyphae also pulled down endothelial cell SEPT7 over a similar time course, suggesting that N-cadherin and SEPT7 interact with each other during endocytosis.
FIG 3 .
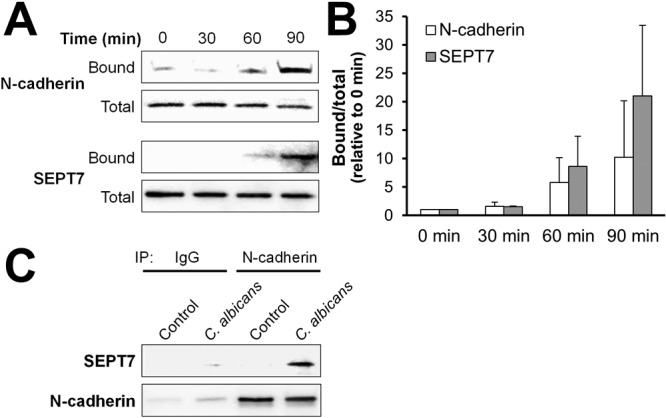
C. albicans binds to a complex of N-cadherin and SEPT7. Endothelial cells were infected with C. albicans for the indicated time points, and the endothelial cell proteins that bound to the organisms, either directly or indirectly (bound), were isolated. The nonbinding proteins (total) were also collected. The bound and total N-cadherin and SEPT7 were identified by immunoblotting. (A) Representative immunoblot. (B) Densitometric analysis of 3 independent immunoblots comparing the relative amounts of bound to total protein. Results are means ± SD. (C) Endothelial cells were infected with C. albicans for 90 min and lysed, after which the lysates were subjected to immunoprecipitation (IP) with either control IgG or an anti-N-cadherin antibody. The presence of SEPT7 and N-cadherin in immunoprecipitated proteins was detected by immunoblotting.
To investigate whether the interaction between N-cadherin and SEPT7 was enhanced by the presence of C. albicans, we performed coimmunoprecipitation experiments. We found that almost no SEPT7 was immunoprecipitated along with N-cadherin in uninfected endothelial cells (Fig. 3C). However, the amount of SEPT7 that was associated with N-cadherin greatly increased when the endothelial cells were infected with C. albicans. As expected, very little N-cadherin or SEPT7 were pulled down when the endothelial cell lysates were immunoprecipitated with control IgG. Collectively, these data suggest that, while N-cadherin and SEPT7 form a complex in uninfected endothelial cells, C. albicans infection enhances the association of these proteins.
Because septins are intracellular proteins that are generally not expressed on the cell surface (10, 11), it is likely that C. albicans did not bind to SEPT7 directly but instead recruited this protein via N-cadherin and possibly additional endothelial cell receptors. To test this hypothesis, we used siRNA to knock down N-cadherin protein expression and then analyzed the number of C. albicans cells that were surrounded by SEPT7. We found that knockdown of N-cadherin significantly reduced the number of hyphae that were surrounded by either SEPT7 alone or SEPT7 plus N-cadherin (Fig. 4A and B). Furthermore, in the affinity purification experiments, knockdown of N-cadherin caused a 50% reduction in the amount of SEPT7 that was pulled down by C. albicans but had no effect on the total cellular content of SEPT7 (Fig. 4C and D). These results support the model that C. albicans binds to N-cadherin, which in turn either directly or indirectly recruits SEPT7. Moreover, the finding that N-cadherin knockdown did not completely block SEPT7 accumulation and binding suggests that an additional receptor(s) must also recruit SEPT7 and mediate the endocytosis of C. albicans.
FIG 4 .
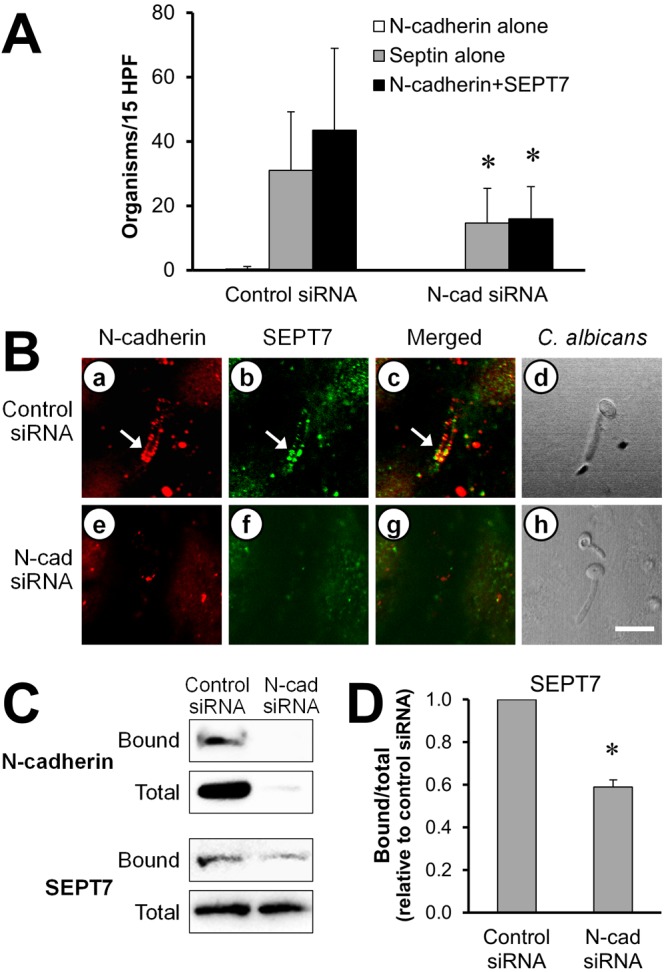
Effects of N-cadherin siRNA on C. albicans recruitment of endothelial cell N-cadherin and SEPT7. Endothelial cells were transfected with either control or N-cadherin siRNA and then infected with wild-type C. albicans for 90 min. (A, B) The cells were fixed and then stained for N-cadherin and SEPT7. (A) The number of organisms in 15 high-power fields that were surrounded by N-cadherin alone, SEPT7 alone, or N-cadherin plus SEPT7. Results are the means ± SD from three experiments, each performed in triplicate. (B) Confocal microscopic images of N-cadherin (a, e) and SEPT7 (b, f) accumulating around the same organism. The merged images are shown in panels c and g, and images of the corresponding microscopic fields viewed by differential interference contrast are shown in panels d and h. Arrows indicate the N-cadherin and SEPT7 that accumulated around the hyphae. Scale bar = 5 µm. (C) Representative immunoblot showing the effects of N-cadherin knockdown on the amount of endothelial cell N-cadherin and SEPT7 that bound to C. albicans hyphae. (D) Densitometric analysis of 3 independent SEPT7 immunoblots. Results are means ± SD. *, P ≤ 0.05 compared to cells transfected with control siRNA.
Formation of the N-cadherin–SEPT7 complex is triggered by C. albicans Als3 and Ssa1.
N-cadherin is bound by the C. albicans invasins Als3 and Ssa1 (5, 6). Therefore, we investigated whether these fungal proteins mediate binding to the N-cadherin–SEPT7 complex. Using our affinity purification assay, we determined that the poorly invasive C. albicans als3Δ/Δ and ssa1Δ/Δ mutants (5, 6) bound weakly to the N-cadherin–SEPT7 complex (Fig. 5). Binding to this complex was restored to wild-type levels by reintegrating an intact copy of ALS3 or SSA1 into the respective deletion mutants. Next, we tested whether the N-cadherin–SEPT7 complex was bound by strains of the normally nonadherent, noninvasive yeast Saccharomyces cerevisiae that expressed either C. albicans ALS3 or SSA1. Both of these strains are avidly endocytosed by endothelial cells (19, 20). As expected, the control strains of S. cerevisiae containing the backbone vector had only low-level, nonspecific binding to the N-cadherin–SEPT7 complex, whereas S. cerevisiae expressing either ALS3 or SSA1 bound avidly to it (Fig. 5). Collectively, these results verify the specificity of the pulldown assay, and they indicate that C. albicans Als3 and Ssa1 bind to a complex containing N-cadherin and SEPT7.
FIG 5 .
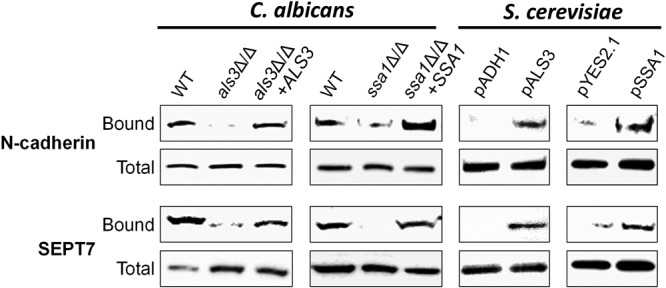
Als3 and Ssa1 mediate binding to the N-cadherin–SEPT7 complex. Endothelial cells were infected for 90 min with the indicated strains of C. albicans or with S. cerevisiae containing the backbone vector (pADH1 or pYES2.1) or expressing C. albicans ALS3 (pALS3) or SSA1 (pSSA1). Next, the endothelial cell proteins that bound to these strains were isolated and then identified by immunoblotting.
SEPT7 is required for maximal N-cadherin accumulation around C. albicans hyphae.
Because septins are involved in the proper localization and function of cell surface proteins (10, 11, 14, 21), we considered the possibility that SEPT7 is required for endothelial cell N-cadherin to accumulate around and bind to C. albicans hyphae. Although siRNA knockdown of SEPT7 did not result in complete depletion of SEPT7, it significantly reduced accumulation of N-cadherin around C. albicans (Fig. 6A and B). Importantly, knockdown of SEPT7 had no effect on the level of surface-expressed N-cadherin (see Fig. S2 in the supplemental material). Furthermore, in the affinity purification assay, SEPT7 knockdown caused a 50% reduction in the amount of N-cadherin that was bound by C. albicans but did not alter the total cellular content of N-cadherin (Fig. 6C and D). Collectively, these results suggest that SEPT7 is necessary for the maximal interaction of N-cadherin with C. albicans.
FIG 6 .
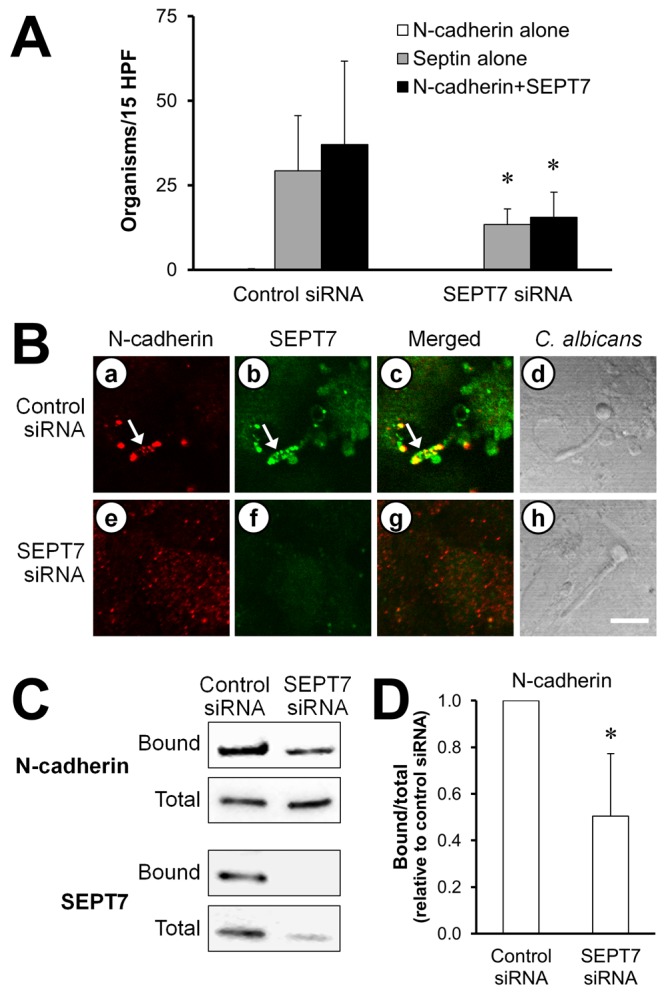
SEPT7 knockdown reduces the accumulation of N-cadherin around C. albicans. Endothelial cells were transfected with either control or SEPT7 siRNA and then infected with C. albicans for 90 min. (A, B) The cells were fixed and then stained for N-cadherin and SEPT7. (A) The number of organisms in 15 high-power fields that were surrounded by N-cadherin alone, SEPT7 alone, or N-cadherin plus SEPT7. Results are the means ± SD from three experiments, each performed in triplicate. (B) Confocal microscopic images of N-cadherin (a, e) and SEPT7 (b, f) accumulating around the same organism. The merged images are shown in panels c and g, and images of the corresponding microscopic fields viewed by differential interference contrast are shown in panels d and h. Arrows indicate the N-cadherin and SEPT7 that accumulated around the hyphae. Scale bar = 5 µm. (C) Representative immunoblot showing the effects of SEPT7 knockdown on the amount of endothelial cell N-cadherin and SEPT7 that bound to C. albicans hyphae. (D) Densitometric analysis of 4 independent N-cadherin immunoblots. Results are means ± SD. *, P < 0.05 compared to cells transfected with control siRNA.
Importantly, SEPT7 knockdown did not cause detectable toxicity to the endothelial cells. The proliferation of endothelial cells transfected with either the control siRNA or SEPT7 siRNA was similar to that of untransfected cells. For example, during the 48 h following transfection, both sets of endothelial cells grew from 60 to 70% confluence to 80 to 90% confluence. In addition, the transfected cells remained attached to the wells of the tissue culture plate, further indicating that cell viability was maintained.
Intact actin microfilaments are necessary for maximal binding of the N-cadherin–SEPT7 complex to C. albicans.
The endocytosis of C. albicans requires intact endothelial cell microfilaments (17, 18). We used the actin assembly inhibitor, cytochalasin D, to investigate whether microfilaments were required for the N-cadherin–SEPT7 complex to bind to C. albicans. Consistent with our previous results (18), cytochalasin D caused a 78% ± 17% reduction in the endocytosis of C. albicans compared to that of control endothelial cells incubated with diluent alone (P < 0.0001). Of note, the concentration of cytochalasin D used in these experiments had no effect on C. albicans hyphal formation. By indirect immunofluorescence, we determined that treatment with cytochalasin D reduced the accumulation of N-cadherin and SEPT7 around C. albicans hyphae by approximately 50% (Fig. 7A and B). In addition, cytochalasin D substantially decreased the binding of both N-cadherin and SEPT7 to C. albicans in the affinity purification assay (Fig. 7C and D). However, cytochalasin D had a significantly greater inhibitory effect on N-cadherin binding (64% reduction) than on SEPT7 binding (36% reduction). Importantly, treatment with cytochalasin D did not significantly reduce the amount of N-cadherin that was expressed on the endothelial cell surface (see Fig. S3 in the supplemental material). Next, we investigated the effects of cytochalasin D on the association between SEPT7 and N-cadherin in coimmunoprecipitation experiments. Treatment with cytochalasin D resulted in a 46% reduction in the amount of SEPT7 that was immunoprecipitated along with N-cadherin in endothelial cells that had been infected with C. albicans (Fig. 7E and F). Collectively, these results demonstrate that intact endothelial cell microfilaments enhance the association of SEPT7 with N-cadherin and facilitate the accumulation of these proteins around C. albicans.
FIG 7 .
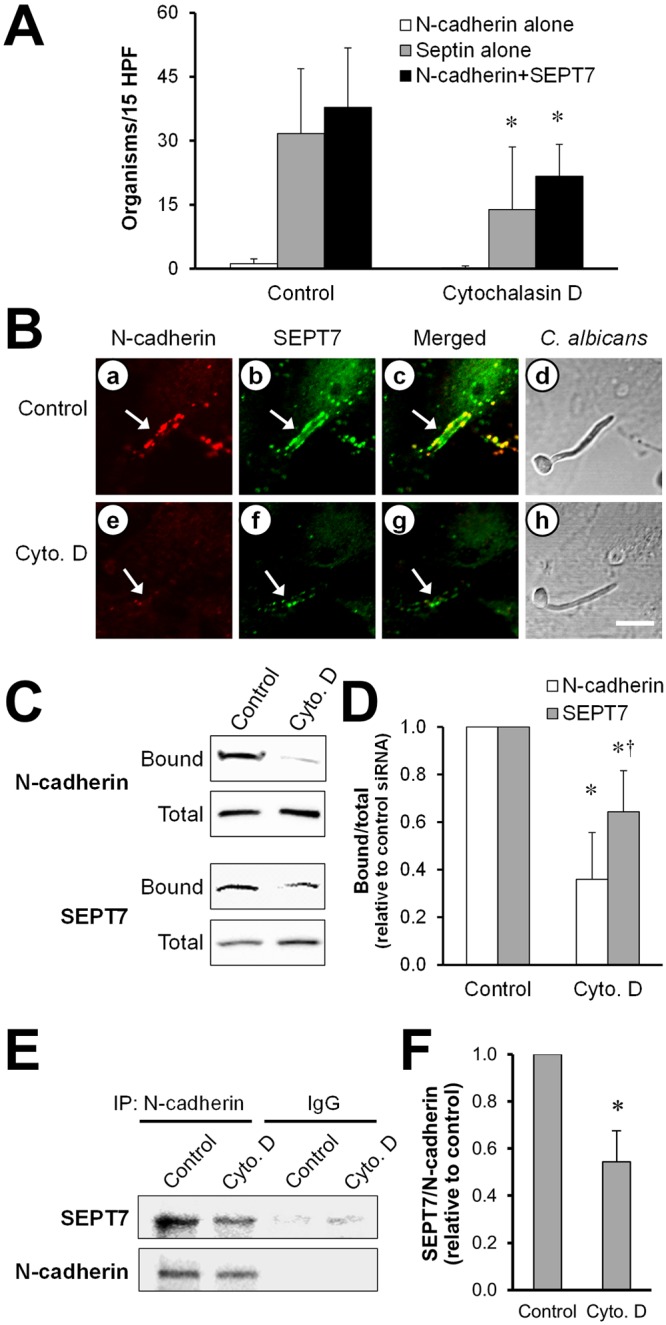
Cytochalasin D inhibits the interaction of N-cadherin and SEPT7 with C. albicans. Endothelial cells were incubated with diluent alone (control) or cytochalasin D (cyto. D) and then infected with C. albicans for 90 min. (A, B) The cells were fixed and then stained for N-cadherin and SEPT7. (A) The number of organisms in 15 high-power fields that were surrounded by N-cadherin alone, SEPT7 alone, or N-cadherin plus SEPT7. Results are the means ± SD from five experiments, each performed in triplicate. (B) Confocal microscopic images of N-cadherin (a, e) and SEPT7 (b, f) accumulating around the same organism. The merged images are shown in panels c and g, and images of the corresponding microscopic fields viewed by differential interference contrast are shown in panels d and h. Arrows indicate the N-cadherin and SEPT7 that accumulated around the hyphae. Scale bar = 5 µm. (C) Representative immunoblot showing the effects of cytochalasin D on the amount of endothelial cell N-cadherin and SEPT7 that bound to C. albicans hyphae. (D) Densitometric analysis of 5 independent N-cadherin and SEPT7 immunoblots. Results are means ± SD. *, P < 0.05 compared to control cells; †, P < 0.05 compared to N-cadherin. (E, F) Coimmunoprecipitation results showing the effects of cytochalasin D on the association between SEPT7 and N-cadherin. (E) Representative immunoblot of cell lysates that were immunoprecipitated with either an anti-N-cadherin antibody or control IgG, separated by SDS-PAGE and then probed for the presence of SEPT7 by immunoblotting. (F) Densitometric analysis of 3 independent immunoblots. Results are means ± SD. *, P = 0.03 compared to control cells that had been infected with C. albicans in the absence of cytochalasin D.
SEPT7 is necessary for maximal endocytosis of C. albicans.
Finally, we analyzed the effects of SEPT7 knockdown on the endocytosis of C. albicans. Endothelial cells transfected with SEPT7 siRNA endocytosed approximately 50% fewer C. albicans cells than endothelial cells transfected with control siRNA (Fig. 8). Thus, SEPT7 expression is necessary for the maximal endocytosis of C. albicans. Taken together, these data support a model in which C. albicans Als3 and Ssa1 bind to a complex containing N-cadherin and SEPT7, which in turn interacts with endothelial cell microfilaments, thereby inducing the endocytosis of the organism.
FIG 8 .
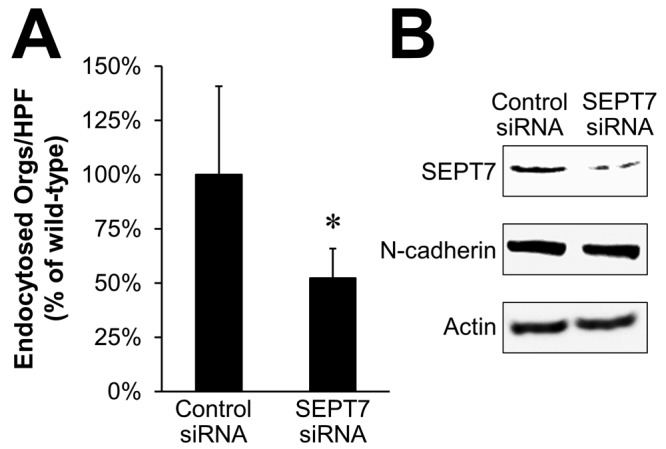
SEPT7 knockdown inhibits endocytosis of C. albicans. (A) Endothelial cells transfected with either control or N-cadherin siRNA were infected with C. albicans for 90 min, after which the number of endocytosed organisms was determined by a differential fluorescence assay. Results are the means ± SD from three experiments, each performed in triplicate. *, P < 0.001 compared to endothelial cells transfected with the control siRNA. (B) Verification by immunoblotting of SEPT7 knockdown, which did not affect total N-cadherin content.
DISCUSSION
To develop new approaches to prevent C. albicans invasion from the blood vessels into the surrounding tissue, we must first understand the mechanism by which this fungus subverts host cell signaling mechanisms and induces its own endocytosis. In this study, we established a link between SEPT7 and N-cadherin-mediated endocytosis of C. albicans. Previous studies have described the key role of SEPT2 during host cell uptake of bacteria such as L. monocytogenes and Shigella flexneri (14, 15). Our study adds new data to existing knowledge of septin function in multiple regards. First, C. albicans invasion of endothelial cells is mediated in part by N-cadherin, which is different from the host cell receptors that L. monocytogenes utilizes to invade epithelial cells (4, 22). Second, because filamentous C. albicans (roughly 30 µm in length) is approximately 60 times larger than its bacterial counterparts (0.5 to 2 µm in length), the mechanism of host cell uptake of this organism is likely to be different. Third, previous studies of the role of septins in endocytosis by mammalian cells have focused on SEPT2, SEPT9, and SEPT11 (15, 23–25) and have not investigated SEPT7. Finally, the role of septins in endothelial cell endocytosis has not been studied previously. Thus, our study offers a novel perspective on septin function during the endocytosis of microbial pathogens by endothelial cells.
N-cadherin functions as a receptor for C. albicans Als3 and Ssa1; binding of these proteins to N-cadherin activates the signaling cascade that results in rearrangement of actin microfilaments, leading to the endocytosis of C. albicans (5, 6). We found that knockdown of N-cadherin reduced the amount of SEPT7 pulled down by C. albicans, while knockdown of SEPT7 decreased binding of N-cadherin to the organism. Furthermore, the coimmunoprecipitation experiments demonstrated that the association of SEPT7 with N-cadherin is increased in response to C. albicans infection. Collectively, these results indicate that N-cadherin and SEPT7 are part of a complex that binds to C. albicans. This link between SEPT7 and a vital C. albicans cell receptor explains why septins are essential for the maximal host cell endocytosis of this fungal pathogen.
The results from these experiments also indicate that additional endothelial cell receptors and signaling pathways likely mediate the endocytosis of C. albicans. For example, in the indirect immunofluorescence experiments, approximately 43% of the organisms were surrounded by SEPT7 but not N-cadherin. In addition, siRNA knockdown of N-cadherin reduced SEPT7 accumulation and binding by approximately 50%. Furthermore, we found previously that knockdown of N-cadherin decreases the endocytosis of C. albicans by approximately 40% (4). Taken together, these results strongly suggest that at least one receptor other than N-cadherin also interacts with SEPT7 and mediates the endocytosis of C. albicans. Experiments to identify additional endothelial cell receptors for C. albicans are currently in progress.
It was notable that inhibiting actin function with cytochalasin D reduced the association of N-cadherin and SEPT7 and decreased binding of the N-cadherin–SEPT7 complex to C. albicans. Septins are known to interact closely with the actin cytoskeleton (11). Also, Hagiwara et al. (21) found that treatment of a neuroblastoma cell line with cytochalasin D retarded septin turnover. These results demonstrate that the actin cytoskeleton influences septin function, consistent with our findings. Hagiwara et al. (21) further discovered that knockdown of SEPT7 increased the turnover of the glutamate aspartate transporter membrane protein. These findings, combined with our data, support the model that SEPT7 interacts with the actin cytoskeleton and acts as a scaffold that prevents the lateral diffusion of N-cadherin and thereby facilitates its interaction with C. albicans.
There are thirteen different human septins (SEPT1 to SEPT14, but SEPT13 is a pseudogene), and different septins function together, forming hetero-oligomers. Although SEPT2, SEPT9, and SEPT11 are known to accumulate around L. monocytogenes (15, 24, 26), these septins have different functions. For example, SEPT2 is required for the maximal uptake of L. monocytogenes, whereas SEPT11 actually restricts the uptake of this bacterium (15, 24). In addition, SEPT2 is required for the Met receptor tyrosine kinase to bind tightly to the L. monocytogenes InlB invasin (14). Our data suggest that SEPT7 similarly controls the binding of N-cadherin to C. albicans Als3 and Ssa1. Although SEPT7 plays a key role in N-cadherin-mediated endocytosis of C. albicans, it is probable that other septins are also involved in this process and form part of the N-cadherin–SEPT7 complex.
From these results, we propose a refined molecular model of C. albicans endocytosis. N-cadherin and SEPT7 form part of a complex, which in turn is linked to the endothelial cell cytoskeleton, either directly or indirectly. This multicomponent complex anchors N-cadherin to specific regions on the endothelial cell surface. When C. albicans Als3 or Ssa1 binds to N-cadherin, there is recruitment of additional N-cadherin molecules, a process that requires SEPT7. In addition, the formation of the N-cadherin–SEPT7 complex results in multivalent binding of N-cadherin to C. albicans, which stabilizes the binding and enhances its affinity. The progressive recruitment of N-cadherin and SEPT7 around the organism induces the accumulation of the actin cytoskeleton, which structurally supports the extending pseudopods. Once the pseudopods have formed, N-cadherin and SEPT7 are no longer recruited. Thus, without the presence of septins, N-cadherin molecules would diffuse away along the plasma membrane, unable to remain bound to C. albicans. This model explains why SEPT7 is necessary for N-cadherin to bind to the fungal hyphae but knockdown of the adaptor protein does not affect the total amount of the cell receptor. In SEPT7-depleted cells, N-cadherin is still present on the cell surface, but this receptor cannot bind in sufficient numbers to C. albicans to initiate pseudopod formation and subsequent endocytosis.
We have found that C. albicans interacts with receptors other than N-cadherin when it invades other host cells. For example, it binds to E-cadherin, HER2, and the epidermal growth factor receptor on oral epithelial cells and to gp96 on brain microvascular endothelial cells (5, 19, 27). Identifying the septins that are required for the proper localization and function of these receptors will be the topic of additional investigations.
MATERIALS AND METHODS
Strains and culture conditions.
The strains of C. albicans and Saccharomyces cerevisiae used in the experiments are listed in Table 1. Yeast-phase C. albicans cells were cultured in liquid YPD medium (1% yeast extract, 2% Bacto-peptone, and 2% d-glucose) in a shaking incubator at 30°C overnight. S. cerevisiae strains expressing C. albicans ALS3 or SSA1 were grown in SC minimal medium containing either glucose (for the ALS3 strains) or galactose (for the SSA1 strains). The next day, the cells were washed twice in Dulbecco’s phosphate-buffered saline (PBS) without calcium or magnesium, sonicated, and counted with a hemacytometer.
TABLE 1 .
Fungal strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| C. albicans | ||
| SC5314 | Wild type | 32 |
| DIC185 | ura3Δ::λimm434::URA3-IRO1/ura3Δ::λimm434 arg4::hisG::ARG4/arg4::hisG his1::hisG::HIS1/his1::hisG | This work |
| CAI-4–CIp10 | ura3Δ::λimm434/ura3Δ::λimm434 rps10::CIp10 (URA3)/RPS10 | 33 |
| CAYF178U | als3::ARG4/als3::HIS1 ura3Δ::λimm434::URA3-IRO1/ura3Δ::λimm434 arg4::hisG/arg4::hisG his1::hisG/his1::hisG | 34 |
| CAQTP178U | als3::ARG4::ALS3/als3::HIS1 ura3Δ::λimm434::URA3-IRO1/ura3Δ::λimm434 arg4::hisG/arg4::hisG his1::hisG/his1::hisG | 34 |
| ssa1Δ/Δ-URA3 | ura3Δ::λimm434/ura3Δ::λimm434 ssa1Δ::FRT/ssa1Δ::FRT SSA2/ssa2::FRT rps10::URA3/RPS10 | 6 |
| ssa1Δ/Δ-SSA1 | ura3Δ::λimm434/ura3Δ::λimm434 ssa1Δ::FRT/ssa1Δ::FRT SSA2/ssa2::FRT rps10::URA3::SSA1/RPS10 | 35 |
| S. cerevisiae | ||
| pADH1 | leu2 his3 trp1 ura3+pADH1 | 20 |
| pALS3 | leu2 his3 trp1 ura3+pALS3 | 20 |
| pYES2.1 | leu2 his3 trp1 ura3+pGAL1 | 19 |
| pSSA1 | leu2 his3 trip1 ura3+pSSA1 | 19 |
C. albicans strain DIC185 was constructed by restoring the URA3-IRO1, HIS1, and ARG4 loci of strain BWP17 (28). This auxotrophic strain was first transformed with a PstI/NotI-digested fragment of pBSK-URA3 (29) to restore the URA3-IRO1 locus. Next, the resulting Ura+ strain was transformed with a KpnI/SacI-digested fragment of pRS-ARG4ΔSpeI (28) to restore the ARG4 locus. Finally, the resulting Ura+ Arg+ strain was transformed with a SalI/SphI-digested fragment of pGEM-HIS1 (28) to restore the HIS1 locus and produce strain DIC185. While constructing these strains, the correct genomic integration of the URA3-IRO1 fragment was verified by whole-cell PCR using the primers 5′ TGCTGGTTGGAATGCTTATTTG 3′ and 5′ TGCAAATTCTGCTACTGGAGTT 3′. The proper integration of ARG4 was confirmed using the primers 5′ GGAATTGATCAATTATCTTTTGAAC 3′ and 5′ TTATCACACATTGGAGTTGTTG 3′, and the correct integration of HIS1 was verified with the primers 5′ CAATATGGTTTCTATGGCC 3′ and 5′ GGTTTGATACCATGGCAAG 3′.
In experiments using the C. albicans als3Δ/Δ mutant, strain DIC185 was used as the wild-type control. In experiments using the C. albicans ssa1Δ/Δ mutant, strain CAI-4-CIp10 was used as the wild-type control.
Endothelial cells.
Endothelial cells were isolated from human umbilical cords and grown at 37°C in a T25 culture flask in M199 medium containing 10% bovine calf serum and 10% fetal bovine serum plus l-glutamine, penicillin, and streptomycin as described previously (30, 31). These cells were grown to confluence in either 6- or 24-well tissue culture plates.
Indirect immunofluorescence.
To analyze the accumulation of SEPT7, actin, and N-cadherin around C. albicans cells, endothelial cells on fibronectin-coated glass coverslips in a 24-well plate were infected with 2 × 105 C. albicans yeast cells in RPMI 1640 medium in 5% CO2 at 37°C for selected times. Cells were then rinsed with Hank’s balanced salt solution (HBSS), fixed with 3% paraformaldehyde in PBS, and then blocked and permeabilized with 0.05% Triton X-100 in 5% goat serum for 30 min. SEPT7 was labeled with rabbit anti-SEPT7 polyclonal antibody (catalog no. 13018-1-AP; Protein Tech Group, Inc.). Actin was detected with Alexa Fluor 568-labeled phalloidin (Invitrogen), and N-cadherin was labeled with a mouse anti-N-cadherin monoclonal antibody (catalog no. C70320; BD Transduction Laboratories). Next, the cells were incubated with the appropriate secondary antibodies conjugated with either green fluorescing Alexa Fluor 488 or red fluorescing Alexa Fluor 568 (Invitrogen), mounted inverted on glass slides, and observed via confocal microscopy. Multiple z-sections of each sample were taken to ensure maximal resolution. The C. albicans cells in each region of interest were imaged by differential interference contrast.
In some experiments, the endothelial cells were incubated with 0.4 µM cytochalasin D (Sigma-Aldrich) in 0.004% dimethyl sulfoxide (DMSO) for 45 min prior to the addition of C. albicans (18). The cytochalasin D and DMSO remained in the medium for the duration of infection.
To determine the number of C. albicans cells that had recruited N-cadherin, SEPT7, and/or actin, the endothelial cells were infected with 3.5 × 105 organisms and then fixed and stained as described above. Next, at least 15 high-powered fields were examined by epifluorescent microscopy, and the fungal cells were scored for the presence of the various endothelial cell proteins. Each experiment was performed in triplicate on at least three separate occasions.
Affinity purification and immunoblotting of endothelial cell proteins.
Endothelial cell protein complexes that were bound by C. albicans were isolated using an affinity purification approach. Endothelial cells in a 6-well plate were infected with 5.25 × 106 yeast-phase C. albicans cells in RPMI 1640. At selected time points, the infected endothelial cells were removed from the wells with a cell scraper and collected by centrifugation at 1,000 × g at 4°C. After the supernatant was aspirated, 1.5% octyl-glucopyranoside in PBS with calcium and magnesium (PBS++) and protease inhibitors was added to the pellet to lyse the endothelial cells but not the C. albicans cells. Following a 20-min incubation on ice, this mixture was centrifuged at 4°C to pellet the intact C. albicans cells and the endothelial cell membrane protein complexes that were bound to these organisms. The proteins that were not bound by C. albicans remained in the supernatant, an aliquot of which was collected. The pellet, consisting of C. albicans cells and the bound endothelial cell protein complexes, was washed three times with ice-cold 1.5% octyl-glucopyranoside in PBS++ and protease inhibitors. Next, the endothelial cell protein complexes that had bound to C. albicans were eluted by incubating the pellet with 6 M urea on ice for 10 min. The eluted proteins were separated from the organisms by centrifugation, after which the resultant supernatant was collected. The proteins in the various samples were separated by SDS-PAGE, and SEPT7 and N-cadherin were detected by immunoblotting with the same primary antibodies that were used in the immunofluorescence experiments.
Coimmunoprecipitation of SEPT7 and N-cadherin.
Coimmunoprecipitation experiments were performed to determine if SEPT7 and N-cadherin form part of a complex. Endothelial cells in 150-mm-diameter tissue culture dishes were incubated with medium alone or infected for 90 min with 4 × 107 C. albicans cells. Next, they were rinsed with ice-cold PBS++ plus protease inhibitors and then scraped from the dishes with a cell scraper. The endothelial cells were pelleted by centrifugation and lysed by incubation in 1.5% octyl-glucopyranoside in PBS++ with protease inhibitors on ice for 30 min. After the cell debris and the intact fungi were removed by centrifugation, the resulting supernatant was precleared by incubating it with protein A/G+ agarose beads (Santa Cruz Biotechnology) on ice for 30 min. Next, the lysate was incubated with either an anti-N-cadherin antibody or control mouse IgG at 4°C overnight followed by the protein A/G+ agarose beads for 1 h. The beads were rinsed twice in 1.5% octyl-glucopyranoside and protease inhibitors, after which they were boiled in sample buffer. After the endothelial cell proteins were separated by SDS-PAGE, SEPT7 and N-cadherin were detected by immunoblotting as described above.
Endocytosis assay.
The number of C. albicans cells that were internalized by the endothelial cells was determined by a differential fluorescence assay as previously described (4, 5). Briefly, confluent endothelial cells on coverslips in a 24-well plate were infected with C. albicans as in the indirect immunofluorescence experiments. After a 90-min incubation, nonadherent organisms were removed by gently rinsing the wells twice with HBSS. The adherent but noninternalized C. albicans cells were stained with a polyclonal rabbit anti-Candida antibody (Biodesign International) that had been conjugated with red fluorescing Alexa Fluor 568 (Invitrogen). The endothelial cells were then permeabilized with 0.05% Triton X-100 in 5% goat serum, after which the cell-associated organisms (adherent plus internalized organisms) were then stained with an anti-Candida antibody that had been conjugated with green fluorescing Alexa Fluor 488 (Invitrogen). When viewed with an epifluorescent microscope, the adherent but noninternalized organisms fluoresced both red and green, while the internalized organisms fluoresced green but not red. At least 100 organisms per coverslip were evaluated, and the experiment was performed in triplicate on three separate occasions.
siRNA.
To knock down the expression of SEPT7 or N-cadherin, endothelial cells were grown to 60 to 70% confluence in 6-well tissue culture plates in M199 medium without antibiotics. These cells were then transfected with SEPT7 siRNA (Santa Cruz Biotech), N-cadherin siRNA (Santa Cruz Biotech), or control siRNA (Qiagen) using Lipofectamine 2000 (Invitrogen) by following the manufacturer’s instructions. In some experiments, the transfected cells were kept in the 6-well plates. In other experiments, the transfected cells were transferred to 24-well plates containing glass coverslips for confocal imaging or to analyze their internalization of C. albicans cells as described above.
Flow cytometry.
Endothelial cells were grown in a 6-well tissue culture plate, rinsed with ice-cold PBS++, and then removed from the plate with a cell scraper. After the cells were collected by centrifugation, they were blocked by incubation on ice with PBS++ containing 5% goat serum. The cells were labeled with an anti-N-cadherin monoclonal antibody (ACAM; Sigma-Aldrich) and then fixed in 3% paraformaldehyde. Following extensive rinsing with PBS++ containing 1% bovine serum albumin (BSA), the cells were incubated with Alexa Fluor 488-labeled goat anti-mouse secondary antibody and rinsed in PBS++ plus 1% BSA. Finally, 10,000 cells were analyzed by flow cytometry for N-cadherin expression. Control cells were processed similarly except that the primary antibody was omitted.
Statistical analysis.
Differences among the experimental groups were assessed using analysis of variance. P values of ≤0.05 were considered significant.
SUPPLEMENTAL MATERIAL
Expression of N-cadherin and SEPT7 in uninfected endothelial cells. Confocal microscopic images of uninfected endothelial cells that were stained for N-cadherin (A) and SEPT7 (B). The merged image is shown in panel C. Scale bar = 10 µm. Download
Knockdown of SEPT7 does not influence N-cadherin content. (A) Flow cytometric analysis of surface expressed N-cadherin in endothelial cells that had been transfected with either control or SEPT7 siRNA. (B) Immunoblots of lysates of endothelial cells that had been transfected with either control or SEPT7 siRNA. Download
Cytochalasin D does not influence N-cadherin content. Flow cytometric analysis of surface-expressed N-cadherin in endothelial cells that had been incubated with either cytochalasin D or diluent (control). Download
ACKNOWLEDGMENTS
We thank David Villareal for assistance with tissue culture.
The endothelial cells used in these studies were isolated from human umbilical cords, which were collected by the pediatric, perinatal, and mobile unit of the UCLA Clinical and Translational Science Institute at LA Biomed/Harbor-UCLA Medical Center (UL1TR000124).
This work was supported in part by grant R01AI054928 from the National Institutes of Health, United States.
S.G.F. is a cofounder and shareholder of Novadigm Therapeutics, Inc. None of the other authors has any financial conflicts.
Footnotes
Citation Phan QT, Eng DK, Mostowy S, Park H, Cossart P, Filler SG. 2013. Role of endothelial cell septin 7 in the endocytosis of Candida albicans. mBio 4(6):e00542-13. doi:10.1128/mBio.00542-13.
REFERENCES
- 1. Horn DL, Neofytos D, Anaissie EJ, Fishman JA, Steinbach WJ, Olyaei AJ, Marr KA, Pfaller MA, Chang CH, Webster KM. 2009. Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin. Infect. Dis. 48:1695–1703 [DOI] [PubMed] [Google Scholar]
- 2. Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39:309–317 [DOI] [PubMed] [Google Scholar]
- 3. Grubb SE, Murdoch C, Sudbery PE, Saville SP, Lopez-Ribot JL, Thornhill MH. 2008. Candida albicans-endothelial cell interactions: a key step in the pathogenesis of systemic candidiasis. Infect. Immun. 76:4370–4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Phan QT, Fratti RA, Prasadarao NV, Edwards JE, Jr, Filler SG. 2005. N-cadherin mediates endocytosis of Candida albicans by endothelial cells. J. Biol. Chem. 280:10455–10461 [DOI] [PubMed] [Google Scholar]
- 5. Phan QT, Myers CL, Fu Y, Sheppard DC, Yeaman MR, Welch WH, Ibrahim AS, Edwards JE, Filler SG. 2007. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 5:e64. 10.1371/journal.pbio.0050064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun JN, Solis NV, Phan QT, Bajwa JS, Kashleva H, Thompson A, Liu Y, Dongari-Bagtzoglou A, Edgerton M, Filler SG. 2010. Host cell invasion and virulence mediated by Candida albicans Ssa1. PLoS Pathog. 6:e1001181. 10.1371/journal.ppat.1001181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Y, Filler SG. 2011. Candida albicans Als3, a multifunctional adhesin and invasin. Eukaryot. Cell 10:168–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moreno-Ruiz E, Galán-Díez M, Zhu W, Fernández-Ruiz E, d’Enfert C, Filler SG, Cossart P, Veiga E. 2009. Candida albicans internalization by host cells is mediated by a clathrin-dependent mechanism. Cell. Microbiol. 11:1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fratti RA, Ghannoum MA, Edwards JE, Jr, Filler SG. 1996. Gamma interferon protects endothelial cells from damage by Candida albicans by inhibiting endothelial cell phagocytosis. Infect. Immun. 64:4714–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hall PA, Russell SE. 2012. Mammalian septins: dynamic heteromers with roles in cellular morphogenesis and compartmentalization. J. Pathol. 226:287–299 [DOI] [PubMed] [Google Scholar]
- 11. Mostowy S, Cossart P. 2012. Septins: the fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 13:183–194 [DOI] [PubMed] [Google Scholar]
- 12. Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ. 2010. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 329:436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma S, Quintana A, Findlay GM, Mettlen M, Baust B, Jain M, Nilsson R, Rao A, Hogan PG. 2013. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca entry. Nature 499:238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mostowy S, Janel S, Forestier C, Roduit C, Kasas S, Pizarro-Cerdá J, Cossart P, Lafont F. 2011. A role for septins in the interaction between the Listeria monocytogenes invasion protein InlB and the Met receptor. Biophys. J. 100:1949–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mostowy S, Nam Tham T, Danckaert A, Guadagnini S, Boisson-Dupuis S, Pizarro-Cerdá J, Cossart P. 2009. Septins regulate bacterial entry into host cells. PLoS One 4:e4196. 10.1371/journal.pone.0004196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Serrão VH, Alessandro F, Caldas VE, Marçal RL, Pereira HD, Thiemann OH, Garratt RC. 2011. Promiscuous interactions of human septins: the GTP binding domain of SEPT7 forms filaments within the crystal. FEBS Lett. 585:3868–3873 [DOI] [PubMed] [Google Scholar]
- 17. Rotrosen D, Edwards JE, Jr, Gibson TR, Moore JC, Cohen AH, Green I. 1985. Adherence of Candida to cultured vascular endothelial cells: mechanisms of attachment and endothelial cell penetration. J. Infect. Dis. 152:1264–1274 [DOI] [PubMed] [Google Scholar]
- 18. Filler SG, Swerdloff JN, Hobbs C, Luckett PM. 1995. Penetration and damage of endothelial cells by Candida albicans. Infect. Immun. 63:976–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y, Mittal R, Solis NV, Prasadarao NV, Filler SG. 2011. Mechanisms of Candida albicans trafficking to the brain. PLoS Pathog. 7:e1002305. 10.1371/journal.ppat.1002305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheppard DC, Yeaman MR, Welch WH, Phan QT, Fu Y, Ibrahim AS, Filler SG, Zhang M, Waring AJ, Edwards JE., Jr. 2004. Functional and structural diversity in the Als protein family of Candida albicans. J. Biol. Chem. 279:30840–30849 [DOI] [PubMed] [Google Scholar]
- 21. Hagiwara A, Tanaka Y, Hikawa R, Morone N, Kusumi A, Kimura H, Kinoshita M. 2011. Submembranous septins as relatively stable components of actin-based membrane skeleton. Cytoskeleton 68:512–525 [DOI] [PubMed] [Google Scholar]
- 22. Veiga E, Cossart P. 2006. The role of clathrin-dependent endocytosis in bacterial internalization. Trends Cell Biol. 16:499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mostowy S, Cossart P. 2011. Septins as key regulators of actin based processes in bacterial infection. Biol. Chem. 392:831–835 [DOI] [PubMed] [Google Scholar]
- 24. Mostowy S, Danckaert A, Tham TN, Machu C, Guadagnini S, Pizarro-Cerdá J, Cossart P. 2009. Septin 11 restricts InlB-mediated invasion by Listeria. J. Biol. Chem. 284:11613–11621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang YW, Yan M, Collins RF, Diciccio JE, Grinstein S, Trimble WS. 2008. Mammalian septins are required for phagosome formation. Mol. Biol. Cell 19:1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mostowy S, Bonazzi M, Hamon MA, Tham TN, Mallet A, Lelek M, Gouin E, Demangel C, Brosch R, Zimmer C, Sartori A, Kinoshita M, Lecuit M, Cossart P. 2010. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8:433–444 [DOI] [PubMed] [Google Scholar]
- 27. Zhu W, Phan QT, Boontheung P, Solis NV, Loo JA, Filler SG. 2012. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc. Natl. Acad. Sci. U. S. A. 109:14194–14199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson RB, Davis D, Mitchell AP. 1999. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J. Bacteriol. 181:1868–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park H, Myers CL, Sheppard DC, Phan QT, Sanchez AA, Edwards JE, Jr, Filler SG. 2005. Role of the fungal Ras-protein kinase A pathway in governing epithelial cell interactions during oropharyngeal candidiasis. Cell. Microbiol. 7:499–510 [DOI] [PubMed] [Google Scholar]
- 30. Jaffe EA, Nachman RL, Becker CG, Minick CR. 1973. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Invest. 52:2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Phan QT, Belanger PH, Filler SG. 2000. Role of hyphal formation in interactions of Candida albicans with endothelial cells. Infect. Immun. 68:3485–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fonzi WA, Irwin MY. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murad AM, Lee PR, Broadbent ID, Barelle CJ, Brown AJ. 2000. CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast 16:325–327 [DOI] [PubMed] [Google Scholar]
- 34. Nobile CJ, Andes DR, Nett JE, Smith FJ, Yue F, Phan QT, Edwards JE, Filler SG, Mitchell AP. 2006. Critical role of Bcr1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo. PLoS Pathog. 2:e63. 10.1371/journal.ppat.0020063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li XS, Sun JN, Okamoto-Shibayama K, Edgerton M. 2006. Candida albicans cell wall Ssa proteins bind and facilitate import of salivary histatin 5 required for toxicity. J. Biol. Chem. 281:22453–22463 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of N-cadherin and SEPT7 in uninfected endothelial cells. Confocal microscopic images of uninfected endothelial cells that were stained for N-cadherin (A) and SEPT7 (B). The merged image is shown in panel C. Scale bar = 10 µm. Download
Knockdown of SEPT7 does not influence N-cadherin content. (A) Flow cytometric analysis of surface expressed N-cadherin in endothelial cells that had been transfected with either control or SEPT7 siRNA. (B) Immunoblots of lysates of endothelial cells that had been transfected with either control or SEPT7 siRNA. Download
Cytochalasin D does not influence N-cadherin content. Flow cytometric analysis of surface-expressed N-cadherin in endothelial cells that had been incubated with either cytochalasin D or diluent (control). Download