

Abstract
The identification of non-canonical (caspase-1 independent) pathways for IL-1β production has unveiled an intricate interplay between inflammatory and death-inducing signaling platforms. We found a heretofore unappreciated role for caspase-8 as a major pathway for IL-1β processing and release in murine bone marrow-derived dendritic cells (BMDC) co-stimulated with TLR4 agonists and pro-apoptotic chemotherapeutic agents such as doxorubicin (Dox) or staurosporine (STS). The ability of Dox to stimulate release of mature (17 kDa) IL-1β was nearly equivalent in wild-type (WT) BMDC, Casp1−/− Casp11−/− BMDC, WT BMDC treated with the caspase-1 inhibitor YVAD, and BMDC lacking the inflammasome regulators ASC, NLRP3, or NLRC4. Notably, Dox-induced production of mature IL-1β was temporally correlated with caspase-8 activation in WT cells and greatly suppressed in Casp8−/−Rip3−/− or Trif−/− BMDC, as well as in WT BMDC treated with the caspase-8 inhibitor, IETD. Similarly, STS stimulated robust IL-1β processing and release in Casp1−/−Casp11−/− BMDC that was IETD-sensitive. These data suggest that TLR4/TRIF activation induce assembly of caspase-8-based signaling complexes that become licensed as IL-1β converting enzymes in response to Dox and STS. The responses were temporally correlated with downregulation of cIAP1 suggesting suppressive roles for this and likely other Inhibitor of Apoptosis Proteins (IAPs) on the stability and/or proteolytic activity of the caspase-8 platforms. Thus, pro-apoptotic chemotherapeutic agents stimulate the caspase-8-mediated processing and release of IL-1β, implicating direct effects of such drugs on a non-canonical inflammatory cascade that may modulate immune responses in tumor microenvironments.
Introduction
Innate and adaptive immune responses play important roles in cancer progression and therapy (1, 2). Interleukin-1β is a pleiotropic proinflammatory cytokine that is predominantly expressed in myeloid leukocytes. Depending on context, IL-1β can contribute to beneficial anti-tumor immune responses or maladaptive responses such as neovascularization leading to tumor survival. Much attention has been focused on the mechanisms of IL-1β production within particular tissue niches including tumor microenvironments. Activation of TLRs or receptors for proinflammatory cytokines (including IL-1β per se) induces the NF-κB-dependent expression of proIL-1β (33kD) as a cytosolic, biologically inactive precursor protein. The canonical cleavage and processing of proIL-1β into mature IL-1β cytokine (17kD) is catalyzed by caspase-1, a pathway regulated by multiprotein inflammasome signaling complexes. Prior to its subclassification within the caspase family of proteases, caspase-1 was originally described as the interleukin-converting enzyme or ICE. The most intensively studied inflammasome comprises an oligomeric complex of procaspase-1 with the NLRP3 and ASC adapter proteins that rapidly assembles in response to diverse stress stimuli such as increased reactive oxygen species (3), mitochondrial dysfunction (4), perturbation of intracellular ion homeostasis (5-7), disruption of lysosomal membrane integrity (8), and activation of deubiquitinases (9-11).
On the one hand, NLRP3/caspase-1 inflammasomes play a central role in the regulation of IL-1β production within tumor loci or in response to chemotherapeutic drugs. In tumor-bearing mice treated with oxaliplatin, NLRP3-dependent IL-1β release from dendritic cells (DCs) occurs via paracrine activation of P2X7 receptors (P2X7R) in response to ATP released from dying tumor cells (5). In this model, DC-derived IL-1β contributes to the tumor-specific immune response by polarizing tumor antigen-reactive CD8+ T cells into IFN-γ-producing cytotoxic T cells (12). Two other drugs, gemcitabine and 5-fluorouracil, have been shown to elicit NLRP3-dependent IL-1β production in response to the accumulation of cytosolic cathepsin B in myeloid-derived suppressor cells (MSDC) with consequent promotion of tumor growth and vascularization (13). Other reports have described the ability of pro-apoptotic chemotherapeutic agents, such as doxorubicin and staurosporine analogs, to directly activate NLRP3- and caspase-1-dependent IL-1β processing in ex vivo LPS-primed macrophages via mechanisms correlated with ribotoxic stress or increased mitochondrial dysfunction (4, 14).
On the other hand, recent studies have indicated that caspase-8 can substitute for caspase-1 and can function as an efficient “ICE” for proteolytic maturation of IL-1β. Dectin-1 engagement by fungal ligands can engage a non-canonical ASC/caspase-8 inflammasome for processing of IL-1β (15). In addition, Maelfait et al. observed that activation of the TRIF (TIR domain-containing adapter-inducing interferon-β) signaling pathway by TLR3 or TLR4 induced a caspase-8 signaling pathway that, when combined with cyclohexamide-mediated inhibition of protein translation, was sufficient to drive efficient IL-1β processing even in caspase-1 knockout macrophages (16). A subsequent study found that treatment of LPS-primed macrophages or DCs with Smac (Second mitochondrial-derived activator of caspases)-mimetic drugs triggered a robust maturation of IL-1β that was mediated in part by caspase-8 (17). Most recently, the engagement of Fas (CD95) signaling in WT or caspase-1 knockout macrophages was shown to induce IL-1β and IL-18 processing via caspase-8 (18). A Fas-induced caspase-8 cascade was also identified as a major pathway for IL-1β and IL-18 production in Listeria-infected peritoneal exudate cells isolated from B6 mice (19). Caspase-8 is best characterized for its multiple roles in the regulation of cell death via apoptosis or necroptosis (20). These reports of non-canonical IL-1β processing via caspase-8 indicate that proinflammatory cytokine production may be mediated by caspase-8, potentially in signaling complexes such as ripoptosomes, known to control apoptotic or necroptotic death cascades.
In this study, we assessed the relative contributions of caspase-1 and caspase-8 to the processing and release of IL-1β in LPS-primed dendritic cells stimulated with the chemotherapeutic drug, doxorubicin, or the pro-apoptotic agent, staurosporine. Our observations support a model wherein TLR4 activation induces upregulation of proIL-1β together with the TRIF-dependent assembly of a latent caspase-8 signaling platform. Pro-apoptotic agents, such as Dox or STS, act to license such caspase-8 signaling complexes for maximal caspase-8-mediated cleavage of proIL-1β, in part by depleting cIAPs (cellular Inhibitor of Apoptosis Proteins). Comparative analyses of DCs isolated from control mice versus Casp1/11−/− or Casp8−/− Rip3−/ − mice indicated that caspase-8 acts as the predominant IL-1β converting enzyme in response to engagement of TLR4 signaling when the intrinsic apoptotic pathway is triggered by chemotherapeutic agents.
Materials and Methods
Reagents
Key reagents and their sources were: Escherichia coli LPS serotype O1101:B4 (List Biological Laboratories), Pam3CSK4 (Invivogen), Ac-YVAD-cmk (Bachem), z-IETD-fmk (R&D), zVAD-fmk (Tocris or Abcam), recombinant murine GM-CSF (Peprotech), recombinant murine TNFα (Peprotech), doxorubicin (LC Laboratories or Sigma-Aldrich), staurosporine (LC Laboratories), UCN-01 (Enzo), and necrostatin-1 (Tocris). Oxaliplatin, cisplatin, TRIzol reagent, nigericin, and ATP were purchased from Sigma-Aldrich. Anti-caspase-1 (p10) rabbit polyclonal (sc-514), anti-actin goat polyclonal (sc-1615), anti-FADD goat polyclonal (sc-6036), and all HRP-conjugated secondary antibodies (Abs) were from Santa Cruz Biotechnology. The monoclonal 3ZD anti-IL-1β Ab, which recognizes both 33-kDa pro-IL-1β and 17-kDa mature IL-1β in western blot analysis, was provided by the Biological Resources Branch, National Cancer Institute, Frederick Cancer Research and Development Center (Frederick, MD). Other antibodies included anti-cIAP1 mouse monoclonal (1E1-1-10) from Enzo, anti-RIP mouse monoclonal (38-RIP) from BD Biosciences, and anti-NLRP3 mouse monoclonal (Cryo-2) from AdipoGen. Anti-caspase-7 (9492), anti-caspase-8 (4927), and anti-PARP (9542) rabbit polyclonal Abs were from Cell Signaling. Murine IL-1β DuoSet ELISA kit was from R&D Systems, and the murine TNFα ELISA kit was from BioLegend. Cell Titer-Glo Luminescent Viability Assay Kit was from Promega. EnzChek Caspase-3 Assay Kit and DEVD-cho were from Invitrogen. RT2 SYBR Green/ROX qPCR Master Mix (PA-012) and predesigned qPCR primers for murine IL-1β (PPM03109E), murine TNFα (PPM03113F), and murine GAPDH (PPM02946E) were from SA Biosciences. Transcriptor first strand cDNA synthesis kit was from Roche.
Murine models
Wild-type C57BL/6 mice were purchased from Taconic. Mice lacking both caspase-1 and caspase-11 on the C57BL/6 background (Casp1−/− Casp11−/−) have been previously described (21-23). Asc−/−, Trif−/−, and Nlrp3−/− Nlrc4−/− double-knockout mouse strains (C57BL/6 background) were obtained from Eric Pearlman (Case Western Reserve University). Femoral and tibial bones for BMDC cultures were also isolated from Casp8−/− Rip3−/−, Casp8+/+Rip3−/−, and litter-matched control Casp8+/+ Rip3+/+ mice which have been described previously (24). All experiments and procedures involving mice were approved by the Institutional Animal Care and Use Committees of Case Western Reserve University.
Isolation, culture, and experimental testing of bone-marrow derived dendritic cells (BMDC)
BMDC from 9-12 week old mice were isolated by minor modification of previously described protocols (25). Mice were euthanized by CO2 inhalation. Femura and tibiae were removed, briefly sterilized in 70% ethanol, and PBS was used to wash out the marrow cavity plugs. The bone marrow cells were resuspended in DMEM (Sigma Aldrich), supplemented with 10% bovine calf serum (HyClone Laboratories), 100 units/ml penicillin, 100 μg/ml streptomycin (Invitrogen), 2mM L-glutamine (Lonza), 15 ng/ml GM-CSF, plated onto 150-mm dishes, and cultured in the presence of 10% CO2. On day 3 post-isolation, 80% of the non-adherent population was removed and centrifuged at 300 g for 5 min at room temperature, and fresh medium was applied. Five days post-isolation, the resulting loosely adherent BMDC were collected, resuspended to a cell density of ~1 × 106/ml in the above differentiation medium, and plated into 6-well (2 ml/well), 12-well (1 ml/well), 24-well (0.5 ml/well), or 96-well (0.1 ml/well) plates as needed for particular experiments, and used between days 7 and 10 post-isolation.
For experimental tests, plated BMDC were centrifuged at 300 × g for 5-10 min, and the differentiation medium was removed and replaced with low-serum DMEM (0.1% bovine calf serum plus penicillin, streptomycin, and L-glutamine). The cells were equilibrated for 15 min at 37°C in 10% CO2 prior to addition of test reagents. BMDC were routinely primed with 1 μg/ml LPS (unless indicated otherwise) for 4 h to activate TLR4 signaling prior to treatment with indicated concentrations of Dox, STS, or other chemotherapeutic drugs for additional periods ranging from 2 to 18 h (with the LPS being present throughout). In some experiments, the cells were primed with 200 ng/ml Pam3CSK4 to induce TLR2, rather than TLR4, signaling cascades. Stimulation of NLRP3/ caspase-1 inflammasome signaling by either P2X7 receptor activation or nigericin treatment was routinely used as a positive control by supplementing the medium of LPS-primed BMDC with either 5 mM ATP or 10 μM nigericin for the final 30 min of test incubations. Where indicated, the BMDC cultures were treated with various pharmacological inhibitors (50-100 μM Ac-YVAD-cmk, 100 μM z-VAD-fmk, 100 μM z-IETD-fmk, 100 μM DEVD-cho, 50 μM necrostatin-1) either before or after the LPS priming steps.
Western blot analysis
BMDC were seeded in 6-well or 12-well plates and treated as described above prior to processing of both extracellular medium and whole cell lysates. Extracellular medium samples were concentrated by tricholoroacetic acid precipitation/ acetone washing, while cell lysates were prepared by detergent-based extractions as described previously (21) prior to standard processing by SDS-PAGE, transfer to PVDF membranes, and western blot analysis. Primary antibodies (Abs) were used at the following concentrations: 5 μg/ml for IL-1β, 1 μg/ml for caspase-1, 1 μg/ml for actin, 0.1 μg/ml for caspase-7, 1.2 μg/ml for caspase-8, 0.05 μg/ml for PARP, 2 μg/ml for cIAP, 1 μg/ml for NLRP3, 0.4 μg/ml for RIP, and 1 μg/ml for FADD. HRP-conjugated secondary Abs were used at a final concentration of 0.13 μg/ml. Where indicated, western blots were probed with anti-caspase-1 and anti-IL-1β Abs simultaneously. Chemiluminescent images of the developed blots were detected, stored, and quantified using a FluorChemE processor (Cell Biosciences).
ELISA analyses for IL-1β or TNFα release
BMDC were seeded in 24-well plates. Extracellular media samples were removed and centrifuged at 10,000 g for 15 s to pellet floating BMDC. The supernatants were then assayed for murine IL-1β or murine TNFα by standard ELISA (R&D Systems for IL-1β or BioLegend for TNFα) according to the manufacturer's protocol. All test conditions for ELISA-based experiments were performed in duplicate.
Cell viability assays
BMDC were seeded in 96-well plates. Viability of BMDC was measured by quantifying intracellular ATP content using the Cell Titer-Glo Luminescent Viability Assay (Promega). Luminescence was quantified in relative light units (RLU) using a BioTek Synergy HT plate-reader and normalized to the values measured in control, untreated BMDC.
Caspase-3/7 activity assays
BMDC seeded in 12-well plates were primed for 4 h with LPS and then incubated with various chemotherapeutic agents plus or minus 100 μM DEVD-cho for an additional 2-18 h prior to preparation of cell lysates for quantification of accumulated caspase-3/7 activity by EnzChek Fluorescence Caspase-3 assay kits (Invitrogen).
qPCR analyses
Casp8+/+ Rip3+/+, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC were treated with 1 μg/ml LPS for 1 or 4 h prior to extraction of total RNA by TRIZol reagent. Transcriptor First Strand cDNA Synthesis kit was used to generate first-strand cDNA from the purified RNA. IL-1β, TNFα, and GAPDH transcripts were quantified using a StepOne-Plus Real-Time PCR System (Applied Biosystems) with reactions performed in 25 μl containing RT2 SYBR Green/ROX qPCR Master Mix (12.5μl), 1:100 dilutions of RT product, and 1 μM PCR primer pair stock that were run in triplicate. Amplification cycle conditions were 95°C for 10 minutes followed by 40 cycles of (95°C, 15 sec; 55°C, 30-40 sec; and 72°C, 30 sec.). Expression of IL-1β or TNFα was calculated using the ΔΔCt method using StepOne software v. 2.1 with values normalized to GAPDH expression.
Preparation of detergent-soluble and detergent-insoluble BMDC lysate fractions
BMDC (4-5 × 106 in 60 mm culture dishes) were stimulated for 2 h with or without LPS (1 μg/ml) or TNFα (50 ng/ml) in the absence or presence of 50 μM zVAD in DMEM containing 10% bovine calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2mM L-glutamine . Other dishes were primed with LPS (1 μg/ml) for 4 h and then stimulated for an additional 3-8 h with 10 μM Dox in the absence or presence of 50 μM zVAD. Incubations were terminated by removal of supernatant medium for sedimentation and isolation of detached cells. Detached cells were centrifuged at 400 × g for 5 min and washed with 1 ml of ice-cold PBS. Whole cell detergent lysates were prepared by addition of 85 μl of RIPA lysis buffer (0.5% Na deoxycholate, 0.1% SDS, 1% Igepal CA630 in PBS, pH 7.4 plus protease inhibitor cocktail) to the adherent cells on the dish and incubated on ice for 20 min. Lysed adherent cells were scraped with a rubber policeman, pooled with detached cells, and extracted for an additional 10 min on ice. The whole cell lysates were separated into detergent-soluble and detergent-insoluble fractions by centrifugation at 15,000 × g for 15 min at 4° C. SDS sample buffer (20 μl) was added to the detergent-soluble fractions, while 58 μl of RIPA lysis buffer (supplemented with 5mM MgCl2) was added to the insoluble lysate pellet. The insoluble lysate pellet was subjected to 5 × freeze- thaw cycles (dry ice/ethanol and 37°C heating blocks) and vortexing after each freeze-thaw cycle. Each sample was then DNase-treated (2 units/sample) and incubated on ice for 10 min prior to addition of SDS sample buffer (12 μl) and extraction at 100°C for 5 min.
Data processing and analysis
All experiments were repeated 2-8 times with separate BMDC preparations. Figures illustrating western blot results are from representative experiments. As indicated, figures illustrating IL-1β ELISA, cell viability, or caspase activity results represent either the mean (± SE) of data from several identical experiments or the average (± range) of duplicate samples from single representative experiments. Experiments with 3 or more repeats were analyzed by one-way ANOVA with Bonferroni post-test comparison using Prism 3.0 software. The absolute magnitudes of maximal IL-1β release as quantified by ELISA could vary by ~2-fold between different BMDC preps. Thus, for some experimental series, IL-1β release measured in different conditions was normalized to the maximal release for the particular BMDC preparation; the normalized values from several identical experiments were then used to generate the mean (± SE) for evaluation by ANOVA.
Results
Pro-apoptotic chemotherapeutic drugs induce the release of IL-1β in LPS-primed murine bone marrow-derived dendritic cells (BMDC)
Fig. 1A illustrates the ability of several pro-apoptotic or chemotherapeutic drugs (12 h stimulation) to induce IL-1β processing and release in LPS-primed BMDC. Robust accumulation of extracellular IL-1β (10-20 ng/ml over 12 h) was elicited in response to: 1) doxorubicin (Dox), an anthracycline inhibitor of topoisomerase II; 2) staurosporine (STS), a broad-specificity kinase inhibitor commonly used to induce intrinsic apoptosis (26, 27); and 3) UCN-01, a 7-OH staurosporine analog and experimental therapeutic for hematopoietic and solid tumors (28, 29). The IL-1β release was comparable in magnitude to that elicited by ATP activation of P2X7 receptors for 30 min. In contrast, oxaliplatin and cisplatin, two widely-used platinum-based chemotherapeutic agents, did not stimulate significant IL-1β production. The ability of Dox, STS, or UCN-01, but not the platinum reagents, to trigger IL-1β release correlated with the relative efficacies of the drugs to induce apoptosis of LPS-primed BMDC as indicated by intracellular accumulation of active caspase-3/7 (Fig. 1B). These results are consistent with the studies of Sauter et al. (14) and Shimada et al. (30) who reported the abilities of Dox and STS, respectively, to stimulate an NLRP3- and caspase-1 dependent IL-1β processing response in LPS-primed murine macrophages. It is important to note that LPS was present throughout the duration of drug stimulation to facilitate sustained TLR4 signaling. We characterized the kinetics (Fig. 1C) and concentration-response relationship (Fig. 1D) for Dox-stimulated IL-1β release. After a lag phase of 4 h, 10 μM Dox induced a progressive increase in IL-1β production over the subsequent 8 h with a plateau at times ≥12 h. Increased IL-1β accumulation (at 12 h) occurred over a narrow range of [Dox] with a threshold at 1 μM and maximal effect at 10 μM; concentrations >10 μM resulted in an attenuated response due likely to a more rapid loss of BMDC viability. Although 1 μg/ml LPS was routinely used for TLR4 activation, equivalent Dox-induced IL-1β release was observed in BMDC primed with 10 or 100 ng/ml LPS while 1 ng/ml LPS was sub-threshold for supporting the response to Dox (Supplemental Fig. 1A). Western blot analysis (Fig. 1E) of BMDC stimulated with 10 μM Dox revealed that: 1) the time-dependent accumulation of extracellular IL-1β assayed by ELISA reflected the release of mature (17 kDa) IL-1β rather than unprocessed (33 kDa) proIL-1β; 2) the release of mature IL-1β was preceded by the processing and release of the p10 subunit of active caspase-1; and 3) the proteolytic maturation of IL-1β was temporally correlated with the proteolytic processing and release of the p20 subunit of caspase-7, another defined substrate for caspase-1 in myeloid lineage leukocytes.
FIGURE 1. Pro-apoptotic chemotherapeutic drugs induce the release of IL-1β in LPS-primed murine bone marrow-derived dendritic cells (BMDC).
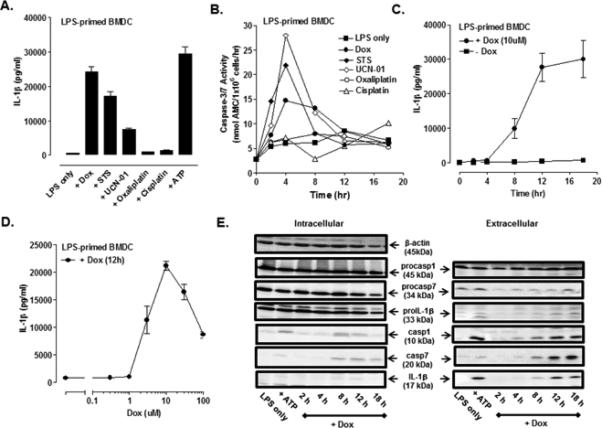
(A) BMDC were primed with LPS (1μg/ml) for 4 h prior to stimulation for 12 h with a panel of pro-apoptotic agents including staurosporine (STS, 5 μM), UCN-01 (10 μM), doxorubicin (Dox, 10 μM), oxaliplatin (Ox, 25 μM) and cisplatin (CDDP, 25 μM). The extracellular medium was collected and assayed for IL-1β by ELISA. BMDC were primed with LPS for 15.5 h prior to ATP (5mM) stimulation for 30 min. Results are the mean ± range of two experiments. (B) The kinetics of drug-induced caspase-3/7 activity in LPS-primed BMDC was measured by proteolytic cleavage of the DEVD-AMC substrate. The concentrations of each drug used were the same as described in (A). Results are from a single experiment. (C) The kinetics of IL-1β release from LPS-primed and Dox-stimulated (10 μM) WT BMDC were assayed by ELISA. Results are the mean ± SE from 4-8 experiments. (D) LPS-primed BMDC were stimulated with varying doses of Dox for 12 h. Results are the mean ± SE of 3 experiments. (E) WT BMDC were stimulated as in (C), and the extracellular medium and cell lysates were collected and processed for western blot analysis for detection of IL-1β, caspase-1, and caspase-7. BMDC were primed with LPS for 5.5 h prior to ATP (5mM) stimulation for 30 min. The data are representative of results from 3 experiments.
Doxorubicin induces caspase-1 independent processing and release of IL-1β in LPS-primed BMDC
Given the strong temporal association of the accumulation of mature IL-1β and active caspase-1 p10 subunit in the Dox-treated BMDC (Fig. 1E), we hypothesized that the IL-1β processing reflected Dox-induced activation of the NLRP3/ caspase-1 inflammasome pathway. This was tested by directly comparing (in the same experiments), the kinetics and magnitudes of Dox-induced IL-1β release in LPS-primed wildtype (WT) versus Casp1/11−/− BMDC. To our surprise, we observed that the rate and magnitude of mature IL-1β production were only modestly delayed (by ~ 2h) and decreased (by ~20%) in the Casp1/11−/− BMDC as monitored by either western blot (Fig. 2A) or ELISA (Fig. 2B). Efficient ablation of caspase-1 signaling in the knockout cells was confirmed by: 1) the absence of p45 procaspase-1 and the p10 subunit of active caspase-1 in the cell lysates and extracellular medium; and 2) suppression of P2X7 receptor-stimulated IL-1β maturation and release. As with WT cells, 1 ng/ml LPS was sub-threshold for supporting Dox-induced IL-1β release in the Casp1/11−/− BMDC, and equivalent secretion was observed when 10 μM Dox was combined with 10, 100, or 1000 ng/ml LPS (Supplemental Fig. 1B). The observation that WT, Asc−/−, and Nlrp3−/− Nlrc4−/− BMDC (all LPS-primed) exhibited similar magnitudes of IL-1β release in response to 12 h stimulation with 10 μM Dox further indicated the presence of an alternative inflammasome-independent pathway for IL-1β processing (Fig. 2C). As with the Casp1/11−/− cells (Fig. 2B inset), the Asc−/− or Nlrp3−/− Nlrc4−/− BMDC exhibited a reduced IL-1β release at 8 h of Dox stimulation but the differences did not reach statistical significance at p<0.05. In contrast, ATP-stimulated IL-1β production was completely suppressed in the same Asc−/− and Nlrp3−/− Nlrc4−/− BMDC preparations.
FIGURE 2. Doxorubicin induces caspase-1 independent processing and release of IL-1β in LPS-primed BMDC.
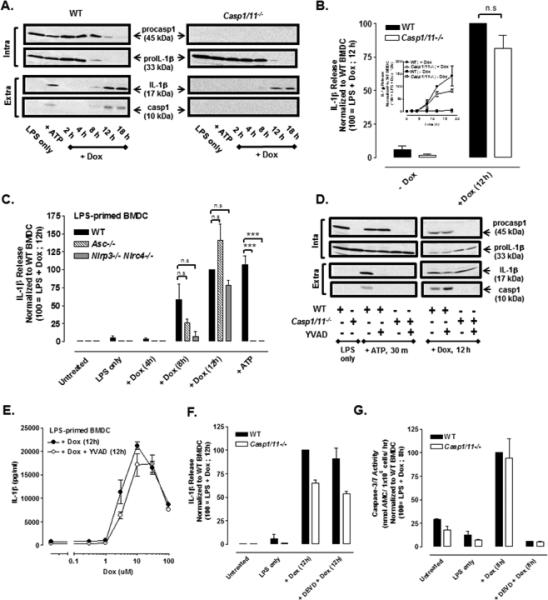
(A) LPS-primed (1 μg/ml) WT or Casp1/11−/− BMDC were stimulated with Dox (10 μM) for 2-18 h, and western blot analysis of IL-1β and caspase-1 from cell lysates and extracellular supernatants was performed. BMDC were LPS-primed for 5.5 h followed by 30 min of ATP (5mM) stimulation. Results are representative of 3 identical experiments. (B) The release of IL-1β from LPS-primed (1ug/ml) WT or Casp1/11−/− BMDC stimulated or not with 10 μM Dox for 12 h was assayed by ELISA. IL-1β release was normalized to WT BMDC treated with LPS + Dox for 12 h and expressed as the mean ± SE of 5 experiments. The differences between WT and Casp1/11−/− BMDC were not significant (n.s, P > .05) by Student's t-test. Inset: Kinetics of Dox-induced IL-1β release (normalized to WT BMDC treated with LPS + Dox for 12 h) in LPS-primed WT versus Casp1/11−/− BMDC as described in part (A). Results are the mean ± SE of 3 experiments. (C) WT, Asc−/−, and Nlrp3−/− Nlrc4−/− BMDC were primed with LPS for 4 h before stimulation with Dox for 4, 8, or 12 h and assayed for release IL-1β release by ELISA. Parallel samples were primed with LPS for 15.5 h prior to ATP (5mM) stimulation for 30 min. IL-1β release was normalized to WT BMDC treated with LPS + Dox for 12 h and expressed as the mean ± SE of 3-5 experiments. ***P < .001 or n.s by ANOVA. (D) Western blot analysis of LPS-primed (1μg/ml) WT or Casp1/11−/− BMDC stimulated with 10 μM Dox for 12 h or 5mM ATP for 30 min in the presence or absence of the caspase-1 inhibitor, YVAD (50 μM). BMDC were LPS-primed for 7.5 h followed by ATP (5mM) stimulation for 30 min. The data are representative of results from 2 experiments. (E) Concentration-response relationship for Dox-stimulated IL-1β release in the presence or absence of YVAD in LPS-primed BMDC. The curve depicted without YVAD treatment is the same as shown in Figure 1D. Results are the mean ± SE of 3 experiments. (F) The release of IL-1β was measured in LPS-primed WT or Casp1/11−/− BMDC stimulated ± 10μM Dox for 12 h in the presence or absence of DEVD (50μM). IL-1β release was normalized to WT BMDC treated with LPS + Dox for 12 h. Results are the mean ± range of 2 experiments. (G) Caspase-3/7 activity was measured in LPS-primed WT and Casp1/11−/− BMDC ± 10 μM Dox in the presence or absence of DEVD. Casp3/7 activity was normalized to WT BMDC treated with LPS + Dox for 8 h. Results are the mean ± range of 2 experiments.
Production of mature IL-1β in response to Dox was also observed in WT LPS-primed BMDC treated with YVAD, an inhibitor of caspase-1 activity (Fig. 2D, 2E). YVAD only modestly reduced (by ~15%) the IL-1β release stimulated by maximally effective (10 μM) Dox while producing a ~50% suppression of the response to submaximal (3 μM) Dox and complete inhibition of P2X7 receptor-induced IL-1β release. Inclusion of YVAD during Dox treatment of Casp1/11−/− BMDC did not reduce IL-1β maturation. Using a murine bone marrow-derived macrophage (BMDM) experimental model, Sauter et al. (14) reported that the ability of shorter term Dox exposure (8 h) to stimulate IL-1β processing was almost completely suppressed in macrophages isolated from Asc−/−, Nlrp3−/−, or Casp1−/− mice. We found that more prolonged (12 h) Dox treatment also induced caspase-1–independent IL-1β release in murine BMDM (Supplemental Fig. 2A). We similarly observed that LPS + Dox elicited IL-1β release in the RAW264.1 murine macrophage cell line (Supplemental Fig. 2B) that lacks expression of functional ASC (31, 32) and cannot process IL-1β in response to canonical inflammasome agonists such as ATP. These data suggest that intracellular signals generated during the initial phases of Dox-induced apoptosis in DCs and macrophages elicit modest assembly of NLRP3/ASC/ caspase-1 inflammasomes but that this canonical pathway is superseded by a caspase-1 independent pathway for IL-1β processing driven by signals which develop with sustained apoptotic stress.
As indicated in Fig. 1B, Dox treatment induces peak accumulation of apoptotic executioner caspase-3/7 activity within 4-8 h, which precedes the major phase of IL-1β processing and release. We tested whether active caspase-3/7 comprises a necessary upstream signal for the caspase-1-independent IL-1β processing by including the caspase-3/7 inhibitor DEVD during LPS + Dox stimulation. DEVD had no inhibitory effect on LPS + Dox-stimulated IL-1β release in wildtype or Casp1/11−/− BMDC (Fig. 2F), but completely suppressed the increased caspase-3/7 activity stimulated by Dox in parallel BMDC samples (Fig. 2G). Thus, Dox-induced accumulation of active apoptotic executioner caspases can be dissociated from its stimulatory action on non-canonical IL-1β processing and release.
TLR4 activation coupled with caspase-8 inhibition induces RIP1/RIP3-dependent necroptosis and release of unprocessed proIL-1β while TLR4 activation coupled with Dox treatment induces caspase-8-dependent apoptosis
We hypothesized that caspase-8 was the alternative IL-1β processing enzyme activated in Dox-treated BMDC given its demonstrated ability to act as an IL-1β convertase in other models of macrophage innate immune response (15-18). However, testing this by utilizing the pan-caspase inhibitor, zVAD, or the caspase-8 selective inhibitor, IETD, in LPS-primed BMDC is complicated by the risk of unleashing RIP1/RIP3-dependent necroptotic death downstream of TLR4 activation (33, 34). Consistent with the induction of necroptosis under such conditions, we observed a time-dependent accumulation of extracellular proIL-1β in BMDC within 4 h after initiation of the co-treatment with LPS and zVAD (Fig. 3A). The ability of zVAD to completely inhibit the caspase-1-mediated IL-1β processing response to P2X7 receptor activation verified the efficacy of zVAD as a pan-caspase inhibitor in these experiments. The LPS + zVAD-induced release of proIL-1β was completely suppressed by the RIP1 inhibitor, necrostatin-1 (Nec-1), consistent with a role for necroptosis in the release of extracellular proIL-1β. Similarly, combined LPS + IETD treatment of WT BMDC, either with or without Dox stimulation (12 h), resulted in the accumulation of extracellular proIL-1β (Fig. 3B). In contrast, IETD completely suppressed the accumulation of extracellular mature IL-1β triggered by LPS + Dox. The induction of necroptosis in BMDC co-treated with LPS + zVAD or LPS + IETD (versus LPS alone) was also verified by observing decreased cell viability (as indicated by total intracellular ATP content) and the ability of Nec-1 to prevent this loss of viability (Fig. 3C).
FIGURE 3. TLR4 activation coupled with caspase-8 inhibition induces RIP1/RIP3-dependent necroptosis and release of unprocessed proIL-1β while TLR4 activation coupled with Dox treatment induces caspase-8-dependent apoptosis.
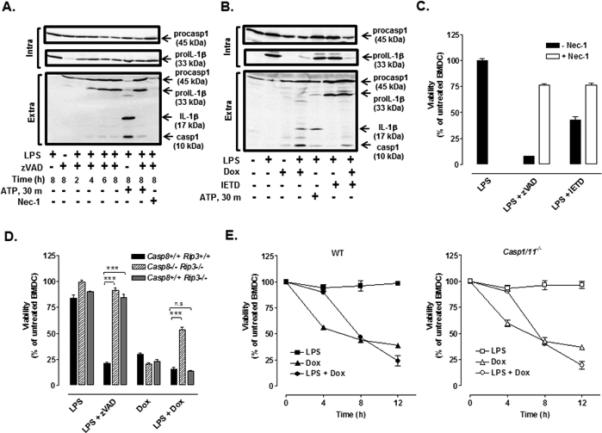
(A) Western blot analysis of IL-1β and caspase-1 in cell lysates and extracellular medium from WT BMDC treated with LPS (1μg/ml) and zVAD (50 μM) for 2-8 h with or without necrostatin-1 (Nec-1, 50 μM). (B) Western blot analysis of IL-1β and caspase-1 in cell lysates and extracellular medium from WT BMDC treated with LPS for 4 h prior to stimulation with Dox (10 μM) in the presence or absence of IETD (100uM) for 12 h. In panels A and B, BMDC were primed with LPS for 7.5 h prior to 30 min of ATP (5mM) stimulation. The data are representative of results from 2-3 experiments. (C) WT BMDC were co-treated with LPS + zVAD or LPS + IETD in the presence or absence of Nec-1, and cell viability was assessed by measuring intracellular ATP content. Results are from a single experiment with each condition performed in quadruplicate. (D) WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC were treated with LPS (16 h), Dox (12h), or LPS for 4 h prior to Dox or zVAD for 12 h, and cell viability was assessed. Results are from a representative experiment (of 2 similar experiments) performed in quadruplicate. ***P < .001 by ANOVA. (E) WT and Casp1/11−/− BMDC were LPS-primed or not for 4 h prior to stimulation or not with Dox for 4, 8, or 12 h, and cell viability was assessed. Results are from a representative experiment of 2 similar experiments with each condition performed in triplicate.
We also assessed the roles of caspase-8 in the LPS + zVAD-induced death response in WT, Casp8−/− Rip3−/− and Casp8+/+ Rip3−/− BMDC. Casp8−/− mice undergo mid-gestational death due to unrestrained necroptosis, but this phenotype is reversed in mice that additionally lack RIP3 (Casp8−/− Rip3−/−) (24, 35). Consistent with this phenotype, the decrease in BMDC viability induced by combined LPS + zVAD was absent in Casp8−/− Rip3−/− or Casp8+/+ Rip3−/− BMDC (Fig. 3D). The ability of Dox treatment alone to elicit similar decreases in BMDC viability, independent of caspase-8 and/or RIP3, was consistent with initiation of the intrinsic apoptotic cascade with likely engagement of caspase-9 as the initiator caspase. Kinetic analysis revealed that Dox alone triggered a rapid decrease (50% within 4 h) in the viability of WT or Casp1/11−/− BMDC (Fig. 3E) that was suppressed in LPS-primed cells, consistent with a protective effect of NF-κB-dependent anti-apoptotic gene expression. However, sustained (≥ 8 h) co-treatment with LPS + Dox resulted in decreased viability indicating engagement of an alternative regulated cell death pathway. The similar kinetics and magnitude of this response to LPS + Dox in WT and Casp1/11−/− BMDC argued against involvement of pyroptosis. In contrast, the rescued viability in Casp8−/− Rip3−/− cells, but not in WT or Casp8+/+ Rip3−/− cells, indicated that TLR4 activation coupled with Dox treatment mediated caspase-8-dependent apoptosis (Fig. 3D).
Doxorubicin induces caspase-8 dependent IL-1β processing and release in LPS-primed BMDC
We also utilized the Casp8−/− Rip3−/− and Casp8+/+ Rip3−/− BMDC to assess the role of caspase-8 as the alternative IL-1β processing enzyme stimulated by LPS + Dox. We first determined how canonical NLRP3/caspase-1 inflammasome signaling pathways are operative in the knockout BMDC. LPS triggered rapid (within 1 h) and quantitatively equivalent accumulation of IL-1β mRNA (and TNFα mRNA, data not shown) in WT (Casp8+/+ Rip3+/+), Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC (Fig. 4A). LPS priming for 8 h induced similar accumulation of proIL-1β protein in the three cell types (Fig. 4B). Inclusion of nigericin (Fig. 4B) or ATP (Fig. 4C) as canonical NLRP3 stimuli during the final 30 min of the 8 h LPS treatment periods also resulted in robust release of mature IL-1β in each BMDC genotype, albeit at modestly lower levels in the Casp8−/− Rip3−/− cells. When the total LPS treatment time was extended to 16 h (Fig. 4D) or 18 h (Fig. 4H), we observed that the proIL-1β protein levels were markedly reduced in Casp8−/− Rip3−/− BMDC relative to those in WT or RIP3-ko cells at these time points, or in Casp8−/− Rip3−/− cells at 8 h post LPS. In contrast, LPS stimulation for 16 h induced equivalent TNFα secretion in the three BMDC genotypes (Fig. 4G). Thus, the combined absence of caspase-8 and RIP3 apparently results in signals (or loss of sustaining signals) that result in gradual attenuation of on-going TLR4-driven transcription and translation of proIL-1β. Consistent with this model, inclusion of nigericin or ATP during the final 30 min of 16 h LPS treatment periods resulted in markedly reduced levels of mature IL-1β production in Casp8−/− Rip3−/− BMDC as assayed by western blot (Fig. 4D) and ELISA (Fig. 4F). Notably, the ability of nigericin to elicit production and release of caspase-1 p10 subunit by Casp8−/− Rip3−/− BMDC was suppressed when these cells were treated with LPS for 16 h (Fig. 4D) but not for 8 h (Fig. 4B); the former effect was correlated with the reduced ability to sustain elevated levels of TLR4-dependent NLRP3 protein expression (Fig. 4H).
FIGURE 4. Doxorubicin induces caspase-8 dependent IL-1β processing and release in LPS-primed BMDC.
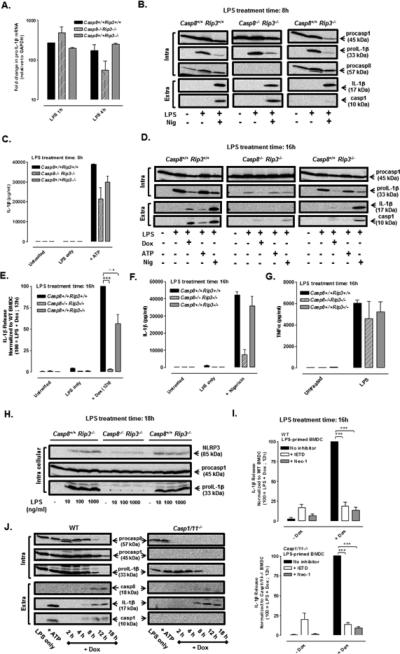
(A) ProIL-1β mRNA (normalized to GAPDH) was measured by qPCR in WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC treated with LPS (1 μg/ml) for 1 h or 4 h. Results are from a single experiment. (B) Western blot analysis of proIL-1β, procaspase-1, and procaspase-8 in cell lysates (intra) and mature IL-1β and caspase-1 p10 subunit in extracellular (extra) supernatants of WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC treated with LPS for 8 h and stimulated ± nigericin (10 μM) for the final 30 min of the LPS treatment period. Data are representative of results from 2 experiments. (C) BMDC of the indicated genotypes were treated with LPS (1 μg/ml) for 7.5 h prior to simulation ± ATP (5 mM) for an additional 30 min; IL-1β release was assayed by ELISA. Results are the mean ± range of 2 experiments. (D) Western blot analysis of pro-IL-1β and procaspase-1 in cell lysates (intra) and mature IL-1β and caspase-1 p10 subunit in extracellular (extra) supernatants of WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC treated with LPS for a total of 16 h and stimulated with either Dox (10 μM) for the final12 h, or with ATP (5 mM) or nigericin (10 μM) for the final 30 min, of the LPS treatment period . Results are representative of 3 similar experiments. (E, F) WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC were treated with LPS (1 μg/ml) for a total of 16 h and stimulated with Dox (10 μM) for the last 12 h (E) or nigericin (10 μM) for the last 30 min (F) of the LPS treatment period. IL-1β release was assayed by ELISA and normalized to WT stimulated with LPS + Dox (E) or WT stimulated with LPS + nigericin (F). Results are the mean ± SE of 3 experiments for (E) or the mean ± SE for 2 experiments (F) ***P < .001 or not significant (n.s) by ANOVA. (G) TNFα release (by ELISA) from WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC treated ± LPS (1 μg/ml) for 16 h. Results are the mean ± range of 2 experiments. (H) Western blot analysis of proIL-1β, procaspase-1, and NLRP3 expression in WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC treated for 18 h with 0, 10, 100, or 1000 ng/ml of LPS. Results are representative of 2 experiments. (I) WT and Casp1/11−/− BMDC were treated with LPS (1 μg/ ml) for a total of 16 h and stimulated ± Dox (10 μM), ± IETD (100 μM), ± Nec-1 (50 μM) for the final 12 h of the LPS treatment period. IL-1β release was assayed by ELISA and normalized to the samples stimulated with LPS + Dox. Results are the mean ± SE of 3 experiments. ***P < .001 by ANOVA. (J) Western blot analysis of procapase-8, procaspase-1, and proIL-1β in cell lysates (intra) and mature IL-1β, caspase-8 p18 subunit, and caspase-1 p10 subunit in the extracellular supernatants (extra) from WT or Casp1/11−/− BMDC treated with LPS (1 μg/ml) for 4 h prior to co-stimulation with Dox (10 μM) for another 2-18 h or with LPS for 5.5 h prior to ATP (5mM) stimulation for 30 min. The data are representative of results from 3 experiments.
These analyses of canonical NLRP3 inflammasome signaling provide an important context for comparison and interpretation of the non-canonical IL-1β release responses to LPS + Dox in WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC. IL-1β release was completely suppressed in Casp8−/− Rip3−/− BMDC compared to WT BMDC treated with LPS and Dox as assayed by ELISA (Fig. 4E) and western blot analysis (Fig. 4D). The LPS + Dox-induced IL-1β release was attenuated by ~50-60% in Casp8+/+ Rip3−/− cells, suggesting a possible modulatory role for RIP3 in the response to Dox. It is important to note that the Dox treatment in these experiments was initiated after 4 h of LPS priming and maintained during the next 12 h of LPS stimulation. Despite the gradual decrease in TLR4-dependent proIL-1β protein expression in Casp8−/− Rip3−/− BMDC, the cells contained high levels of this cytokine precursor during the initial several hours of Dox treatment. Thus, the complete inhibition of Dox-induced mature IL-1β release in these cells most likely reflects the absence of caspase-8 as a major non-canonical IL-1β converting enzyme rather than reduced levels of the proIL-1β substrate per se.
Consistent with a role for caspase-8 as an IL-1β converting enzyme, procapase-8 processing and accumulation of mature extracellular caspase-8 was detected in WT and Casp1/11−/− BMDC treated with LPS + Dox for >4 h (Fig. 4J). Interestingly, the clearance of procaspase-8 induced by Dox occurred more rapidly in Casp1/11−/− BMDC compared to WT BMDC. With recognition of the caveats in using pharmacological inhibitors of caspase-8 to assess its role in IL-1β processing in LPS-primed BMDC, we observed that the ability of LPS + Dox to stimulate extracellular accumulation of ELISA-measurable IL-1β was greatly suppressed by IETD in both WT and Casp1/11−/− BMDC (Fig. 4I). The RIP1 inhibitor Nec-1 also suppressed these IL-1β release responses. These results implicate caspase-8 signaling platforms, possibly in RIP1-containing ripoptosomes, as key mediators of LPS + Dox stimulated non-canonical IL-1β processing.
LPS + Dox-induced IL-1β release is TRIF-dependent and correlated with IAP downregulation and recruitment of caspase-8/ FADD to a detergent-insoluble compartment
Maelfait et al. demonstrated that the ability of TLR4 and TLR3 activation (combined with cyclohexamide-mediated suppression of protein turnover) to induce caspase-8 maturation of IL-1β was compromised in macrophages from TRIF-knockout mice (16). Kaiser and Offermann found that TRIF possesses a RHIM (receptor interacting protein (RIP) homotypic interaction motif) domain that facilitates association with RIP1 (and RIP3) (36). Thus, activation of TLR4 in macrophages and DCs results in NF-κB-dependent up-regulation of proIL-1β, via both the MyD88 and TRIF adapters, as well as the assembly of latent RIP1/ FADD/ caspase-8 ripoptosome complexes, orchestrated by the TRIF adapter (Fig. 5A). Because TLR4 activation drives the NF-κB-mediated upregulation of NLRP3 expression (Fig. 4H) (37), LPS-treated cells will also be primed for induction of canonical caspase-1-mediated IL-1β maturation in the presence of appropriate signal 2 stress stimuli. In contrast, while TLR2 activation can also trigger the NF-κB-dependent up-regulation of proIL-1β and NLRP3 that facilitates utilization of caspase-1 as an IL-1β converting enzyme, this pathway will not engage TRIF-dependent signaling cascades, such as those leading to possible assembly of RIP1/ FADD/ caspase-8 ripoptosomes (Fig. 5A). To test the differential contributions of MyD88- versus TRIF-signaling to LPS + Dox-induced IL-1β processing, WT and Trif−/− BMDC were primed either with the TLR4 agonist, LPS, or the lipopeptide TLR2 agonist, Pam3CSK4, for 4 h before stimulation with Dox for 12 h. LPS + Dox-induced IL-1β maturation and release as assayed by western blot (Fig. 5B) and ELISA (Figs. 5C and 5D) was significantly reduced in Trif−/− BMDC. Prolonged LPS treatment resulted in lower levels of proIL-1β in the Trif−/− cells (Fig 5B), but this partial reduction in proIL-1β substrate contrasted with the near-complete suppression of IL-β maturation in response to Dox. Inclusion of YVAD did not further suppress the residual LPS + Dox-triggered IL-1β release observed in Trif−/− BMDC (Fig. 5D). Although Pam3CSK4 and LPS induced equivalent accumulation of proIL-1β in WT cells (Fig. 5B), combined stimulation by Pam3CSK4 + Dox resulted in ~4-fold less production of mature IL-1β compared to LPS + Dox treatment (Fig 5C). Regardless, the magnitude of Pam3CSK4 + Dox -induced IL-1β release was similar in Trif−/− and WT BMDC. These data support a role for TRIF in mediating a TLR4-dependent activation of caspase-8 and consequent IL-1β maturation when combined with Dox.
FIGURE 5. LPS + Dox-induced IL-1β release is TRIF-dependent and correlated with IAP downregulation and recruitment of caspase-8/ FADD to a detergent-insoluble compartment.
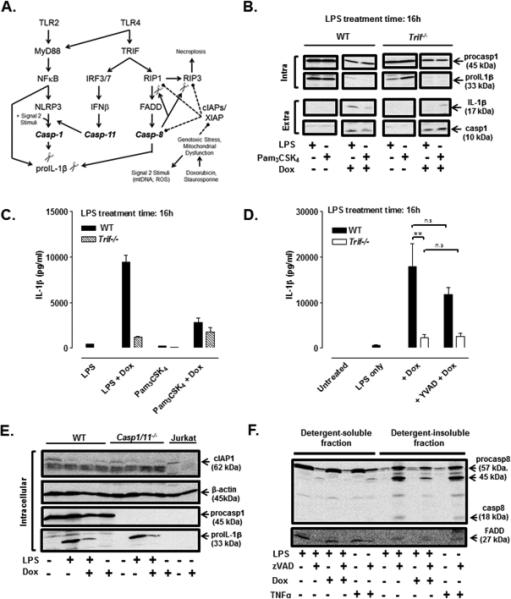
(A) A model for parallel pathways of canonical NLRP3 inflammasome activation and TRIF-induced caspase-8 signaling complexes that may mediate IL-1β processing induced by LPS + doxorubicin. (B) Western blot analysis of proIL-1β and procaspase-1 in cell lysates (intra) and mature IL-1β and caspase-1 p10 subunit in extracellular supernatants (extra) from WT and Trif−/− BMDC stimulated Pam3CSK4 (200 ng/ml) ± Dox (10 μM) or LPS (1 μg/ml) ± Dox for 12 h. The data are representative of 2 experiments. (C, D) IL-1β release (by ELISA) from WT or Trif−/− BMDC treated with Pam3CSK4 (200 ng/ml) or LPS (1 μg/ml) for a total of 16 h and stimulated ± Dox (10 μM) (C, D) or Dox + YVAD (50 μM) (D) for the final 12 h of the LPS or Pam3CSK4 treatment periods). Data in (C) are representative of results from 2 experiments. Data in (D) are the mean ± SE of 3 experiments. **P < .01 or not significant (n.s) by ANOVA. (E) Western blot analysis of cIAP1, proIL-1β, procaspase-1, and actin in cell lysates from WT BMDC or Casp1/11−/− BMDC treated ± LPS (1 μg/ml) for 4 h prior to stimulation ± Dox (10 μM) for 12 h. As a positive control, lysates from control or Dox-treated Jurkat leukemic T cells were also analysed. Results are from a single experiment. (F) WT BMDC were treated with LPS (1 μg/ml) or TNFα (50 ng/ml) for 2 h in the presence or absence of zVAD (50 μM). Alternatively, BMDC were treated with LPS for 4 h prior to stimulation with Dox (10 μM) or Dox + zVAD for another 8 h. Cell lysates were separated into detergent-soluble versus detergent-insoluble fractions for western blot analysis of caspase-8 and FADD. Results are representative of 2 experiments.
We next explored the mechanism by which Dox may license the activation of caspase-8 within the presumed signaling platforms assembled in response to the TLR4-TRIF cascade. Tenev et al. reported that genotoxic stressors such as etoposide (a topoisomerase II inhibitor like doxorubicin) facilitate ripoptosome assembly and caspase-8-mediated cell death in non-myeloid tumor cells by inducing the degradation of cellular Inhibitor of Apoptosis Proteins (cIAPs) (38). Furthermore, Vince et al. showed that Smac mimetics, which induce degradation of cIAPs, activated IL-1β processing in LPS-primed BMDC via a caspase-8 pathway that required coordinate down-regulation/inhibition of all three major IAPs including cIAP1, cIAP2, and XIAP (17). We assessed the levels of cIAP1 in WT and Casp1/11−/− BMDC treated (or not) with LPS only, Dox only, or LPS + Dox. The amount of cIAP1 was greatly reduced in LPS + Dox-treated BMDC and attenuated in the LPS only or Dox only treatment conditions compared to untreated cells (Fig. 5E). As a positive control, we showed that Dox treatment reduced cIAP1 levels in Jurkat leukemic T cells.
We initially assessed whether LPS + Dox stimulated the formation of RIP1/ FADD/ caspase-8 ripoptosome complexes in BMDC by adapting co-immunoprecipitation protocols developed for the characterization of such platforms in tumor cells or non-myeloid cell types challenged with pro-apoptotic stimuli, TLR3 agonist, or TNFα (38-40). WT BMDC were treated with various combinations of LPS, Dox, and zVAD, or TNFα ± zVAD, prior to generation of whole lysates by standard detergent extraction, removal of detergent-insoluble material by centrifugation, incubation of the detergent-soluble lysates with anti-FADD, collection of anti-FADD immune complexes, and western blot analysis of the immune complexes for caspase-8, FADD, and RIP1. ZVAD is commonly used in such analyses to stabilize caspase-8-containing signaling complexes (38-40). No caspase-8 or RIP1 was observed in FADD immunoprecipitates prepared from cells treated with any combination of those stimuli. However, we consistently found that the detergent-soluble faction (i.e., the fraction subjected to subsequent immunoprecipitation) of lysates from BMDC treated for 2 h with LPS + zVAD or TNFα + zVAD contained markedly lower levels of procaspase-8 and FADD (Fig. 5F). To our surprise, analysis of the detergent-insoluble lysate pellets from cells stimulated with LPS + zVAD or TNFα + zVAD revealed robust enrichment of 57 kDa procaspase-8 and 45 kDa partial cleavage fragment of caspase-8, as well as lower amounts of 18 kDa processed caspase-8 subunit; FADD was also enriched in the same detergent-insoluble fractions. This redistribution of caspase-8 and FADD was not observed in lysates from cells treated with only zVAD (data not shown) or in BMDC that were primed with LPS for 4 h followed by treatment with Dox for an additional 8 h (Fig. 5F) or 3 h (data not shown. However, inclusion of zVAD during the Dox treatment did facilitate caspase-8 and FADD partitioning into the insoluble lysate pellets (Fig. 5F). Analysis of RIP1 recruitment to these pellets was limited by the finding that total RIP1 levels in BMDC (but not BMDM or J774.1 murine macrophages) were below the detection limit of the antibody used (Fig. 6E). These data indicate that TLR4 activation in BMDC induces the recruitment of caspase-8 and FADD to signaling pltaforms that may underlie non-canonical IL-1β processing when combined with pro-apoptotic stimuli that down-regulate IAPs.
FIGURE 6. Staurosporine induces caspase-8-dependent IL-1β processing and release in LPS-primed BMDC.
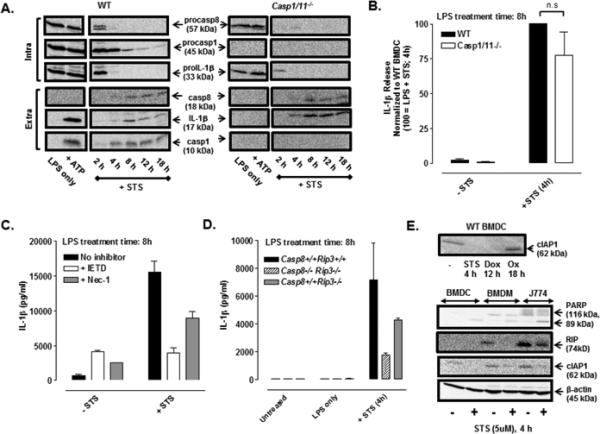
(A) Western blot analysis of procapase-8, procaspase-1, and proIL-1β in cell lysates (intra) and mature IL-1β, caspase-8 p18 subunit, and caspase-1 p10 subunit in the extracellular supernatants (extra) from WT or Casp1/11−/− BMDC were treated ± LPS (1 μg/ml) for 4 h and then co-stimulated with STS (5 μM) for an additional 2-18 h or with LPS for 5.5 h prior to ATP (5mM) stimulation for 30 min. The data are representative of results from 3 experiments. (B) LPS-primed (1 μg/ml, 4h) WT and Casp1/11−/− BMDC were stimulated ± STS (5 μM) for 4 h; IL-1β release was assayed by ELISA and normalized to WT BMDC treated with LPS + STS (4 h). Results are the mean ± SE of 4-5 experiments. The differences between WT and Casp1/11−/− BMDC were not significant (n.s, P > .05) by Student's t-test. (C) WT BMDC were primed with LPS (1 μg/ml, 4 h) prior to stimulation ± STS (5 μM, 4 h) in the presence or absence of IETD (100 μM) or Nec-1 (50 μM); IL-1β release was assayed by ELISA. Results are from a single representative experiment of 3 experiments. (D) WT, Casp8−/− Rip3−/−, and Casp8+/+ Rip3−/− BMDC were treated ± LPS (1 μg/ml) for 4 h prior to co-stimulation ± STS (5 μM) for an additional 4 h. Results are from a single experiment with each condition was performed in duplicate. (E) Upper panel: Western blot analysis of cIAP1 of lysates from untreated WT BMDC or WT BMDC treated with STS (5 μM, 4 h), Dox (10 μM, 12 h), or oxaliplatin (Ox) (25 μM, 18 h). Lower panel: Western blot analysis of cIAP1, PARP (full length and cleaved forms), and RIP1 in lysates in control versus STS-treated (5 μM,4 h) BMDC, BMDM, and J774 murine macrophages. Results are from a single experiment.
Staurosporine induces caspase-8-dependent IL-1β processing and release in LPS-primed BMDC
Staurosporine (STS) has been extensively studied with regard to its ability to induce a very rapid apoptotic response in most cell types via incompletely understood mechanisms that include suppression of multiple pro-survival kinases (41, 42), disruption of mitochondrial integrity (30), and allosteric modulation of caspase-9 independently of Apaf-dependent apoptosome assembly (43). Shimada et al. demonstrated that STS also stimulates a rapidly developing NLRP3- and caspase-1 dependent IL-1β processing response in LPS-primed murine macrophages via a mechanism linked to disruption of mitochondrial integrity (30). Fig. 6A shows that STS triggers a similarly rapid (near-maximal within 4 h) increase in IL-1β processing and release in LPS-primed WT BMDC. Notably, STS also induced a robust accumulation of mature extracellular IL-1β in Casp1/11−/− BMDC. The STS-stimulated processing and release of IL-1β in both WT and Casp1/11−/− cells was temporally correlated with production and release of the p18 subunit of active caspase-8 (Fig. 6A). As with LPS + Dox treatment, LPS + STS-induced clearance of procaspase-8 occurred more rapidly in Casp1/11−/− BMDC compared to WT BMDC even though mature 18 kD caspase-8 accumulation appeared to be equivalent. ELISA measurements indicated that STS-induced IL-1β release (at 4 h) was non-significantly attenuated by ~25% in either Casp1/11−/− BMDC (Fig. 6B) or in WT BMDC treated with YVAD (data not shown). STS-induced IL-1β release from either WT (Fig. 6C) or Casp1/11−/− (data not shown) BMDC was markedly suppressed by IETD and partially (~50%) decreased by Nec-1 (Fig. 6C). A 75% reduction in STS-triggered IL-1β secretion was also observed in Casp8−/− Rip3−/− BMDC (Fig. 6D). Similarly to treatment with 12 h of Dox, a 4 h exposure to STS suppressed the expression of cIAP1 in WT BMDC (Fig. 6E, upper panel). In contrast, 18 h of oxaliplatin induced no change in cIAP1 expression relative to untreated BMDC. Additionally, a 4-h treatment with STS was sufficient to produce marked depletion of cIAP1 levels in other murine myeloid cell types including BMDC, BMDM, and the J774.1 murine macrophage line (Fig. 6E, lower panel). Rapid STS-triggered depletion of cIAP1 in these cells was correlated with apoptotic induction as indicated by depletion of intact 116 kDa PARP (poly-ADP-ribosyltransferase) and accumulation of 89 kDa PARP cleavage product. STS treatment also decreased RIP1 protein levels in these detergent-solubilized cell lysate samples. Despite the complexity of STS action, these data indicate that this apoptosis inducer also triggers caspase-8 dependent IL-1β processing and release in LPS-primed BMDC.
Discussion
This study describes a novel and robust mechanism of caspase-8-dependent processing of IL-1β induced by the chemotherapeutic drug, Dox, in LPS-primed BMDC. This release did not require caspase-1 based on the fact that mature IL-1β was produced by Casp1/11−/− BMDC as well as by WT BMDC treated with YVAD (Fig. 2). The pro-apoptotic agent, STS, triggered caspase-1 independent IL-1β release in LPS-primed BMDC even more rapidly than Dox (Fig. 6). Dox and STS-induced IL-1β production in LPS-primed WT BMDC was attenuated in the presence of the caspase-8 inhibitor, IETD, or the RIP1 inhibitor, Nec-1, while Dox- and STS-induced IL-1β release was greatly suppressed in LPS-primed Casp8−/−Rip3−/− BMDC, implicating a caspase-8 signaling platform for IL-1β processing in TLR4-activated DCs (Figs. 4 and 6). The marked attenuation of LPS + Dox-induced IL-1β release from Trif−/− BMDC relative to WT cells supported an important role for TRIF in coupling TLR4 stimulation to caspase-8 activation and non-canonical IL-1β processing (Fig. 5). In the presence of a pan-caspase inhibitor, TLR4 activation induced recruitment of caspase-8 and FADD to a detergent-insoluble compartment (Fig. 5) and also triggered RIP1-dependent necroptotic death (Fig. 3). The ability of both Dox and STS to trigger IL-1β release in LPS-primed BMDC was correlated with the degradation of cIAP1 (Figs. 5 and 6) which, together with cIAP2 and XIAP, is known to restrain ripoptosome activity by destabilizing such complexes. Taken together, these results support a model wherein TLR4 drives TRIF-dependent assembly of caspase-8 ripoptosomes that are licensed as IL-1β converting enzyme platforms in the presence of pro-apoptotic stimuli, likely via modulated expression of IAPs and/or other factors (e.g. cFLIP) which control ripoptosome assembly, stability, and activity. However, a rapidly expanding literature is identifying roles for caspase-8 as a component of multiple signaling complexes or pathways linked to innate immune response (15, 18, 19, 44, 45). Thus, ripoptosomes may comprise only one of several caspase-8-based pathways that participate in non-canonical IL-1β processing.
Relationships between caspase-1 and caspase-8 signaling in IL-1β processing and release
Consistent with the reported role for the NLRP3 inflammasome in LPS + Dox-induced IL-1β production by bone marrow-derived macrophages (14), we observed reduced IL-1β release at the early (≤ 8 h) stages of LPS + Dox stimulation in BMDC lacking caspase-1 (Figs. 2A, 2B), ASC (Fig. 2C), or NLRP3 (Fig. 2C). However, when Dox stimulation was sustained, the relative contribution of this NLRP3/caspase-1 cascade to IL-1β accumulation was superseded by an alternative pathway that was attenuated in the absence of caspase-8 or TRIF (Figs. 2, 4, and 5). Robust proteolytic maturation of caspase-1 per se was invariably observed in WT BMDC treated with LPS + Dox (Figs. 1, 2 and 4) or LPS + STS (Fig. 6) despite the fact that caspase-1 was dispensable for maximal IL-1β processing. We initially expected that the activation of caspase-1 and caspase-8 in response to Dox or STS comprised independent IL-1β processing pathways operating in parallel. However, our observations and recent studies by others suggest a linked but asymmetric relationship in the ability of pro-apoptotic stimuli to activate the caspase-1 versus caspase-8 pathways coupled to IL-1β maturation and release. Although the caspase-8 pathway was strongly activated by LPS + Dox in BMDC that lack caspase-1, caspase-11, ASC, or NLRP3 (Fig. 2), activation of caspase-1 and caspase-1 mediated IL-1β processing was greatly reduced in Casp8−/−Rip3−/− BMDC (Fig. 4) but only modestly attenuated in cells lacking only RIP3 (Figs. 4D and 4E). This was mediated in part by the reduced ability of the Casp8−/−Rip3−/−cells to sustain high expression of proIL-1β and NLRP3 protein during prolonged TLR4 activation (Figs. 4D and 4H). Notably, the ability of ATP or nigericin, as rapidly acting signal 2 stimuli for NLRP3 inflammasome assembly, to trigger caspase-1 processing (Fig. 4B) and IL-1β release (Fig. 4B, 4C) was normal in Casp8−/−Rip3−/− BMDC during the earlier (< 8 h) stages of TLR4 activation but was markedly reduced when the Casp8−/−Rip3−/− cells were primed with LPS for longer durations (>12 h) before being challenged with nigericin or ATP (Figs. 4D and 4F). These data indicate that caspase-8 in DCs and macrophages can act directly as an alternative IL-1β converting enzyme and indirectly as a modulator of the expression of proIL-1β and NLRP3. Although the underlying mechanism for this latter role of caspase-8 remains to be defined, the dependence on prolonged TLR4 activation suggests feedback regulation by autocrine or paracrine pathways that fine-tune ongoing transcription, translation, and turnover of proIL-1β and NLRP3, and possibly other proinflammatory gene products.
Other investigators have recently described complex roles for caspase-8 in the direct and indirect regulation of IL-1β production (17, 46). Kang et al. observed that accumulation of active RIP3 and RIP1 in BMDC isolated from mice with a DC-restricted deletion of Casp8 was sufficient to license rapid assembly of NLRP3 inflammasomes in response to TLR4 activation in the absence of a signal 2 stimulus, such as ATP or nigericin (46). This ability of caspase-8 deletion to potentiate TLR4 induction of NLRP3 inflammasomes was independent of significant RIP1/RIP3-induced cell death. Kang et al. additionally noted that caspase-8 and FADD were co-immunoprecipitated with NLRP3 in lysates from LPS-stimulated WT BMDC. We found that caspase-8 and FADD were recruited to a detergent-insoluble compartment in DCs treated with LPS and zVAD (Fig. 5F). Vince et al. characterized additional interactions between caspase-8, RIP3, and NLRP3 signaling in their model of Smac-mimetic-induced IL-1β accumulation by murine BMDM and BMDC (17). Activation of RIP3 was correlated with production of reactive oxygen species that stimulated IL-1β processing via both caspase-1 and caspase-8. Those investigators also reported recruitment of caspase-8 and RIP1 to detergent-insoluble fraction in lysates of the Smac-mimetic stimulated BMDM. Another study found that caspase-8 interacted with ASC and co-localized with AIM2/ASC specks during cell death triggered by Francisella tularensis infection of caspase-1 deficient BMDM (45). Finally, Sagulenko et al. have described a direct interaction between the death effector domain (DED) of procaspase-8 and the pyrin doman of ASC that can occur in the context of NLRP3- or AIM2-inflammasome activation in macrophages (44).
These various findings underscore complex interactions between caspase-8 and proteins/pathways associated with canonical caspase-1 inflammasome assembly and activity. Our data suggest that pro-apoptotic agents, in conjunction with TLR4 activation, elicit a temporally defined hierarchy of reactions that utilize various combinations of TRIF, FADD, RIP1/3, and conventional inflammasome proteins for the assembly of caspase-8 and caspase-1 platforms that catalyze IL-1β processing. Although both Dox and STS induced proteolytic processing of both caspase-1 and caspase-8 (Figs. 4J and 6A), STS stimulated these responses much more rapidly with significant activation of caspase-1 activation preceding that of caspase-8. This more rapid engagement of the caspase-1 pathway may underlie the lower efficacy of IETD or caspase-8 deletion in attenuating STS-induced IL-1β processing (Figs. 6C and 6D) relative to Dox-induced IL-1β production (Figs. 4E and 4I). The relative rate at which a particular “signal 2” stimulus triggers activation of the NLRP3/caspase-1 pathway likely determines whether the more slowly developing caspase-8 pathway will, or will not, comprise a major route for IL-1β processing. ATP acting directly via P2X7 non-selective cation channel receptors, and nigericin, acting as a direct K+ ionophore, trigger dissipation of the normal trans-plasma membrane K+ gradient within minutes and the resulting decrease in cytosolic [K+] elicits exceptionally rapid (complete within 30 min) and efficient assembly of NLRP3 inflammasomes (Figs. 1E, 2D, 4B, 4D, 4J) (7). Thus, very robust processing of proIL-1β occurs prior to any substantial activation of caspase-8. STS also triggers K+ efflux-dependent NLRP3 activation (4), but the K+ efflux likely occurs as a secondary and more slowly induced consequence of mitochondrial dysfunction and reduced ATP generation. As a result, STS-induced activation of caspase-1 occurs only modestly faster than induction of the caspase-8 pathway (Fig. 6A). Finally, because changes in mitochondrial function (and apoptotic signaling) are even more slowly induced by Dox, the activation of both the caspase-1 and caspase-8 pathways develop over similar time periods (Fig. 6J).
Pro-apoptotic stimuli and IAPs as regulators of IL-1β production
We observed a strong correlation between the relative efficacies of pro-apoptotic agents to induce IL-1β production and their abilities to stimulate apoptotic executioner caspase-3/7 activity in DCs (Fig. 1A, 1B). Perturbation of mitochondrial function comprises one possible link by which pro-apoptotic agents can engage both the caspase-1 and caspase-8 pathways for IL-1β production. STS elicits exceptionally rapid changes in mitochondrial function and apoptotic progression in macrophages (30) and most other cell types by incompletely understood mechanisms. Given its ability to inhibit topoisomerase II, Dox can induce conventional DNA damage-dependent apoptosis in tumor cells and other rapidly dividing cells (47). In quiescent or slowly proliferating cells (e.g., cardiomyocytes), Dox also triggers apoptosis via activation of iron-dependent redox cycling reactions in mitochondria that result in accumulation of reactive oxygen species (ROS) (47). Accumulation of ROS together with release of mitochondrial DNA has been linked to assembly of the NLRP3 inflammasome pathway for IL-1β production (3). However, another hallmark of the intrinsic apoptotic pathway is the release of Smac from dysfunctional mitochondria. Binding of Smac to IAPs either directly inhibits their ability to bind to caspases, as with XIAP, or induces their autoubiquitination and proteasome-mediated clearance as with the cIAP1/cIAP2 E3 ubiquitin ligases (48). The cIAPs per se target pro-apoptotic proteins and components of the ripoptosome, including RIP1 and RIP3, for ubiquitin-mediated degradation (Fig. 5A). Genotoxic stressors, such as etoposide, or the degradation of cIAPs using Smac mimetics, promote ripoptosome assembly (17, 38). We found that Dox and STS, but not oxaliplatin, induced marked downregulation of cIAP1 levels in BMDC (Figs. 5E and 6E); this likely comprises one mechanism by which these agents trigger caspase-8 activation when combined with TLR4 stimulation. A limitation of our study is that we assayed only changes in cIAP1 levels. Vince et al. found that the ability of Smac-mimetics to maximally induce IL-1β production by LPS-primed BMDM or BMDC required coordinated targeting of cIAP2 and XIAP, in addition to cIAP1 (17). Thus, it will be important to assess the effects of Dox and other pro-apoptotic agents on the expression/ activity of cIAP2 and XIAP in future studies of non-canonical IL-1β production. In addition to IAPs, other E3 ubiquitin ligases may contribute to caspase-8 regulation in our model. Jin et al. identified a critical role for cullin-3 in mediating the polyubiquitination, aggregation, and full activation of caspase-8 during TRAIL-mediated apoptosis of cancer cells (49). Notably, those investigators also observed the recruitment of caspase-8 and FADD to a detergent-insoluble compartment similar to our findings in LPS + zVAD-stimulated BMDC (Fig. 5F).
The role of cIAPs in inflammasome assembly and caspase-1 signaling is more complex. Labbé et al. found that cIAP1 and cIAP2 were required for efficient caspase-1 activation and IL-1β processing in response to multiple stimuli for NLRP3 and NLRC4 inflammasome assembly (50). This reflected a direct interaction of the cIAPs with caspase-1 that results in an activating and non-degradative K63-linked ubiquitination of caspase-1. In contrast, Vince et al. showed that the loss of cIAPs upon stimulation with Smac mimetics promoted RIP3-dependent caspase-1 and caspase-8 activation for IL-1β processing (17). These nominally conflicting observations are particularly germane given the E3 ubiquitin ligase activity of IAPs and recent findings that the ubiquitination status of NLRP3 per se (9-11) and caspase-1 (50) strongly modulate the assembly and activity of caspase-1 inflammasomes. Our observations that the caspase-8 pathway progressively predominates over the caspase-1 pathway during Dox and STS treatment may reflect the opposing consequences of IAP suppression on these signaling cascades. Progressive loss of the cIAPs may act to bias the signaling network by reversing the stimulatory effect of K63-ubiquitination on caspase-1 while enhancing ripoptosome assembly/activity. Further investigation is required to assess the differential roles of IAPs and other E3 ubiquitin ligases in the engagement of the canonical and non-canonical pathways for IL-1β processing by various cell death and inflammatory stimuli.
Roles for TRIF, RIPs, and other adapters in regulating caspase-8 mediated processing of IL-1β
The ability of LPS + Dox to stimulate IL-1β maturation and release was greatly reduced in Trif−/− BMDC (Figs. 5B, 5C, and 5D). The absence of TRIF could reduce the contribution from the TLR4→ TRIF→ IRF3/7→ IFN-β → caspase-11 signaling cascade that amplifies NLRP3/caspase-1 inflammasome activation (Fig. 5A) (51). However, another major role for TRIF is recruitment of RIP1 via RHIM domain interactions with consequent induction of RIP1/FADD/caspase-8 complexes. The assembly, activity, and stability of caspase-8-containing ripoptosomes are additionally modulated by association with isoforms of cFLIP (cellular FLICE inhibitory protein), a non-catalytic paralogue of caspase-8 (20). Maximal ripoptosome-associated caspase-8 activity (sufficient to drive apoptosis) is restrained via association of caspase-8 with cFLIPL (long isoform). However, the low-level ripoptosome-associated caspase-8 activity of the cFLIPL-containing heterodimeric complex is sufficient to either cleave RIP1 and/or RIP3 directly or to eliminate another process that impacts function and thereby suppresses the assembly of the necrosome complexes necessary for necroptosis. When the activity of this heterodimeric complex is completely inhibited, either by pharmacological inhibition of caspase-8 or when the complex contains cFLIPs (short isoform), the suppression of RIP1 and RIP3 is relieved; RIP1 then phosphorylates RIP3 which regulates an incompletely understood necroptotic signaling cascade (35, 39). Inhibition of RIP1 kinase activity by Nec-1 blocks this cascade. This latter necroptotic pathway was operative in our BMDC experimental model as indicated by morphology as well as the loss of DC viability in response to LPS stimulation in combination with either pan-caspase inhibition or selective caspase-8 inhibition (Fig. 3A-C). The inhibitory effects of Nec-1 on LPS + Dox or STS-induced IL-1β release illustrated in Figs. 4I and 6C further suggested that this IL-1β production involves a RIP1-regulated pathway for engagement of caspase-8. The accumulation of extracellular IL-1β was temporally correlated with caspase-8 activation in WT and Casp1/11−/− BMDC stimulated with Dox (Fig. 4J) or STS (Fig. 6A) and markedly suppressed in Casp8−/−Rip3−/− BMDC (Figs. 4D, 4E, and 6D).
There are several limitations in our experimental results and their implications regarding the possible involvement of RIP1/FADD/caspase-8 ripoptosomes as the major signaling platform for non-canonical IL-1β processing. The genetic support for caspase-8 as the alternative IL-1β converting enzyme utilized cells deficient in both caspase-8 and RIP3. Experiments with cells lacking only caspase-8 would eliminate potentially confounding effects of additional RIP3 ablation on the Dox-stimulated responses. However, unlike Casp8−/−Rip3−/− BMDC, cells deficient in only RIP3 were characterized by only modestly attenuated IL-1β processing and release in response to LPS + Dox (Figs. 4D and 4E) or LPS + STS (Fig. 6D). This suggests that the absence of caspase-8 rather than RIP3 underlies the very strong suppression of IL-1β production in Casp8−/−Rip3−/− cells. Our results also suggest stimulus-specific roles for RIP1 because Nec-1 almost completely suppressed Dox-stimulated IL-β production (Fig. 4I) but only partly attenuated the response to STS (Fig. 6C). However, an additional caveat is that Nec-1 inhibits indoleamine 2,3-dioxygenase (IDO), a known immunomodulatory enzyme (52). Finally, the isolation of RIP1, FADD, and caspase-8 in co-precipitating immune complexes would provide stronger biochemical support for ripoptosomes as an underlying signaling platform for non-canonical IL-1β processing. Studies of ripoptosome assembly have predominantly utilized cancer cells or established cell lines that may express higher levels of the interacting signaling proteins (36-38). Moreover, the accumulation of RIP1/FADD/caspase-8 ripotosomes as soluble protein complexes within the cytosol may be an intermediate state superseded by the recruitment or aggregation of such complexes into higher-order macromolecular ensembles. This possibility is supported by our observations (Fig. 5F) and those of Vince et al.(17) demonstrating the recruitment of caspase-8 into a detergent-insoluble compartment of BMDC treated with LPS + zVAD or BMDM treated with LPS + Smac mimetic, respectively. Additional studies are necessary to address how RIP1, RIP3, and cFLIP isoforms modulate the ability of various pro-apoptotic stimuli to induce specific caspase-8 signaling complexes that mediate processing of IL-1β.
Context-dependent modes of dendritic cell death triggered by doxorubicin
The differential viabilities of Casp8−/− Rip3−/−, Casp8+/+ Rip3−/−, and control Casp8+/+ Rip3+/+ BMDC treated with Dox only, LPS only, or LPS + Dox indicated that Dox may engage either a conventional caspase-9-mediated intrinsic apoptotic program in the absence of TLR4 co-stimulation or a caspase-8 mediated apoptosis in the context of TLR4 activation (Fig. 3D). The viability of Casp8−/−Rip3−/− BMDC treated with LPS + Dox, but not Dox only, was sustained thereby implicating caspase-8-dependent apoptosis as the mode of cell death in this instance. Pyroptosis was eliminated as a major mode of cell death or IL-1β release under LPS + Dox treatment conditions because similar time courses of reduced viability (Fig. 3E) and extracellular IL-1β accumulation (Fig. 2B) were observed in WT and caspase-1-deficient BMDC. Dox-induced accumulation of active executioner caspase-3/7 activity could also be dissociated from the parallel IL-1β processing response to this pro-apoptotic agent (Figs. 2F and 2G). Notably, mature IL-1β rather than proIL-1β was the predominant form in the extracellular medium of cells treated with LPS + Dox (Fig. 1E). This indicated that the production of mature IL-1β and its release from the DCs was tightly coordinated with the decrease in cell viability orchestrated by apoptotic signaling. In contrast, induction of necroptosis by LPS + zVAD or LPS + IETD resulted in a massive release of proIL-1β that was suppressed by Nec-1 (Fig. 3A).
Relevance to the anti-tumor chemotherapeutic and immunogenic actions of doxorubicin
It will be important to determine whether doxorubicin and related anthracyclines can engage this caspase-8 mediated pathway for IL-1β production under in vivo conditions wherein these agents exert their anti-tumor chemotherapeutic and immunogenic actions. In this regard, two questions are particularly germane. First, might dendritic cells or macrophages within tumor loci be exposed to the micromolar doxorubicin concentrations that trigger this signaling cascade in cell culture conditions? Both the tumor cells and immune cells within tumor sites can be exposed to high local concentrations of chemotherapy drugs depending on the method and site of drug delivery. There is increasing development of new modes of doxorubicin therapy that involve tumor-directed delivery via encapsulation within liposomes or conjugation with nanoparticles and antibody complexes (53, 54). Such particulate-based therapies can result in phagocytosis of Dox-containing liposomes or conjugates by tumor-resident DCs and macrophages. The second question concerns possible in vivo mediators within the tumor microenvironment that might act as TLR4 agonists to facilitate the TRIF-dependent signals that are also required for caspase-8 activation. Several studies have identified HMGB1 released from dying tumor cells or other host cells as a relevant DAMP agonist for TLR4 that supports activation of IL-1β processing and release (12, 55-58). Moreover, advanced stages of cancer progression or cytotoxic cancer therapies can result in compromised barrier function of the gut epithelia and increased circulating levels of PAMPs derived from commensal bacteria (59, 60). This study further validates the importance of exploring the direct effects of pro-apopoptic chemotherapeutic drugs on IL-1β production by tumor-resident immune cells in addition to previously described models wherein other DAMPs, such as ATP, released from dying chemotherapy-treated tumor cells initiate canonical inflammasome signaling (61). Moreover, the overall consequences of the myeloid-driven IL-1β production response to various cancer chemotherapy drugs may vary with the stage of tumor progression (62). For example, IL-1β production elicited in the early stages of cancer therapy may be advantageous to the tumor-bearing host in initiating an IL-1β-dependent immunogenic anti-tumor response (12), but at more advanced stages, may contribute to tumor growth (13). Overall, better understanding of the relative contributions of various IL-1β processing pathways elicited by cancer chemotherapeutics may aid in the development of refined therapeutic approaches that limit tumor progression.
Supplementary Material
Acknowledgements
We thank Eric Pearlman and Mausita Karmakar for providing Asc−/−, Trif−/−, and Nlrp3−/− Nlrc4−/− double-knockout mouse strains.
Footnotes
Grant support: NIH-R01-GM36387 (GRD); NIH-R01-AI020211 (ESM); OD012198 (WJK)
References
- 1.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Metivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, Zitvogel L, Kroemer G. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, Mendiboure J, Pignon JP, Jooste V, van Endert P, Ducreux M, Zitvogel L, Piard F, Kroemer G. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 3.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat. Immunol. 2012;13:333–332. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994;269:15195–15203. [PubMed] [Google Scholar]
- 6.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 7.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 9.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Mol. Cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S, Kirov A, Prudovsky I, Swanton E, Brough D. Deubiquitinases regulate the activity of caspase-1 and interleukin-1beta secretion via assembly of the inflammasome. J. Biol. Chem. 2013;288:2721–2733. doi: 10.1074/jbc.M112.422238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 13.Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, Kanellopoulos J, Martin F, Rebe C, Apetoh L, Ghiringhelli F. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013;19:57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- 14.Sauter KAD, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1 beta through activation of the NLRP3 inflammasome. Cancer Biol. Ther. 2011;11:1008–1016. doi: 10.4161/cbt.11.12.15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TB. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012;13:246–254. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 16.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J. Exp. Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vince JE, Wong WWL, Gentle I, Lawlor KE, Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, Hacker G, Silke J, Tschopp J. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, Mocarski ES, Subramanian D, Green DR, Silverman N, Fitzgerald KA, Marshak-Rothstein A, Latz E. Cutting Edge: FAS (CD95) Mediates Noncanonical IL-1beta and IL-18 Maturation via Caspase-8 in an RIP3-Independent Manner. J. Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchiyama R, Yonehara S, Tsutsui H. Fas-mediated inflammatory response in Listeria monocytogenes infection. J. Immunol. 2013;190:4245–4254. doi: 10.4049/jimmunol.1203059. [DOI] [PubMed] [Google Scholar]
- 20.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 2012;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu Y, Ramachandra L, Mohr S, Franchi L, Harding CV, Nunez G, Dubyak GR. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J. Immunol. 2009;182:5052–5062. doi: 10.4049/jimmunol.0802968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu JF, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–U146. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 23.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 24.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 2002;296:1873–1876. doi: 10.1126/science.1071065. [DOI] [PubMed] [Google Scholar]
- 26.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 27.Gescher A. Staurosporine analogues - pharmacological toys or useful antitumour agents? Crit. Rev. Oncol. Hematol. 2000;34:127–135. doi: 10.1016/s1040-8428(00)00058-5. [DOI] [PubMed] [Google Scholar]
- 28.Marti GE, Stetler-Stevenson M, Grant ND, White T, Figg WD, Tohnya T, Jaffe ES, Dunleavy K, Janik JE, Steinberg SM, Wilson WH. Phase I trial of 7-hydroxystaurosporine and fludararbine phosphate: in vivo evidence of 7-hydroxystaurosporine induced apoptosis in chronic lymphocytic leukemia. Leuk. Lymphoma. 2011;52:2284–2292. doi: 10.3109/10428194.2011.589547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jimeno A, Rudek MA, Purcell T, Laheru DA, Messersmith WA, Dancey J, Carducci MA, Baker SD, Hidalgo M, Donehower RC. Phase I and pharmacokinetic study of UCN-01 in combination with irinotecan in patients with solid tumors. Cancer Chemother. Pharmacol. 2008;61:423–433. doi: 10.1007/s00280-007-0485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang YS, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X(7) receptor differentially couples to distinct release pathways for IL-1 beta in mouse macrophage. J. Immunol. 2008;180:7147–7157. doi: 10.4049/jimmunol.180.11.7147. [DOI] [PubMed] [Google Scholar]
- 32.Verhoef PA, Kertesy SB, Lundberg K, Kahlenberg JM, Dubyak GR. Inhibitory effects of chloride on the activation of caspase-1, IL-1 beta secretion, and cytolysis by the P2X7 receptor. J. Immunol. 2005;175:7623–7634. doi: 10.4049/jimmunol.175.11.7623. [DOI] [PubMed] [Google Scholar]
- 33.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SO, Ono K, Han J. Apoptosis by pan-caspase inhibitors in lipopolysaccharide-activated macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;281:L1095–1105. doi: 10.1152/ajplung.2001.281.5.L1095. [DOI] [PubMed] [Google Scholar]
- 35.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 2005;174:4942–4952. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 37.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frelin C, Imbert V, Bottero V, Gonthier N, Samraj AK, Schulze-Osthoff K, Auberger P, Courtois G, Peyron JF. Inhibition of the NF-kappaB survival pathway via caspase-dependent cleavage of the IKK complex scaffold protein and NF-kappaB essential modulator NEMO. Cell Death Differ. 2008;15:152–160. doi: 10.1038/sj.cdd.4402240. [DOI] [PubMed] [Google Scholar]
- 42.Dudgeon C, Wang P, Sun X, Peng R, Sun Q, Yu J, Zhang L. PUMA induction by FoxO3a mediates the anticancer activities of the broad-range kinase inhibitor UCN-01. Mol. Cancer Ther. 2010;9:2893–2902. doi: 10.1158/1535-7163.MCT-10-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manns J, Daubrawa M, Driessen S, Paasch F, Hoffmann N, Loffler A, Lauber K, Dieterle A, Alers S, Iftner T, Schulze-Osthoff K, Stork B, Wesselborg S. Triggering of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine: activation of caspase-9 in the absence of Apaf-1. FASEB J. 2011;25:3250–3261. doi: 10.1096/fj.10-177527. [DOI] [PubMed] [Google Scholar]
- 44.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013 doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, Henry T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012;19:1709–1721. doi: 10.1038/cdd.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 47.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 48.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 49.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 50.Labbe K, McIntire CR, Doiron K, Leblanc PM, Saleh M. Cellular Inhibitors of Apoptosis Proteins cIAP1 and cIAP2 Are Required for Efficient Caspase-1 Activation by the Inflammasome. Immunity. 2011;35:897–907. doi: 10.1016/j.immuni.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 51.Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barenholz Y. Doxil(R)--the first FDA-approved nano-drug: lessons learned. J. Control. Release. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 54.Ali I, Rahis U, Salim K, Rather MA, Wani WA, Haque A. Advances in nano drugs for cancer chemotherapy. Curr. Cancer Drug Targets. 2011;11:135–146. doi: 10.2174/156800911794328493. [DOI] [PubMed] [Google Scholar]
- 55.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, Andre F, Tursz T, Kroemer G, Zitvogel L. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 56.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brenchley JM, Douek DC. Paul WE, editor. Microbial Translocation Across the GI Tract. Annu. Rev. Immunol. Vol 30. 2012:149–173. doi: 10.1146/annurev-immunol-020711-075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Vliet MJ, Harmsen HJ, de Bont ES, Tissing WJ. The role of intestinal microbiota in the development and severity of chemotherapy-induced mucositis. PLoS Pathog. 2010;6:e1000879. doi: 10.1371/journal.ppat.1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 62.Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012;13:343–351. doi: 10.1038/ni.2224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.