

Abstract
AIM: To determine the molecular mechanisms involved in experimental hepatic fibrosis prevention by caffeine (CFA).
METHODS: Liver fibrosis was induced in Wistar rats by intraperitoneal thioacetamide or bile duct ligation and they were concomitantly treated with CFA (15 mg/kg per day). Fibrosis and inflammatory cell infiltrate were evaluated and classified by Knodell index. Inflammatory infiltrate was quantified by immunohistochemistry (anti-CD11b). Gene expression was analyzed by quantitative reverse transcription-polymerase chain reaction for collagen I (Col-1), connective tissue growth factor (CTGF), transforming growth factor β1 (TGF-β1), tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), IL-6, superoxide dismutase (SOD) and catalase (CAT). Activation of Nrf2 and Snail-1 was analyzed by Western-blot. TNF-α expression was proved by enzyme-linked immunosorbant assay, CAT activity was performed by zymography.
RESULTS: CFA treatment diminished fibrosis index in treated animals. The Knodell index showed both lower fibrosis and necroinflammation. Expression of profibrogenic genes CTGF, Col-1 and TGF-β1 and proinflammatory genes TNF-α, IL-6 and IL-1 was substantially diminished with CFA treatment with less CD11b positive areas. Significantly lower values of transcriptional factor Snail-1 were detected in CFA treated rats compared with cirrhotic rats without treatment; in contrast Nrf2 was increased in the presence of CFA. Expression of SOD and CAT was greater in animals treated with CFA showing a strong correlation between mRNA expression and enzyme activity.
CONCLUSION: Our results suggest that CFA inhibits the transcriptional factor Snail-1, down-regulating profibrogenic genes, and activates Nrf2 inducing antioxidant enzymes system, preventing inflammation and fibrosis.
Keywords: Liver fibrosis, Caffeine, Thioacetamide, Bile duct ligation, Profibrogenic genes, Proinflammatory cytokines, Antioxidant enzymes
Core tip: This paper shows the protective effect of caffeine in the liver to the constant aggressiveness of a hepatotoxic. Here we present evidence not published before of some molecular mechanisms like inhibition of Snail-1 and activation of Nrf2 that could be involved in this beneficial effect down-regulating pro-fibrogenic genes and up-regulating antioxidant molecules.
INTRODUCTION
The liver performs essential functions in the body[1]. Hepatic stellate cells (HSC) are key in the fibrogenic process[2]. After stimulation of liver damage, HSC undergo a process called “activation”; characterized by synthesis of type I and III collagens[3-5]. This state of activation is maintained by growth factors such as transforming growth factor β1 (TGF-β1)[6], connective tissue growth factor (CTGF)[7], and pro-inflammatory molecules such as tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), interleukin 6 (IL-6)[8] and reactive oxygen species (ROS).
Epidemiological studies had associated caffeine (CFA) consumption with protection against development of chronic liver disease or reduction of disease severity[9-11]. In vitro studies have shown beneficial effects of CFA, that can be useful in preventing HSC activation and perpetuation of this state[12,13]. Among CFA effects observed in vitro are: inhibition of expression of CTGF[14-16]; reduction of pro-inflammatory cytokines expression such as TNF-α, IL-1 and IL-6 by mechanisms not yet defined[8], and CFA antioxidant effect[17-21].
On the other hand, a potent natural antioxidant, quercetin increases the transcriptional and translational activity of the transcriptional factor Nrf2 which has potent antioxidant activity[22].
Activation of HSC is a complex process where the transcriptional factor Snail-1 has an important role. Several authors have reported the overexpression of Snail-1 in pathological conditions associated with extracellular matrix (ECM) deposition[23,24]. Snail-1 expression has been shown in cholangiocytes and hepatocytes of fibrotic livers[14,16], and recently, Snail-1 has been published as a central transcription factor on the activation of HSC demonstrating its essential role in regulating the liver fibrosis process[25].
According to our findings, CFA-mediated molecular mechanisms comprise in part down-regulation of profibrogenic genes, diminishing of inflammatory cell infiltrate, down-regulation of pro-inflammatory cytokines, and up-regulation of antioxidant enzymes. Our results suggest that these events could be mediated, at least in part, by Nrf2 activation and inhibition of Snail-1 which are key factors in the development of this process.
MATERIALS AND METHODS
Materials
CFA was acquired from Sigma Aldrich Co., (St Louis Missouri). Thioacetamide (TAA) was purchased from Merck Company, (Darmstadt, Germany). CD11b antibody was obtained from Biolegend (San Diego, CA, United States). Biotinylated secondary antibody and avidin-conjugated peroxidase were obtained from Vector Laboratories (Burlingame, CA, United States).
DuoSet enzyme-linked immunosorbant assay (ELISA) Development kit was acquired from R and D Systems, (Minneapolis, United States). Primers and probes to design real time polymerase chain reaction (PCR) were acquired from Applied Biosystems (Hammonton, NJ, United States). Poly vinylidene fluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules CA, United States). Nrf2, Snail-1 and secondary antibodies were purchased from Avcam Inc (Cambridge MA, United States).
Animals and experimental design
Wistar rats used in this study were obtained from Charles Rivers (Boston, MA, United States) and housed according to the Animal Care protocol established by University of Guadalajara. Thirty male Wistar rats, weighing 250-280 g were divided into three groups (10 rats in each group) as follows: (1) healthy (n = 10); (2) TAA (n = 10), rats with intraperitoneal TAA to develop liver fibrosis; and (3) bile duct ligation (BDL) (n = 20), rats that underwent a laparotomy and BDL. Finally 5 rats of each group were treated with CFA and other 5 rats received vehicle only (fibrotic rats).
CFA administration in TAA-intoxicated and BDL rats
Two in vivo models were intended to assess fibrosis prevention via CFA administration, TAA and BDL. TAA-induced fibrosis was achieved using a dose of 200 mg/kg administrated intraperitoneally 3 times a week for 7 wk, as described previously[26-28]. BDL-induced fibrosis was achieved under general anesthesia and laparotomy was made, the common bile duct was localized, doubly ligated and cut between these two ligatures[29]. CFA administration was carried out concomitantly with BDL and TAA intoxication regimen once a day with a dose of 15 mg/kg by the orogastric route. Rats sacrifice was performed at the seventh week for the TAA model, and at the fourth week for the BDL model. Representative liver sections were excised and either fixed with 4% buffered paraformaldehyde for histological examination, or frozen for RNA and protein extraction.
Biochemical assays
Blood was obtained from animals immediately before sacrifice, and serum transaminases, alanine transaminase (ALT) and aspartate transaminase (AST), were determined in automated Vitros DT 60 equipment (Johnson and Johnson, New Jersey, United States).
Histological examination of liver sections
For histological studies, livers were removed and fixed by immersion in 4% paraformaldehyde diluted in PBS, dehydrated in graded ethylic alcohol, and embedded in paraffin.
Assessment of liver inflammatory activity and fibrosis: The Modified Histological Activity Index of Knodell was used to grade the severity of the necroinflammatory process (0-18 scale) and fibrosis (0-6 scale), and was performed blindly by two experienced pathologists[30-32]. Additionally, liver fibrosis was also quantitatively assessed by Masson’s trichromic staining in 4-m liver sections by light microscopy as described previously[33,34] using a computer-assisted morphometric analyzer (Image-ProPlus 6.0; Media Cybernetics, Inc., Bethesda, MD, United States) by analyzing ten random fields per slide and calculating the ratio of connective tissue to the whole liver area, expressed as fibrosis percentage.
Immunohistochemical determination of CD11b: Hepatic tissue sections were deparaffinized and rehydrated with xylene and decreasing graded ethanol. Slides were incubated in 3% H2O2 for 30 min, followed by incubation with polyclonal anti-rat against purified CD11b/c (Biolegend, Cat. No. 201801, San Diego, CA, United States) diluted in PBS (1:100).
The primary antibody was incubated at 4 °C overnight, followed by incubation with biotinylated secondary antibody (Vectastain, Universal Quick Kit, Cat. No. PK-8800). Secondary antibodies were complexed individually with avidin-conjugated peroxidase Vectastain ABC-Elite reagent (Vector Laboratories, Burlingame, CA, United States) and resulting peroxidase activity was detected with 3,30-diaminobenzidine in sections that were briefly counterstained with hematoxylin. Positive areas were analyzed in 20 random fields of pericentral, mid-zonal and periportal areas. Counting was carried out using automated software (Image-Pro plus Analyzer, Qwin-Leica, United States). Results were expressed as a percentage of the positive area.
ELISA assay for TNF-α
Liver tissue was homogenized with Polytron (Janke Kunkel IKA-WERK, Staufen im Breisgau, Germany) and centrifugated at 4 °C for 4 min at 12000 g in lysis buffer with protease inhibitors [50 mmol/L Tris (hydroxymethyl) aminomethane-HCI buffer, pH 7.4, containing 0.02% sodium azide, 150 mmol/L NaCl, 0.1% Tween-20, 150 mmol/L NaCl, 10 g/mL aprotinin, 5 g/mL pepstatin, 5 g/mL leupeptin, 1 mmol/L phenyl-methylsulfonyl fluoride and 25 g/mL E64][35].
Protein concentration of cleared tissue lysates were determined by Bradford method. After quantitation samples were stored at -80 °C until analysis.
We used the kit DuoSet for ELISA for rat TNF-α/TNFSF1A (DuoSet ELISA Development kit, rat TNF-α Cat. No. DY510, R and D Systems, Minneapolis, United States), following the protocol provided by the manufacturer. Finally, the reaction was stopped and the optical density of each well was determined at 450 nm.
Quantitative real-time reverse transcriptase-PCR
RNA was isolated from the liver from different groups of rats with Trizol reagent (Invitrogen, Carlsbad, CA, United States)[36]. Retrotranscription using 2 g of total RNA was achieved using moloney-murine leukemia virus reverse transcriptase (Invitrogen). Then, 2 μL of cDNAs were subjected to real-time PCR using a Rotor Gene Termocycler under the following conditions: 2 min at 50 °C, 5 min at 94 °C, and 45 cycles of 30 s at 94 °C and 40 s at 60 °C. Specific primers and probes designed to align in collagen α1 (I), CTGF, TGF-β1, TNF-α, IL-1, IL-6, superoxide dismutase (SOD) and catalase (CAT) rat RNAs were acquired from Applied Biosystems (Hammonton, NJ, United States). Gene amplification was normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. Relative quantification by the 2-∆∆CT method was realized by comparing to control groups as an internal calibrator[37,38]. Expression gene levels are shown as expression relative units.
Catalase activity
Reported CAT activity was determined according to the zymographic method by Gennady P Manchenko (1994). The method is based on the starch-iodine reaction. Thiosulfate in the staining solution is inactivated by hydrogen peroxide except at the sites of CAT activity, where hydrogen peroxide is destroyed enzymatically. The iodide is oxidized by hydrogen peroxide to iodine which forms a chromatophore with the starch and sites of CAT localization remain achromatic[29].
Western-blot assays
Western blot assays of tissue homogenates were performed to analyze the activation of Nrf-2 and Snail-1. Proteins were extracted from 100 mg of liver tissue using lysis buffer (50 mmol/L Tris-HCl pH 8.0, 150 nmol/L NaCl, 0.02% NaN3). After centrifugation at 13000 rpm/5 min/4 °C, supernatant was collected and quantified by Bradford assay. Briefly, 30 μg of total proteins were separated by 10% sodium dodecyl sulfate psulfate polyacrylamide gel electrophoresis under reducing conditions and transferred to PVDF membranes (Bio-Rad Laboratories, Hercules CA, United States). Blocking was carried out using 3% dry milk for 2 h; primary antibody dilution was 1:500 for GAPDH (loading control) and 1:800 for Nrf2 and Snail-1 antibodies. (Abcam Biotechnology, Santa Cruz CA, United States). Antibody binding was revealed with a secondary anti-antibody diluted 1:5000-1:6000 using BM Chemiluminiscence kit (Roche Diagnostics, Indianapolis IN, United States). Densitometric analysis was realized with a Kodak 1D 3.5 Image analyzer (Eastman Kodak Co., Rochester NY) GAPDH was used as a cell fractionation control.
Statistical analysis
Normally distributed data were analyzed using t test, where statistical significance was P < 0.05. Data are shown as the mean ± SD. For real-time PCR experiments, results are shown as the 2-∆∆CT value (mean ± SD), where the standard deviation was calculated as: s = [s(GAPDH)2 + s(target gene)2]1/2, according to user bulletin 2 from Applied Biosystems.
RESULTS
CFA prevents weight loss in TAA-intoxicated rats
Basal weight and weight at the end of treatment were registered in all groups. As shown in Table 1, CFA prevented weight loss of rats in the TAA model, which suggested that CFA had an effect in improving the nutritional status of rats measured solely by weight.
Table 1.
Weight at the beginning of the treatment and after caffeine treatment, serum markers enzymes in bile duct ligation and thioacetamide-intoxicated rats
| Group | Healthy | TAA | TAA + CFA | BDL | BDL + CFA |
| Rats weight at the beginning of the treatment (g) | 296.0 ± 9.73 | 284.0 ± 22.1 | 282.2 ± 9.1 | 280.1 ± 18.3 | 275.0 ± 19.0 |
| Rats weight after CFA treatment (g) | 321.0 ± 8.5 | 223.1 ± 14.0b | 253.7 ± 5.7 | 219.4 ± 15.3 | 221.0 ± 17.5 |
| AST (U/L) | 226.0 ± 61.0 | 379.7 ± 179.8 | 318.2 ± 144.3 | 576.7 ± 70.0 | 329.5 ± 41.4c |
| ALT (U/L) | 63.0 ± 1.0 | 132.7 ± 7.5 | 98.2 ± 28.7 | 214.7 ± 37.0 | 76.0 ± 10.6d |
Treatment duration: 7 wk for thioacetamide (TAA) and 4 wk for bile duct ligation (BDL).
P < 0.01 vs TAA group;
P < 0.05,
P < 0.01 vs BDL group. Data are shown as the mean ± SD (n = 10). CFA: Caffeine; AST: Aspartate transaminase; ALT: Alanine transaminase.
CFA dosed groups had less hepatocellular damage
AST and ALT levels were higher in TAA-intoxicated (1.7- and 2.1-fold respectively) and BDL (2.6- and 3.4-fold respectively) groups compared with the healthy rats group. The BDL + CFA group showed lower levels in AST compared to the BDL group (1.8-fold) (P < 0.05). The TAA + CFA group only showed a tendency to lower levels. Similarly, ALT levels in the BDL + CFA group were lower (2.8-fold) when compared against the BDL group (P < 0.01) (Table 1).
CFA treatment reduced both BDL and TAA-induced liver fibrosis
To test the antifibrogenic effect of CFA, morphological analysis of liver sections stained with Masson’s was performed. Looking at the histology of the healthy group, we observed a normal morphology, with scarce ECM and hepatocytes arranged in a radial pattern. Histology of TAA and BDL groups showed an altered morphology, with thick collagen bundles, much more noticeable in the BDL group. In contrast, the treated groups TAA + CFA and BDL + CFA showed lower ECM content (Figure 1A). Quantification of ECM demonstrated a potent antifibrogenic effect of CFA. In the TAA + CFA group, fibrosis was lower by 80% compared to the TAA group (P < 0.0001). Likewise, in the BDL + CFA group fibrosis was lower by 38% compared to the BDL group (P < 0.0001) (Figure 1B).
Figure 1.
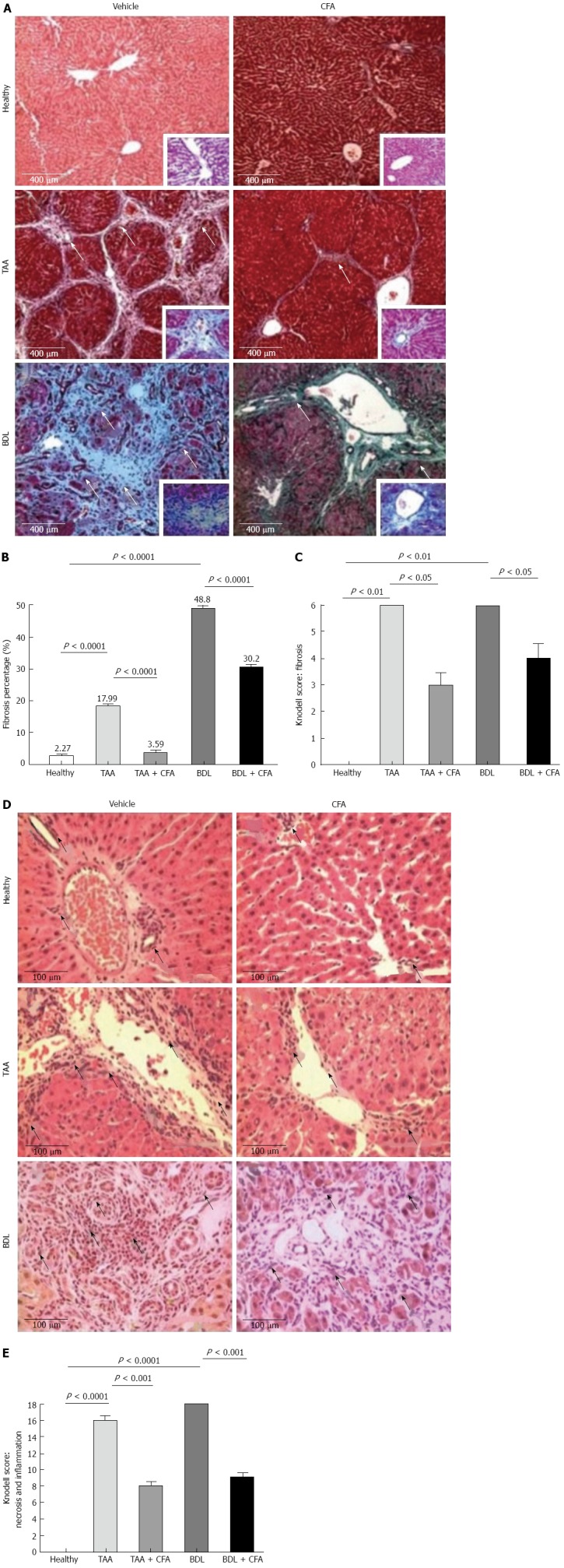
Macroscopic and histological differences and fibrosis index with caffeine treatment. A: Liver fibrosis percentage of healthy rats, caffeine (CFA)-treated, thioacetamide (TAA)-intoxicated, TAA-intoxicated treated with CFA, bile duct ligation (BDL) rats, BDL rats treated with CFA. Sections, 4 μm thick, stained with Masson’s trichrome, × 10. White arrows show the extracellular matrix (ECM) (fibrosis); B: Fibrosis quantification. Fibrosis percentages are shown, they were obtained by computer-assisted morphometric analysis (Software Image pro plus 6.3); C: Knodell Index for fibrosis, sections 4 μm thick, stained with Masson’s trichrome, × 10; D: Inflammatory infiltrate amount. Sections 4 μm thick, stained with hematoxylin and eosin, × 40. Black arrows show inflammatory cells; E: Knodell Index for fibrosis, sections 4 μm thick, stained with hematoxylin and eosin, × 40.
The Knodell score indicated lower fibrosis in the TAA + CFA and BDL + CFA groups (3 ± 0.5 and 4 ± 0.5 points respectively) compared with the TAA and BDL cirrhotic groups (6 ± 0 and 6 ± 0 points respectively) (both P < 0.05) (Figure 1C).
Fewer inflammatory cells infiltrate in CFA groups
It was noted that CFA groups had a low amount of inflammatory infiltrate. TAA and BDL groups had a large number of inflammatory cells, especially the BDL group. In contrast, both cirrhotic groups treated with CFA had a lower amount of inflammatory infiltrate, more evident in the TAA + CFA group (Figure 1D). The Knodell score resulted in lower necroinflammation in the treated groups, TAA + CFA and BDL + CFA (8 ± 0.5 and 9 ± 0.5 points), compared with the cirrhotic groups, TAA and BDL (16 ± 0 and 18 ± 0 points) (both P < 0.001) (Figure 1E).
Fibrogenic genes expression decrease with CFA treatment even in the continuous presence of liver fibrosis inducers
In addition to the histologic analysis we analyzed expression of the fibrogenic genes.
As expected, cirrhotic groups showed an increase in fibrogenic genes expression. In the TAA group there was a 5.3-fold increase for CTGF (P < 0.01), a 10.5-fold increase for collagen I (Col-1) (P < 0.01) and and a 4.3-fold increase for TGF-α1 (P < 0.05). In the BDL group fibrogenic genes expression also showed an increase; this increase was 11.6-fold for CTGF (P < 0.01), 21.5-fold for Col-1 and 3.5-fold for TGF-α1 (P < 0.05), compared with healthy rat group levels (Figure 2A-C). Treatment with CFA also induced a lower expression of fibrogenic genes; in the TAA + CFA group this was 3.5-fold lower for CTGF (P < 0.01), 3.5-fold lower for Col-1 (P < 0.05) and 3.1-fold lower for TGF-β1 (P < 0.01) compared with the TAA group. In the BDL+CFA group the reduction in gene expression was 5.0-fold lower for CTGF (P < 0.01), 3.0-fold lower for Col-1 (P < 0.01), and 1.5-fold lower for TGF-β1, indicating only a declining trend but no statistical significance, compared with BDL group (Figure 2A-C).
Figure 2.
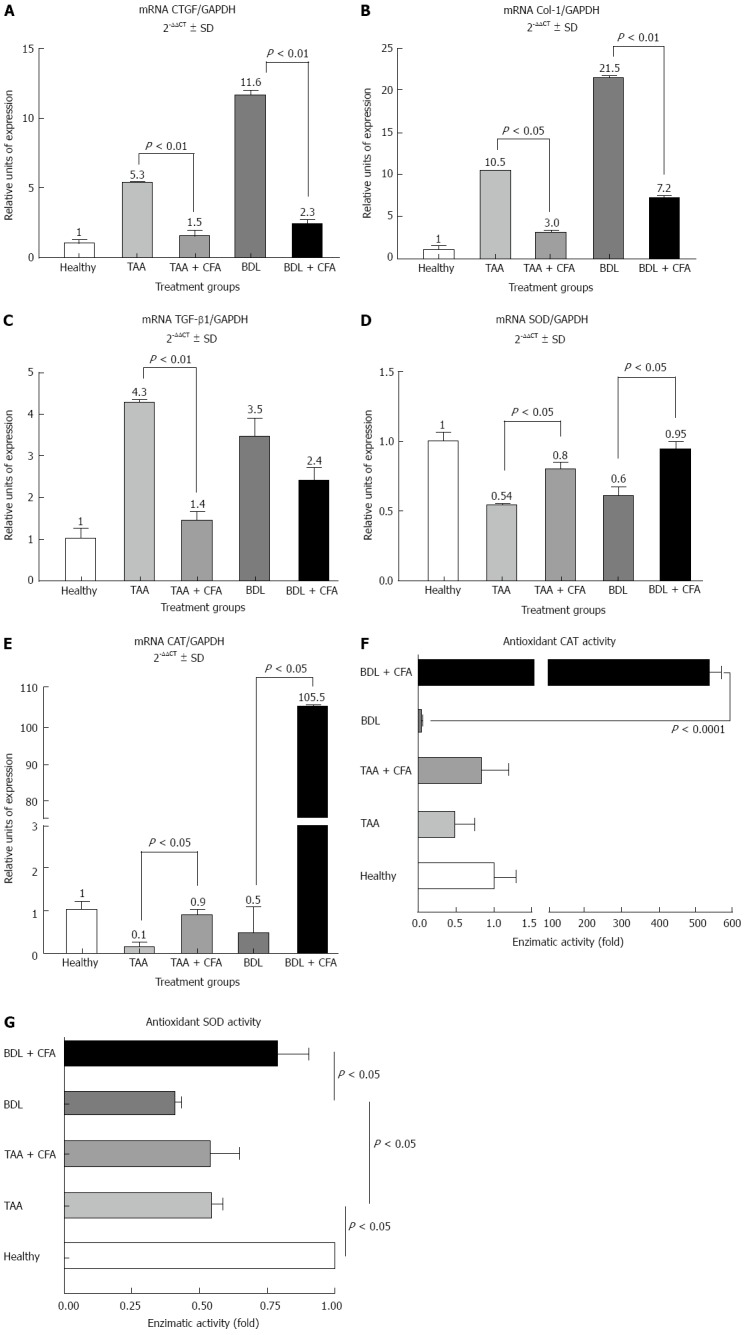
Expression of fibrogenic and antioxidant genes in liver. Reverse transcription-polymerase chain reactions were performed for connective tissue growth factor (CTGF) (A), collagen I (Col-1) (B), and transforming growth factor β1 (TGF-β1) (C), superoxide dismutase (SOD) (D) and catalase (CAT) (E). Gene amplification was normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. CAT (F) and SOD antioxidant activity (G) were analyzed by zymography in acrylamide gels. TAA: Thioacetamide; BDL: Bile duct ligation; CFA: Caffeine.
CFA limits pro-inflammatory genes expression in experimental liver fibrosis models
We performed an immunohistochemical determination of CD11b in hepatic tissue sections. We observed that CD11b positive areas in the TAA + CFA versus the TAA group were lower by 65.5% (P < 0.01), and the BDL group treated with CFA versus the BDL group were lower by 60.8% (P < 0.05) (Figure 3A). In addition to testing the anti-inflammatory effect of CFA at the protein level, we analyzed TNF-α expression by ELISA. Both liver cirrhotic groups showed an increase in TNF-α levels, 560.2 ± 67.8 pg/mL (P < 0.0001) for the TAA group, and 590.3 ± 71.3 pg/mL (P < 0.0001) for the BDL group, compared with healthy rat group levels (140.4 ± 3.4 pg/mL). We observed lower levels in the CFA treated groups; for the TAA + CFA group 313.1 ± 56.6 pg/mL (P < 0.01), and for the BDL + CFA group 420.6 ± 166.1 pg/mL (Figure 3B).
Figure 3.
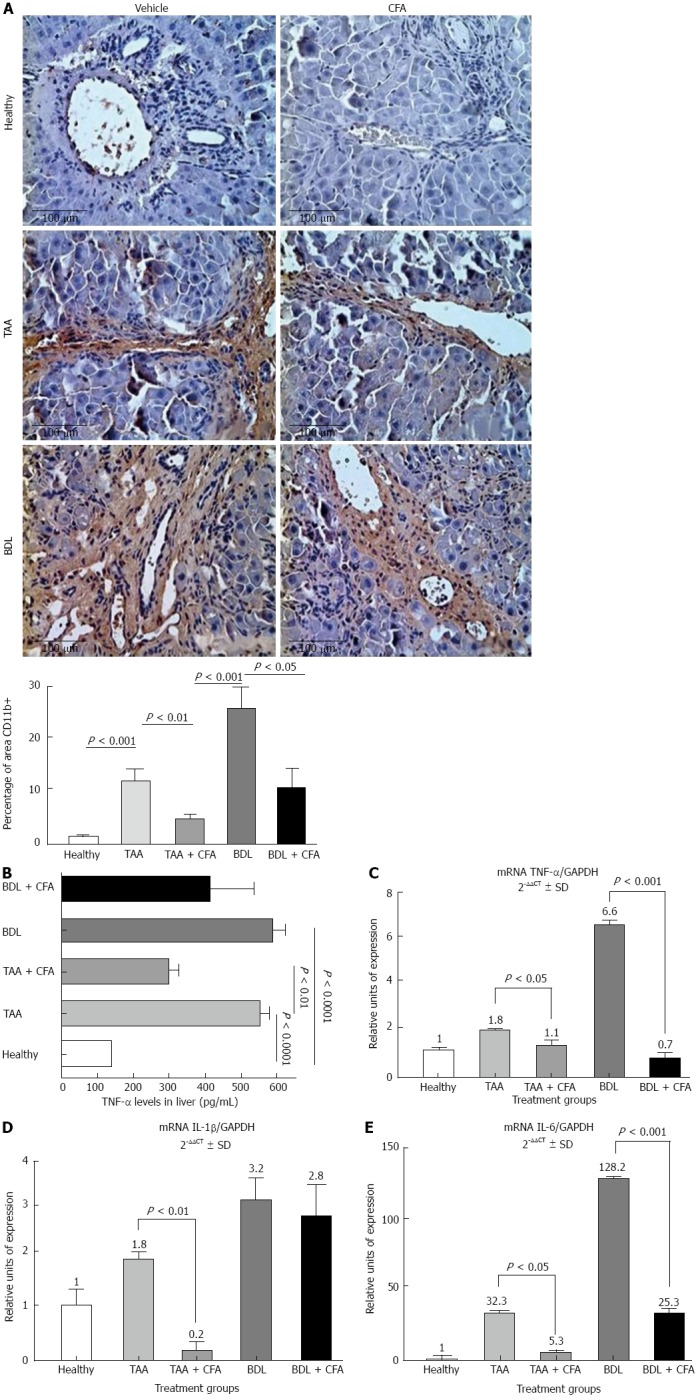
Expression of inflammatory genes in liver. A: Immunohistochemistry for CD11b, sections 4 μm thick, stained with Masson’s trichrome, × 40. Results for CD11b positive area are shown as percentage; B: Tumor necrosis factor alpha (TNF-α) liver levels in different groups of treatment, performed by enzyme-linked immunosorbant assay; C-E: Reverse transcription-polymerase chain reaction were performed for TNF-α, interleukin-1β (IL-1β), IL-6. Gene amplification was normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. TAA: Thioacetamide; BDL: Bile duct ligation. CFA: Caffeine.
Then, we analyzed at the molecular level the pro-inflammatory genes expression of TNF-α, IL-1β and IL-6. Both cirrhotic groups showed a significant increase in all proinflammatory genes expression; in TAA group this was 1.8-fold for TNF-α (P < 0.05), 1.8-fold for IL-1 (P < 0.05) and 32.3-fold for IL-6 (P < 0.001). In the BDL group it was 6.6-fold for TNF-α (P < 0.001), 3.2-fold for IL-1β (P < 0.01) and 128.2-fold for IL-6 (P < 0.0001) compared with the healthy rats group (Figure 3C-E). In groups treated with CFA we observed a decrease of expression; in the TAA + CFA group this was 1.6-fold for TNF-α (P < 0.05), 9-fold for IL-1β (P < 0.01); and 6.1-fold for IL-6 (P < 0.05); and in the BDL + CFA group there was a decrease of 9.4-fold for TNF-α (P < 0.001), 1.1-fold for IL-1 and 5.1-fold for IL-6 (P < 0.001) (Figure 3C-E).
Antioxidant enzymes gene expression and activity is modified by CFA intake
It is known that both liver fibrosis models course with an oxidative stress state. Thus, antioxidant enzymes expression levels were analyzed. We noticed that hepatocellular expression of SOD increased 1.5 (P < 0.05) and 1.6 (P < 0.05)-fold in TAA and BDL models, respectively, when they received CFA (Figure 2D). Likewise, CAT enzyme expression was significantly increased, showing an increase of 1.5-fold (P < 0.05) in the TAA model, and an increase of 211-fold (P < 0.05) in the BDL model (Figure 2E). To explore this last effect we performed an assay to measure SOD and CAT antioxidant activities, where we found a strong correlation between mRNA expression and enzyme activity; in the BDL + CFA group antioxidant CAT activity was significantly increased (535-fold) (P < 0.0001) (Figure 2F) and SOD activity increased twice compared with BDL group (Figure 2G).
Activity of Snai-1 and Nrf2 by Western blot
Protein levels of the antioxidant transcription factor Nrf2 were significantly higher in both animal models compared to healthy rats. Treatment with CFA in liver-injured rats increased these levels significantly in both BDL and TAA models (Figure 4A). This increase suggests that Nrf2 could be inducing SOD and CAT expression, thus preventing liver damage.
Figure 4.
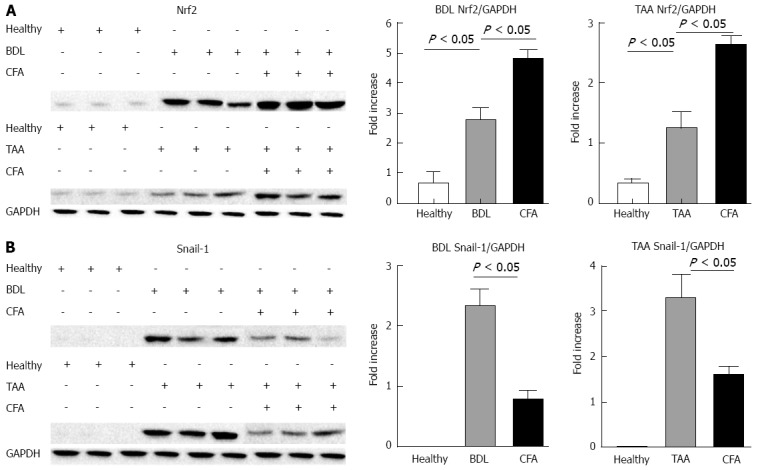
Expression of transcriptional factors. Western-blots were performed for the transcriptional factor Nrf2 (A) and Snail-1 (B). Densitometric values were normalized against the constitutive protein glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and represented like fold increase respect to healthy animals. TAA: Thioacetamide; BDL: Bile duct ligation. CFA: Caffeine.
On the other hand, the pro-fibrogenic transcription factor Snail-1 reduced its protein levels when the animals were treated with CFA in both animal models. These values were 2.33 and 3.25 times higher than healthy animals for BDL and TAA respectively, where animals treated with CFA presented values only of 0.77 and 1.58 times higher with respect to healthy animals (Figure 4B).
DISCUSSION
There are epidemiological data indicating that consumption of CFA protects against development of chronic liver disease or reduces the severity of the disease[12-14]. In vitro studies have shown beneficial effects of CFA useful in preventing HSC activation and perpetuation of this state[15,16]. Although there is a recent preliminary report describing the effect of coffee on liver fibrosis[39], here we describe a more comprehensible mechanism for CFA action on the most important molecules implicated in liver fibrosis. Our experiments were designed to compare CFA effects in two experimental liver fibrosis models, BDL and chronic TAA intoxication, to test whether the preventive effects of CFA were independent of the etiology of liver damage. Dose was chosen based on positive effects of CFA regarding liver disease in epidemiological studies. These studies suggested a CFA consumption of 274 mg/d approximately in humans (2 cups of coffee)[12,40-43]. In our study we used a CFA dose of 15 mg/kg per day, because it is known that rat metabolism is approximately 10 times more accelerated than humans[42,43]. This animal dose is translated to a human dose using the body surface area (calculated with Du Bois formula)[44-47], and an adjustment of rat km (9) to human km (41), resulting in a human dose of 2.4 mg/kg (1.5 cups of coffee)[46], and expecting the same beneficial effects observed in the animal model and diminishing the probability of secondary effects.
It was observed that TAA-intoxicated rats treated with CFA had a higher body weight indicating overall improvement, probably because CFA reduces liver damage (as seen in histological analysis) and thus prevents the loss of appetite. Liver metabolic functions could be less altered, indicating that BDL and liver damage are the most important factors in weight loss.
In a previous experimental study of acute liver damage induced by a single dose of D-galactosamine/lipopolysaccharide, CFA pretreatment correlated with lower levels of AST and ALT. Also, it has been reported in an animal model of liver damage with alcohol that transaminase levels are diminished by the effect of CFA. However these experimental models are different than the ones used in this communication[48,49].
Our results also show that CFA-treated groups had lower levels of AST and ALT. It was observed that levels of both enzymes were similar for both animal models (BDL + CFA and TAA + CFA), and comparing these levels with healthy rats, no significant differences were found. These results suggest that CFA treatment prevents hepatocellular damage resulting in normal levels of these enzymes with prevention of liver fibrosis.
A study in patients with chronic hepatitis C shows that a daily CFA consumption above 308 mg (approximately 2.25 cups of coffee) was significantly associated with reduced liver fibrosis, and the protective association persisted after controlling for age, sex, race, liver disease, body mass index and alcohol intake in all patients[12].
Our results presented in this report are similar; we showed that fibrosis was successfully prevented in the liver of rats treated with CFA, finding a strong effect of CFA on ECM content in rat liver, showing 80% reduction in the TAA + CFA group and 38% reduction in the BDL + CFA group. Both results show that CFA had a powerful preventive effect on the development of fibrosis. The Modified Histological Activity Index of Knodell resulted in significantly lower fibrosis in both treated groups.
In a previous in vitro study, it was found that CFA increases intracellular cAMP, resulting in inhibition of CTGF via Smads proteosomal degradation[18]. CTGF has similar effects to TGF-β1 as ECM production stimulation, chemotaxis, proliferation and integrin expression. Our in vivo data shows that CFA has a strong effect on hepatic CTGF expression, resulting in lower expression of profibrogenic and pro-inflammatory genes. TGF-β1 is a major fibrogenic mediator in which expression is increased in inflamed liver and it is considered the principal fibrogenic component[47]. It has been suggested that TGF-β1 up-regulates gene expression of connective tissue, and Col-1 in activated HSC[6,47]. Results obtained in CFA-treated groups are very interesting, since in both animal models Col-1 expression was significantly lower, a result that correlates with fibrosis percentage shown for each group with CFA.
It has been observed that liver fibrosis process development is accompanied by inflammation, in which pro-inflammatory cytokines play an important role in the perpetuation of signaling pathways[47,50]. Furthermore, one report in alcoholic liver injury shows that CFA decreased serum and tissue inflammatory cytokines levels[48]. In this study we found that induction of both liver fibrosis models had a large amount of inflammatory cell infiltrate; in contrast, CFA-treated groups showed decreased number of inflammatory cells, necroinflammation, CD11b positive areas and TNF-α levels. These results at cellular and molecular levels match with serum and tissue inflammatory cytokines levels in other studies about CFA[48].
IL-1β expression was reduced in CFA-treated groups. ECM signaling is of great importance as it serves as a reservoir of various cytokines such as TGF-β1, TNF-α, platelet-derived growth factor (PDGF), IL-6 and IL-1β, protecting these factors for proteolysis and modulating its bioactivity and bioavailability. In this microenvironment, the cytokines might have a key role in the onset of fibrosis, and perpetuating inflammation[47], where CFA treatment could be useful to break this inflammatory circle, as demonstrated in our different experiments.
HSC have an important role in fibrosis and fibrosis development. HSC activation and proliferation, and collagen synthesis are influenced by factors derived from Kupffer cells (TGF-β1, TNF-α, IL-1β, IL-6 and IL-4), endothelial cells (PDGF) and hepatocytes (insulin-like growth factor). Also, TNF-α, IL-1β, TGF-β1, IL-6 and IL-4, and PDGF are regulated by NF-κB and this promotes inflammatory signaling pathway perpetuation[9-11]. We found that CFA promotes lower levels of pro-inflammatory cytokines expression. These findings could be due to the fact that CFA prevents HSC activation and ECM production.
ROS activate the NF-κB pathway. It is known that both liver fibrosis induction models used here, course with an oxidative stress state. Because of this, we measured antioxidant enzymes gene expression levels to monitor them with CFA treatment[51].
As expected, untreated groups showed lower expression of antioxidant enzymes SOD and CAT, indicating indirectly an oxidative stress state. CFA-treated rats showed higher levels of antioxidant enzymes, especially of CAT in the BDL + CFA group, that could be explained by the type of substrate metabolized (hydrogen peroxide). SOD catalyzes O2- dismutation into O2 and H2O2. In contrast, CAT catalyzes decomposition of H2O2 into O2 and H2O[45]. Considering this, we assume that CAT was much higher in the BDL + CFA group, due to accumulation of H2O2 at 4 wk of treatment by SOD action. To verify this last effect we performed an assay to measure CAT antioxidant activity, where we found a strong correlation between mRNA expression and enzyme activity, especially in the BDL + CFA group; antioxidant CAT activity was significantly increased (P < 0.0001) compared with BDL group.
Along with these results, the significant higher expression of transcription factor Nrf2 in the CFA treated groups supports the evidence of the potent antioxidant effect of CFA acting as an important hepatoprotector agent in the presence of a chronic organ aggression. These results agree with the report by Boettler et al[52] where they found higher expression of Nrf2 in humans with consumption of coffee with respect to normal diet where Nrf2 expression was reduced. In response to oxidative stress Nrf2 is activated, translocates to the nucleus and binds to the promoter of its target genes such as CAT and SOD inducing their expression. Nrf2 half life is around 13-20 min. In oxidative stress and in the presence of antioxidant molecules like quercetin, the half-life is duplicated[53]. Thus, given that CFA is also an antioxidant molecule, we believe the same thing may be taking place, though it would require additional experiments to test this hypothesis. Nguyen et al[54] have suggested that in oxidative stress, Nrf2 diminishes its degradation accumulating in nucleus increasing its transcriptional activity.
On the other hand, activation of HSC is a complex process where the transcriptional factor Snail-1 has an important role.
In vertebrates Snail-1 is activated by a different signal pathway from ERK2, NF-κB and phosphatidylinositol 3-kinase[55-57]. All these pathways have been involved in activation of HSC. Several authors have reported the overexpression of Snail-1 in pathological conditions associated with ECM deposition[23,24]. In vitro studies showed that Snail-1 is expressed by HSC and its transcription is augmented in vitro and in vivo in activated HSC compared with quiescent HSC. At the protein level, the nuclear translocation of Snail-1 in activated HSC was observed[58].
Scarpa et al[25] reported that the use of an adenovector expressing Snail-1 small-interfering (sh) RNA to silence Snail expression in HSC isolated from mouse, dramatically reduced activation-related genes α-smooth muscle actin (α-SMA) and Col-1 and increased quiescence-related gene peroxisome proliferator-activated receptor, evidencing the important role of Snail-1 in HSC activation (Snail-1 transcription factor Am J Physiol 2011). However other studies suggest a multiple cell-type origin of cell source for Snail-1 in human liver fibrosis; thus, this fact should be analyzed. Indeed, it was reported that Snail-1 overexpression induces epithelial mesenchymal transition and siRNA against Snail-1 attenuated this epithelial mesenchymal transition. Immunostaining of fibrotic livers from mice treated with CCl4 revealed the presence of Snail-1+, α-SMA+ cells as well as Snail-1+ α-SMA- and Snail-1-α-SMA+ cells along the fibrotic septa. This staining pattern could be explained by the epithelial mesenchymal transition process where hepatocytes transdifferentiate to mesenchymal cells resulting in new HSC[59-61].
In the same way, Dooley et al[62] observed a hepatic marker at the border of the inflamed region from human liver Snail-1+ cells lacking transferrin and they hypothesize that these cells are hepatocytes in a later stage of transition to mesechymal cells.
In our results, CFA treatment diminished Snail-1 expression in rats with chronic liver injury suggesting that CFA prevents HSC activation and suggesting its protector effect on fibrosis development. Our results together allow us to propose CFA use in pathologies with early chronic damage before the establishment of fibrosis.
The observed effect of CFA in this work on necrosis of hepatocytes and on HSC activation could be explained by an indirect effect of CFA. This might be taking place through a decrease of oxidative stress in the liver produced principally by Kupffer cells which secret cytokines activating HSC.
From the very beginning of its administration, CFA neutralizes free radicals and induces antioxidant molecules production which protect hepatocytes from CCl4 damage; this means there is less hepatocyte death, reflected in there being lower levels of ALT and AST found in CFA treatment groups. TGF-β and TNF-α production is decreased rendering a drop in HSC activation, and consequently, less fibrosis. On the other hand, ECM deposition and loss of microvilli on hepatocytes caused by CCl4 intoxication blocks the free flow of nutrients causing hepatocytes death. It was found in this paper that CFA treatment yields less fibrosis, less block of nutrients and less hepatocyte death. However, a direct effect of CFA on hepatocytes and HSC cannot be ruled out.
COMMENTS
Background
Hepatic stellate cells (HSCs) activation is a major hallmark in liver fibrosis, which is perpetuated by growth factors and pro-inflammatory molecules. Caffeine (CFA) modifies these events in vitro.
Research frontiers
CFA inhibits the transcriptional factor Snail-1, down-regulating profibrogenic genes, and activates the Nrf2 inducing antioxidant enzymes system, preventing inflammation and fibrosis.
Innovations and breakthroughs
CFA treatment diminished Snail-1 expression in rats with chronic liver injury suggesting that CFA prevents HSC activation and provides a protector effect on fibrosis development. Their results together allow the authors to propose CFA use in pathologies with early chronic damage before the establishment of fibrosis.
Peer review
The present manuscript provides a detailed study on the effect of CFA on experimental liver fibrosis in rats. Overall, this is a very interesting paper. The presented data is throughout of very good quality and the conclusions drawn are supported by sufficient data.
Footnotes
Supported by Conacyt grant No. 25474 to Juan Armendáriz-Borunda
P- Reviewers: Di Costanzo GG, Liedtke AC, Tsuchiya A, Xu J S- Editor: Gou SX L- Editor: O’Neill M E- Editor: Ma S
References
- 1.Nedredal GI, Elvevold K, Ytrebø LM, Fuskevåg OM, Pettersen I, Bertheussen K, Langbakk B, Smedsrød B, Revhaug A. Significant contribution of liver nonparenchymal cells to metabolism of ammonia and lactate and cocultivation augments the functions of a bioartificial liver. Am J Physiol Gastrointest Liver Physiol. 2007;293:G75–G83. doi: 10.1152/ajpgi.00245.2006. [DOI] [PubMed] [Google Scholar]
- 2.Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427–436. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38 Suppl 1:S38–S53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 5.Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–335. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 6.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 7.Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61) Angiogenesis. 2002;5:153–165. doi: 10.1023/a:1023823803510. [DOI] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 9.Modi AA, Feld JJ, Park Y, Kleiner DE, Everhart JE, Liang TJ, Hoofnagle JH. Increased caffeine consumption is associated with reduced hepatic fibrosis. Hepatology. 2010;51:201–209. doi: 10.1002/hep.23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gressner OA, Gressner AM. Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008;28:1065–1079. doi: 10.1111/j.1478-3231.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 11.Ruhl CE, Everhart JE. Coffee and tea consumption are associated with a lower incidence of chronic liver disease in the United States. Gastroenterology. 2005;129:1928–1936. doi: 10.1053/j.gastro.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 12.Muriel P, Arauz J. Coffee and liver diseases. Fitoterapia. 2010;81:297–305. doi: 10.1016/j.fitote.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Chan ES, Montesinos MC, Fernandez P, Desai A, Delano DL, Yee H, Reiss AB, Pillinger MH, Chen JF, Schwarzschild MA, et al. Adenosine A(2A) receptors play a role in the pathogenesis of hepatic cirrhosis. Br J Pharmacol. 2006;148:1144–1155. doi: 10.1038/sj.bjp.0706812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gressner OA. Less Smad2 is good for you! A scientific update on coffee’s liver benefits. Hepatology. 2009;50:970–978. doi: 10.1002/hep.23097. [DOI] [PubMed] [Google Scholar]
- 15.Gressner OA, Lahme B, Rehbein K, Siluschek M, Weiskirchen R, Gressner AM. Pharmacological application of caffeine inhibits TGF-beta-stimulated connective tissue growth factor expression in hepatocytes via PPARgamma and SMAD2/3-dependent pathways. J Hepatol. 2008;49:758–767. doi: 10.1016/j.jhep.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 16.Uemura M, Swenson ES, Gaça MD, Giordano FJ, Reiss M, Wells RG. Smad2 and Smad3 play different roles in rat hepatic stellate cell function and alpha-smooth muscle actin organization. Mol Biol Cell. 2005;16:4214–4224. doi: 10.1091/mbc.E05-02-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C, Liu P, Hu Y, Zhu D. Effects of salvianolic acid-B on TGF-beta 1 stimulated hepatic stellate cell activation and its intracellular signaling. Zhonghua Yixue Zazhi. 2002;82:1267–1272. [PubMed] [Google Scholar]
- 18.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Okano J, Nagahara T, Matsumoto K, Murawaki Y. Caffeine inhibits the proliferation of liver cancer cells and activates the MEK/ERK/EGFR signalling pathway. Basic Clin Pharmacol Toxicol. 2008;102:543–551. doi: 10.1111/j.1742-7843.2008.00231.x. [DOI] [PubMed] [Google Scholar]
- 20.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37:139–143. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 21.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanigawa S, Fujii M, Hou DX. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic Biol Med. 2007;42:1690–1703. doi: 10.1016/j.freeradbiomed.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Yáñez-Mó M, Lara-Pezzi E, Selgas R, Ramírez-Huesca M, Domínguez-Jiménez C, Jiménez-Heffernan JA, Aguilera A, Sánchez-Tomero JA, Bajo MA, Alvarez V, et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med. 2003;348:403–413. doi: 10.1056/NEJMoa020809. [DOI] [PubMed] [Google Scholar]
- 24.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scarpa M, Grillo AR, Brun P, Macchi V, Stefani A, Signori S, Buda A, Fabris P, Giordani MT, De Caro R, et al. Snail1 transcription factor is a critical mediator of hepatic stellate cell activation following hepatic injury. Am J Physiol Gastrointest Liver Physiol. 2011;300:G316–G326. doi: 10.1152/ajpgi.00141.2010. [DOI] [PubMed] [Google Scholar]
- 26.Chilakapati J, Shankar K, Korrapati MC, Hill RA, Mehendale HM. Saturation toxicokinetics of thioacetamide: role in initiation of liver injury. Drug Metab Dispos. 2005;33:1877–1885. doi: 10.1124/dmd.105.005520. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Benjamin IS, Alexander B. Reproducible production of thioacetamide-induced macronodular cirrhosis in the rat with no mortality. J Hepatol. 2002;36:488–493. doi: 10.1016/s0168-8278(02)00011-9. [DOI] [PubMed] [Google Scholar]
- 28.Dashti H, Jeppsson B, Hägerstrand I, Hultberg B, Srinivas U, Abdulla M, Bengmark S. Thioacetamide- and carbon tetrachloride-induced liver cirrhosis. Eur Surg Res. 1989;21:83–91. doi: 10.1159/000129007. [DOI] [PubMed] [Google Scholar]
- 29.Salazar-Montes A, Ruiz-Corro L, López-Reyes A, Castrejón-Gómez E, Armendáriz-Borunda J. Potent antioxidant role of pirfenidone in experimental cirrhosis. Eur J Pharmacol. 2008;595:69–77. doi: 10.1016/j.ejphar.2008.06.110. [DOI] [PubMed] [Google Scholar]
- 30.Rahn SB. Liver biopsy interpretation in chronic hepatitis. J Insur Med. 2001;33:110–113. [PubMed] [Google Scholar]
- 31.Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, Denk H, Desmet V, Korb G, MacSween RN. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 32.Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, Kiernan TW, Wollman J. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–435. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 33.Siller-López F, Sandoval A, Salgado S, Salazar A, Bueno M, Garcia J, Vera J, Gálvez J, Hernández I, Ramos M, et al. Treatment with human metalloproteinase-8 gene delivery ameliorates experimental rat liver cirrhosis. Gastroenterology. 2004;126:1122–1133; discussion 949. doi: 10.1053/j.gastro.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 34.Feldmann G. Critical analysis of the methods used to morphologically quantify hepatic fibrosis. J Hepatol. 1995;22:49–54. [PubMed] [Google Scholar]
- 35.Sajic T, Hopfgartner G, Szanto I, Varesio E. Comparison of three detergent-free protein extraction protocols for white adipose tissue. Anal Biochem. 2011;415:215–217. doi: 10.1016/j.ab.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 36.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Yuan JS, Reed A, Chen F, Stewart CN. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arauz J, Moreno MG, Cortés-Reynosa P, Salazar EP, Muriel P. Coffee attenuates fibrosis by decreasing the expression of TGF-β and CTGF in a murine model of liver damage. J Appl Toxicol. 2013;33:970–979. doi: 10.1002/jat.2788. [DOI] [PubMed] [Google Scholar]
- 40.Carrillo JA, Benitez J. Clinically significant pharmacokinetic interactions between dietary caffeine and medications. Clin Pharmacokinet. 2000;39:127–153. doi: 10.2165/00003088-200039020-00004. [DOI] [PubMed] [Google Scholar]
- 41.Benowitz NL, Jacob P, Mayan H, Denaro C. Sympathomimetic effects of paraxanthine and caffeine in humans. Clin Pharmacol Ther. 1995;58:684–691. doi: 10.1016/0009-9236(95)90025-X. [DOI] [PubMed] [Google Scholar]
- 42.Kot M, Daniel WA. The relative contribution of human cytochrome P450 isoforms to the four caffeine oxidation pathways: an in vitro comparative study with cDNA-expressed P450s including CYP2C isoforms. Biochem Pharmacol. 2008;76:543–551. doi: 10.1016/j.bcp.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 43.Kot M, Daniel WA. Relative contribution of rat cytochrome P450 isoforms to the metabolism of caffeine: the pathway and concentration dependence. Biochem Pharmacol. 2008;75:1538–1549. doi: 10.1016/j.bcp.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 44.Du Bois D, Du Bois EF. The measurement of the surface area of man. Arch Intern Med. 1915;15:868–881. [Google Scholar]
- 45.Du Bois D, Du Bois EF. A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition. 1989;5:303–311; discussion 312-313. [PubMed] [Google Scholar]
- 46.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 47.Saile B, Ramadori G. Inflammation, damage repair and liver fibrosis--role of cytokines and different cell types. Z Gastroenterol. 2007;45:77–86. doi: 10.1055/s-2006-927395. [DOI] [PubMed] [Google Scholar]
- 48.Lv X, Chen Z, Li J, Zhang L, Liu H, Huang C, Zhu P. Caffeine protects against alcoholic liver injury by attenuating inflammatory response and oxidative stress. Inflamm Res. 2010;59:635–645. doi: 10.1007/s00011-010-0176-6. [DOI] [PubMed] [Google Scholar]
- 49.Akashi I, Kagami K, Hirano T, Oka K. Protective effects of coffee-derived compounds on lipopolysaccharide/D-galactosamine induced acute liver injury in rats. J Pharm Pharmacol. 2009;61:473–478. doi: 10.1211/jpp/61.04.0009. [DOI] [PubMed] [Google Scholar]
- 50.Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 51.Tobar N, Villar V, Santibanez JF. ROS-NFkappaB mediates TGF-beta1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol Cell Biochem. 2010;340:195–202. doi: 10.1007/s11010-010-0418-5. [DOI] [PubMed] [Google Scholar]
- 52.Boettler U, Volz N, Teller N, Haupt LM, Bakuradze T, Eisenbrand G, Bytof G, Lantz I, Griffiths LR, Marko D. Induction of antioxidative Nrf2 gene transcription by coffee in humans: depending on genotype? Mol Biol Rep. 2012;39:7155–7162. doi: 10.1007/s11033-012-1547-6. [DOI] [PubMed] [Google Scholar]
- 53.Dinkova-Kostova AT, Massiah MA, Bozak RE, Hicks RJ, Talalay P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc Natl Acad Sci USA. 2001;98:3404–3409. doi: 10.1073/pnas.051632198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen T, Sherratt PJ, Huang HC, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J Biol Chem. 2003;278:4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- 55.Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barberà MJ, Puig I, Domínguez D, Julien-Grille S, Guaita-Esteruelas S, Peiró S, Baulida J, Francí C, Dedhar S, Larue L, et al. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene. 2004;23:7345–7354. doi: 10.1038/sj.onc.1207990. [DOI] [PubMed] [Google Scholar]
- 57.Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, Du J, Zhang J, Weng M, Li X, Pu D, Gao L, Deng S, Xia S, She Q. Snail1 is involved in de novo cardiac fibrosis after myocardial infarction in mice. Acta Biochim Biophys Sin (Shanghai) 2012;44:902–910. doi: 10.1093/abbs/gms085. [DOI] [PubMed] [Google Scholar]
- 59.Pagan R, Martín I, Llobera M, Vilaró S. Epithelial-mesenchymal transition of cultured rat neonatal hepatocytes is differentially regulated in response to epidermal growth factor and dimethyl sulfoxide. Hepatology. 1997;25:598–606. doi: 10.1002/hep.510250318. [DOI] [PubMed] [Google Scholar]
- 60.Spagnoli FM, Cicchini C, Tripodi M, Weiss MC. Inhibition of MMH (Met murine hepatocyte) cell differentiation by TGF(beta) is abrogated by pre-treatment with the heritable differentiation effector FGF1. J Cell Sci. 2000;113(Pt 20):3639–3647. doi: 10.1242/jcs.113.20.3639. [DOI] [PubMed] [Google Scholar]
- 61.Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 62.Dooley S, Hamzavi J, Ciuclan L, Godoy P, Ilkavets I, Ehnert S, Ueberham E, Gebhardt R, Kanzler S, Geier A, et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology. 2008;135:642–659. doi: 10.1053/j.gastro.2008.04.038. [DOI] [PubMed] [Google Scholar]