

Significance
Glucocorticoids are well known to play a major role in obesity, but underlying mechanisms have been obscure. We demonstrate that the small G protein Dexras1, first identified based on its dramatic induction by glucocorticoids, mediates adipogenic differentiation of preadipocytes, as well as diet-induced obesity in intact rodents. Thus, the adipogenesis of preadipocytes is abolished by Dexras1 deletion and selectively induced by Dexras1 expression. Relevance to intact animals is evident from our experiments wherein diet-induced obesity is prevented in mice with knockout of Dexras1. Thus, pharmacotherapy involving Dexras1 may afford a promising approach to the therapy of obesity.
Keywords: insulin, cyclic AMP, nitric oxide, Cushing disease
Abstract
Adipogenesis, the conversion of precursor cells into adipocytes, is associated with obesity and is mediated by glucocorticoids acting via hitherto poorly characterized mechanisms. Dexras1 is a small G protein of the Ras family discovered on the basis of its marked induction by the synthetic glucocorticoid dexamethasone. We show that Dexras1 mediates adipogenesis and diet-induced obesity. Adipogenic differentiation of 3T3-L1 cells is abolished with Dexras1 depletion, whereas overexpression of Dexras1 elicits adipogenesis. Adipogenesis is markedly reduced in mouse embryonic fibroblasts from Dexras1-deleted mice, whereas adiposity and diet-induced weight gain are diminished in the mutant mice.
Obesity presents a major public health problem, with its widespread occurrence leading to increases in diabetes, hypertension, and cardiovascular disability (1, 2). Obesity is associated with hypertrophy of adipocytes, as well as increases in adipogenesis, which reflects the differentiation of precursor cells into adipocytes (3–6). The adipogenic process involves multiple factors, especially cAMP, insulin, and glucocorticoids (7, 8). Thus, in the best-characterized model of adipogenesis, 3T3-L1 cells, a fibroblast line, are treated with the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX) to increase cAMP levels, insulin, and the synthetic glucocorticoid dexamethasone. When exposed to a hormonal mixture of IBMX, insulin, and dexamethasone, 3T3-L1 cells accumulate lipid and develop the characteristic morphology of mature adipocytes (7–9).
Adipocyte differentiation is controlled by a complex network of transcription factors that temporally regulate adipocyte gene expression (4). An early response to hormonal stimuli of adipogenesis is activation of two members of the CCAAT/enhancer binding protein (C/EBP) family of transcription factors, C/EBPβ and C/EBPδ, which induce the expression of C/EBPα and peroxisome proliferator-activated receptor (PPAR)γ, the two principal adipogenic transcription factors. Although transcriptional regulation of adipogenic differentiation has been well characterized, less is known about how hormonal inducers promote this process. In particular, how glucocorticoids induce adipogenesis is poorly understood.
Dexras1 (also known as Rasd1) is a small G protein of the Ras family discovered on the basis of its marked induction by the synthetic glucocorticoid dexamethasone (10). Dexras1 interacts with neuronal nitric oxide synthase via the scaffolding protein CAPON, with nitric oxide serving as a guanine nucleotide exchange factor for Dexras1 (11). Dexras1 also participates in the glutamate–NMDA neurotransmission cascade that leads to cellular iron entry and neurotoxicity (12). Dexras1 also influences circadian rhythms (13). Disruption of circadian rhythms leads to the development of metabolic disorders, including obesity and diabetes (14–17).
Here, we show that Dexras1 mediates adipogenesis and diet-induced obesity. Dexras1, which is induced by glucocorticoids during adipogenic differentiation, is essential for adipogenesis. Overexpression of Dexras1 rescues impaired adipogenesis in mouse embryonic fibroblasts (MEFs) from Dexras1-deleted mice. Dexras1 knockout mice display impaired adiposity and are resistant to diet-induced weight gain. Accordingly, agents impacting Dexras1 may offer benefit in the treatment of obesity.
Results
Dexras1 Is Induced by Glucocorticoids During Adipogenic Differentiation.
Preliminary microarray analysis sought genes altered in 3T3-L1 cells with adipogenesis initiated by treatment with IBMX, dexamethasone, and insulin (designated as MDI), as well as genes highly expressed in murine or human adipose tissue. These experiments revealed high levels of Dexras1. Among diverse organs, we observe highest levels of Dexras1 in fat-enriched organs, especially white adipose tissue (WAT) (Fig. 1A). Dexamethasone treatment markedly augments Dexras1 levels in mouse tissues (Fig. 1B). Adipogenic differentiation of 3T3-L1 cells is also associated with a striking induction of Dexras1, with peak sevenfold enhancement at 4–8 h (Fig. 1C and Fig. S1A). Omission of dexamethasone from the MDI mixture abolishes Dexras1 mRNA expression (Fig. S1B), indicating that Dexras1 expression is transcriptionally regulated by interactions of dexamethasone and the glucocorticoid receptor.
Fig. 1.
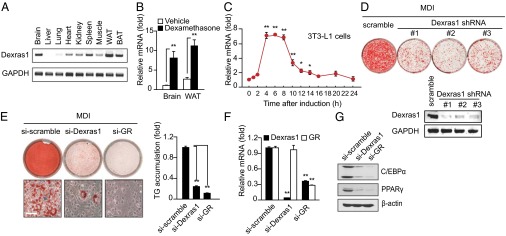
Dexras1 is required for adipogenic differentiation in 3T3-L1 cells. (A) Expression of Dexras1 in various mouse tissues. Total RNA was prepared and analyzed by RT-PCR. (B) Dexamethasone induces Dexras1 expression in brain and WAT. C57BL/6 mice were injected with dexamethasone (0.5 mg/kg) intraperitoneally and killed after 4 h. Expression of mRNA was analyzed by real-time qPCR. (C) Induction of Dexras1 during 3T3-L1 differentiation. Total RNA was isolated at the indicated time points after induction of differentiation and analyzed by qPCR. (D) Knockdown of Dexras1 abolishes 3T3-L1 differentiation; 3T3-L1 cells were infected with lentivirus expressing shRNA targeting Dexras1. After infection, cells were selected with puromycin, induced to differentiate, and stained by oil red O at day 8 (Left). Knockdown efficiency was monitored by semiquantitative RT-PCR (Right). (E) Dexras1 depletion abolishes accumulation of fat droplets and triglycerides comparable to deletion of glucocorticoid receptors. Cells were transfected with control, Dexras1 or glucocorticoid receptor (GR) siRNAs and differentiation induced by MDI. Eight days later, differentiated cells were stained with oil red O, and triglyceride content was measured by spectrometric analysis. (Scale bar: 50 μm.) (F) Dexras1 and GR mRNA expression after knockdown experiments. Total RNA was analyzed by qPCR. (G) Western blot analysis reveals comparable loss of C/EBPα and PPARγ after knockdown of Dexras1 or glucocorticoid receptor. Error bars represent means ± SD. *P < 0.05; **P < 0.01.
Dexras1 Is Required for Adipogenic Differentiation.
To determine the impact of Dexras1 upon adipogenesis, we depleted Dexras1 by lentiviral shRNA transduction. MDI-elicited adipogenesis, monitored in terms of staining for fat droplets, is virtually abolished with Dexras1 knockdown by shRNA (Fig. 1D). Depleting glucocorticoid receptors and Dexras1 by siRNA also produces similar, substantial decrements in adipogenesis (Fig. 1E and Fig. S2A). Knockdown of Dexras1 does not affect mRNA expression of glucocorticoid receptors, whereas knockdown of glucocorticoid receptors blocks Dexras1 induction by MDI mixture (Fig. 1F). MDI-elicited induction of PPARγ and C/EBPα, transcription factors in the adipogenic program (18–20), is virtually abolished by depletion of Dexras1 or glucocorticoid receptors, which also diminishes the induction of adipocyte-specific genes such as aP2/422 and FAS (7) (Fig. 1G and Fig. S2B). In contrast, inhibitory factors of adipogenesis (4, 21, 22) are either unchanged (GATA2, GATA3) or remain high (KLF2, Pref-1) with comparable treatment (Fig. S2B). These data indicate that Dexras1 is required for MDI-induced adipogenic differentiation.
Dexras1 Mediates Actions of Glucocorticoid in the Adipogenic Mixture.
We wondered whether Dexras1 itself is sufficient to elicit adipogenesis. First, we compared different elements of the MDI mixture. Of the three MDI constituents, dexamethasone alone notably increases fat deposition, whereas IBMX and insulin (MI) produce negligible effects (Fig. 2A). The combination of dexamethasone and IBMX elicits more adipogenesis than combinations of dexamethasone with insulin or IBMX with insulin, whereas the full MDI mixture produces maximal adipogenesis. Accordingly, dexamethasone appears to be the most critical component of the mixture, because, in its absence, adipogenesis is not demonstrable. Whereas the combination of IBMX and insulin barely elicits adipogenesis, overexpressing Dexras1 in cells treated with these two agents leads to robust adipogenesis, comparable to the full MDI mixture (Fig. 2B and Fig. S3A). Thus, Dexras1 is sufficient to account for the actions of dexamethasone in the MDI mixture and so is a major regulator of adipogenesis. These conclusions are supported by experiments monitoring expression of PPARγ and C/EBPα. Dexras1 overexpression restores the diminished induction of PPARγ and C/EBPα associated with omission of dexamethasone from the MDI mixture (Fig. 2C). Consistent with these observations, overexpression of Dexras1 enhances expression of PPARγ, C/EBPα, aP2/422, and FAS, marker genes for adipogenesis (Fig. S3B). Depletion of glucocorticoid receptors fails to diminish the stimulation of adipogenesis elicited by Dexras1, consistent with Dexras1 functioning downstream of the receptors (Fig. S3C).
Fig. 2.

Dexras1 mediates actions of glucocorticoid in the adipogenic mixture. (A) Differentiation of 3T3-L1 cells in response to various differentiation mixtures. (B) Overexpression of Dexras1 rescues cells in which dexamethasone was omitted from the MDI mixture. Cells were transfected with either pcDNA3 vector or pcDNA3-Dexras1-FLAG and treated with MI or MDI as indicated (Left). (Scale bar: 50 μm.) (Right) Spectrophotometric quantification of staining from three independent experiments. (C) Overexpression of Dexras1 rescues induction of C/EBPα and PPARγ in cells in which dexamethasone was omitted from the MDI mixture. Error bars represent mean ± SD. **P < 0.01.
The Unique C-Terminal Extension of Dexras1 Is Critical for Adipogenic Differentiation.
What features of Dexras1 might account for its unique role in adipogenesis? Dexras1 differs from most members of the Ras family in the presence of a C-terminal extension of about 50 amino acids (Fig. 3A). The C-terminal four amino acids, CVIS, correspond to the canonic CAAX (where C indicates cysteine; A, aliphatic; and X, terminal) signal for membrane attachment, which in Dexras1 involves farnesylation (23). Deletion of amino acids 223–276 while retaining the CAAX motif abolishes the stimulation of adipogenesis elicited in 3T3-L1 cells by overexpressed Dexras1 (Fig. 3 B and C). Deletion of the entire C-terminal 52 aa, including CVIS, also abolishes adipogenesis. Selective deletion of the C-terminal 4 aa or their mutation moderately reduces adipogenesis. Thus, the C-terminal extension of Dexras1 appears critical for its actions. The uniqueness of Dexras1’s actions is exemplified by the failure of the classical Ras proteins H-Ras and K-Ras to elicit adipogenesis.
Fig. 3.

C-terminal domain of Dexras1 is required for Dexras1-mediated adipogenesis. (A) Schematic drawing of wild type and mutants of Dexras1 used in this study. Other Ras family proteins are depicted along with Dexras1, based on amino acid homology. (B) Mapping of Dexras1 domains mediating adipogenic differentiation; 3T3-L1 cells expressing indicated proteins were induced to differentiate by MI. Protein expression of adipogenic regulators, C/EBPα and PPARγ, was analyzed by Western blot. (C) Dexras1 domains mediating lipid accumulation; 3T3-L1 cells expressing indicated proteins were induced to differentiate with MI. Oil red O staining for fat droplets was performed at day 8 (Left). (Right) Spectrophotometric quantification of staining from 3 independent experiments. Data represent means ± SD. **P < 0.01.
Dexras1 Knockout Mice Exhibit Reduced Adipogenesis.
We extended analysis of Dexras1’s role in adipogenesis to Dexras1-deleted mice. Fat droplet levels are profoundly reduced in MEFs from Dexras1 knockouts (Fig. 4A). Moreover, PPARγ and C/EBPα expression is substantially lower in Dexras1 knockout MEFs. Overexpression of Dexras1 rescues the loss of fat droplets and triglyceride levels associated with Dexras1 deletion (Fig. 4B). Consistent with these observations, PPARγ and C/EBPα expression is significantly reduced in WATs from Dexras1 knockout mice (Fig. 4C). Thus, the adipogenic program is markedly diminished in the absence of Dexras1.
Fig. 4.
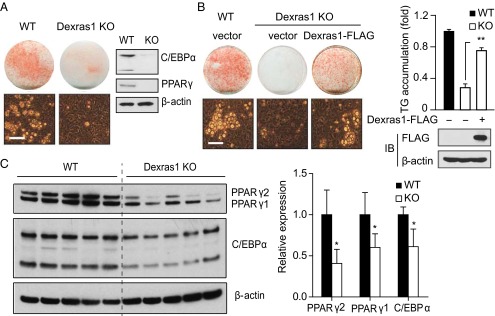
Impaired adipogenesis in Dexras1 knockout mice. (A) Loss of adipogenesis in MEFs of Dexras1 mutants. Wild-type (WT) or Dexras1 knockout (KO) MEFs were induced to differentiate into adipocytes and stained for fat droplets with oil red O at day 8. Induction of C/EBPα and PPARγ expression was analyzed by Western blot analysis. (B) Overexpression of Dexras1 rescues adipogenic defects associated with Dexras1 deletion. WT or Dexras1 KO MEFs were transfected with pcDNA3 vector or pcDNA3-Dexras1-FLAG and subjected to adipogenic stimuli (Left). (Scale bar: 50 μm.) (Right) Triglyceride accumulation and Dexras1-FLAG expression. (C) Relative expression of C/EBPα and PPARγ in epididymal WAT of 12-wk old male Dexras1 knockout and wild-type mice. All data are means ± SD. *P < 0.05; **P < 0.01.
Dexras1 Knockout Mice Are Resistant to Diet-Induced Weight Gain.
To determine whether Dexras1 mediates diet-induced obesity, we examined the influence of high-fat diet (HFD) on the Dexras1 knockout mice. With normal chow, weight gain in the knockouts is modestly but significantly less than in wild type. On HFD, the mutants gain substantially less weight than wild-type animals (Fig. 5 A and B). QNMR analysis reveals a modest but significant decrease in body fat of mutants on normal diet with a corresponding increase in lean body mass. The reduction of body fat composition in knockouts is more pronounced with HFD (Fig. 5C). The weight of WAT (epididymal) is substantially decreased in knockouts both on normal diet and HFD, whereas the weight of brown adipose tissue (BAT), heart, lung, and kidney is not significantly altered (Fig. 5 D and E). On HFD, Dexras1 knockouts have smaller adipocyte diameter than wild type (Fig. 5 F and G).
Fig. 5.
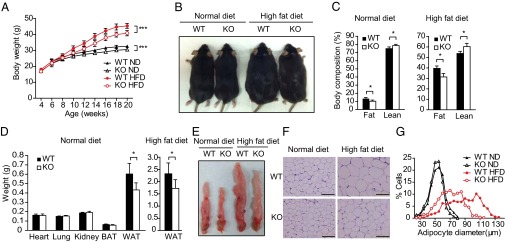
Dexras1 knockout mice are resistant to HFD-induced obesity. (A) Body weight gain of WT and Dexras1 KO mice on normal diet (ND) or HFD (n = 10 per group). (B) Dexras1 KO mice are resistant to diet-induced weight gain. (C) Body composition (% body weight) analysis of WT and KO mice on normal diet (n = 7 per group) or HFD for 11 wk (n = 7 per group). (D) Weight of various tissues from WT and KO mice on normal diet (n = 4 per group) (Left). Weight of epididymal WAT from WT and KO mice fed a HFD (n = 6 per group) (Right). (E) Decreased adipose tissue mass in knockouts on normal diet or HFD. (F) H&E staining of epididymal WAT from wild-type or Dexras1 KO mice on normal diet or HFD for 10 wk. (Scale bar: 100 μm.) (G) Distribution of adipocyte size in epididymal WAT from wild-type or Dexras1 KO mice on normal diet (n = 3 per group) or HFD (n = 3 per group). Mean diameter: WT on normal diet, 50.31 ± 10.25 μm; KO on normal diet, 48.4 ± 8.81 μm; WT on HFD, 85.44 ± 21.43 μm; and KO on HFD, 67.53 ± 15.79 μm. The diameters of epididymal white adipocyte were determined by ImageJ, and more than 500 adipocytes were examined for each group. All data are means ± SD. *P < 0.05; ***P < 0.001.
Discussion
Our findings establish a major adipogenic role for Dexras1. Deleting Dexras1 by RNA interference or gene knockout markedly diminishes adipogenesis and reduces weight gain in mice on HFD. Dexras1 mediates the influences of glucocorticoids upon adipogenesis. Thus, 3T3-L1 cells expressing Dexras1 do not require glucocorticoid treatment for full differentiation. Regulation of glucocorticoid-associated obesity by Dexras1 does not appear to be directly related to behavioral alterations. We detected negligible alterations in gross behavior in our Dexras1 mutants. Cheng et al. (13) characterized behavioral features of Dexras1 knockout mice and observed few if any changes in a battery of tests involving learning and memory, anxiety, social aggression, locomotor responses under stressful conditions, and acute pain responses.
Since the 3T3-L1 cell line was first used to characterize mechanisms of adipogenesis (9), many hormonal factors and their signaling pathways have been implicated in preadipocyte commitment or terminal differentiation. Of these, the glucocorticoid signaling system has not been well characterized, with little investigation of downstream targets for the glucocorticoid receptor in adipogenesis. The ability of Dexras1 to fully replace dexamethasone in the differentiation protocol indicates that glucocorticoids act primarily through Dexras1 in this cascade. Interestingly, Dexras1 is most highly expressed in fat-enriched organs such as WAT and BAT where, presumably, it may influence the development of obesity.
The changes in body weight of Dexras1 mutants are not as dramatic as the influences of Dexras1 upon adipogenesis in 3T3-L1 cells. The 3T3-L1 preadipocytes represent a selective line of cells devoted largely to differentiation into adipocytes under the influence of a limited number of stimuli. Glucocorticoids are one of the principal agents mediating adipogenesis in these cells. Thus, deletion of glucocorticoid receptors markedly diminishes adipogenesis. Our data establish that Dexras1 is primarily responsible for these actions of glucocorticoids. By contrast, body weight of intact mice is determined by a wide range of physiologic processes. Although glucocorticoids, acting through Dexras1, participate in weight control, their impact is likely diluted by diverse other regulatory systems.
The selective influence of Dexras1 upon the weight of WAT as contrasted to BAT and several other organs is consistent with a unique action of Dexras1 upon the adipogenic program. This selectivity indicates that the impact of Dexras1 upon body weight is not attributable in a major way to processes other than the transformation of preadipocytes to adipocytes.
Very recently, Lindroos et al. (24) reported that the adaptor protein LMO3 is induced by glucocorticoids and modulates adipogenesis in human but not mouse tissues. Whether LMO3 and Dexras1 interface in regulating adipogenesis is unclear. DEPTOR, a component of the mTOR system (25), may also interface with the Dexras1 system, because DEPTOR is induced by glucocorticoids and promotes adipogenesis (26).
Our findings may be relevant to clinical instances of glucocorticoid-associated adiposity. Thus, Cushing’s syndrome, reflecting excessive glucocorticoid release, is associated with visceral adiposity, which we speculate to involve Dexras1. Dexras1 may participate in other forms of obesity, especially of dietary origin, because its deletion diminishes dietary adiposity.
Dexras1 has been reported to influence circadian rhythms (13) and nitric oxide-related neurotransmission (11, 12), processes not linked to glucocorticoids. Our findings represent actions of glucocorticoids that are clearly mediated by Dexras1. Whether other actions of glucocorticoids involve Dexras1 remains to be determined.
Glucocorticoids have diverse therapeutic applications, especially as anti-inflammatory agents. Their use has been limited by a variety of adverse effects. Conceivably, drugs influencing Dexras1 will convey some of the therapeutic actions of glucocorticoids with lesser side effects. In the case of obesity, our observations indicate that drugs which reduce effects of Dexras1 may diminish obesity along with its myriad adverse sequelae.
Materials and Methods
Cell Culture and in Vitro Differentiation.
The 3T3-L1 preadipocytes were cultured and differentiated into adipocytes as described previously (27). Briefly, 3T3-L1 preadipocytes were maintained in DMEM containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 8 μg/mL biotin, supplemented with 10% (vol/vol) heat-inactivated calf serum at 37 °C, in an atmosphere of 90% air and 10% CO2. To induce differentiation, 2-d postconfluent 3T3-L1 cells (designated day 0) were incubated in DMEM containing 10% FBS, 0.5 mM IBMX, 1 μM dexamethasone, and 1 μg/mL insulin for 2 d. Cells were then cultured in DMEM containing 10% FBS and insulin for another 2 d, after which they were grown in DMEM containing 10% FBS. Lipid accumulation in the cells was detected by oil red O staining at day 8. For quantification, stained cells were washed extensively with water to remove unbound dye, and 1 mL isopropanol was added to the six-well culture plate. After 5 min, absorbance at 500 nm of the oil red O extract and serially diluted oil red O standards were assayed spectrophotometrically.
Western Blot Analysis.
Protein concentrations were assessed using the BCA assay kit (Pierce). Protein samples of equal amount were separated by SDS/PAGE and transferred to nitrocellulose membranes. Western blot analysis was performed using the following antibodies: polyclonal antibodies against PPARγ (Cell Signaling Technology) and C/EBPα (27); and mouse monoclonal antibodies against PPARγ, β-actin (Santa Cruz Biotechnology), and FLAG (Sigma).
Gene Expression Analysis.
Gene expression was measured either by semiquantitative RT-PCR or real-time quantitative (q) PCR using total RNA from 3T3-L1 cells, MEF cells, or mice as indicated. In some studies, C57BL/6 mice were injected with dexamethasone intraperitoneally (0.5 mg/kg) and killed after 4 h. Total RNA was isolated from cells or tissues using TRIzol (Invitrogen) according to the manufacturer’s instructions. For quantitative RT-PCR, cDNA was synthesized from 5 μg of total RNA using random hexamer primers and SuperScript reverse transcriptase II (Invitrogen). qPCRs were performed with SYBR Green PCR Master Mix (Applied Biosystems) using an ABI PRISM 7300 RT PCR System (Applied Biosystems). All data were normalized to GAPDH and quantitative measures were obtained using the ΔΔ-Ct method (Applied Biosystems). qPCR was performed using the following primers: Dexras1, 5′-CGCCT CTCTA TCCTC ACAGG-3′ and 5′-GGTCC AAGCT GCTGT TCTTC-3′; C/EBPα, 5′-TGGAC AAGAA CAGCA ACGAG-3′ and 5′-TCACT GGTCA ACTCC AGCAC-3′; PPARγ, 5′-TCCGT GATGG AAGAC CACTC-3′ and 5′-CCCTT GCATC CTTCA CAAGC-3′; aP2/422, 5′-TCTCC AGTGA AAACT TCGAT-3′ and 5′-TTACG CTGAT GATCA ATGTT-3′; FAS, 5′-AAGCC GTTGG GAGTG AAAGT-3′ and 5′-CAATC TGGAT GGCAG TGAGG-3′; GATA2, 5′-GGCAC CTGTT GTGCA AATTG-3′ and 5′-CCTTC TTCAT GGTCA GTGGC-3′; GATA3, 5′-AACAG ACCCC TGACT ATGAA-3′ and 5′-GGTTG AAGGA GTTGC TGCTC-3′; KLF2, 5′-GGCCC CAGCT TCGGC GGC-3′ and 5′-TGAGG AGACC CGCGC GTGGC-3′; Pref-1, 5′-GACCC ACCCT GTGAC CCC-3′ and 5′-CAGGC AGCTC GTGCA CCC-3′; and GAPDH, 5′-ACCAC AGTCC ATGCC ATCAC-3′ and 5′-TCCAC CACCC TGTTG CTGTA-3′.
Lentivirus Production and Infection of 3T3-L1 Adipocytes.
Control and Dexras1 lentiviral shRNAs were obtained from Sigma (MISSION shRNA Lentiviral Transduction Particles). Recombinant lentiviruses were produced by cotransfecting 293FT cells with the lentivirus expression plasmid and packaging plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Infectious lentiviruses were harvested at 48 h after transfection and filtered through 0.45-μm cellulose acetate filters; 3T3-L1 cells were plated in 6-well plates at 50% confluence. After overnight incubation, medium containing viral particles was removed and replaced with fresh medium containing 2 μg/mL puromycin. Cells were grown to 90% confluence and subcultured in medium containing puromycin. Dexras1 knockdown efficiency was confirmed by RT-PCR. The sequences for lentiviral shRNAs were as follows: sh-Dexras1, #1, 5′-CCGGG CTGGT CATTT GCGGT AACAA CTCGA GTTGT TACCG CAAAT GACCA GCTTT TTG-3′; #2, 5′-CCGGC AAGCG CTCT GAACT GAGTA CTCGA GTACT CAGTT CAGAG TCGCT TGTTT TTG-3′; and #3, 5′-CCGGG ACCTC ATGTA CATTC GTGAA CTCGA GTTCA CGAAT GTACA TGAGG TCTTT TTG-3′.
siRNA.
The 3T3-L1 cells were plated into 60-mm-diameter dishes 18–24 h before transfection. Cells were transfected with control or gene-specific siRNA at 50 nM (Dharmacon) in OPTI-MEM medium using Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer’s protocol. The next day, the medium was replaced with fresh DMEM containing 10% calf serum, and the cells were incubated for 24 h before the induction of differentiation. Cellular total RNA and protein were assayed at the indicated time points respectively by RT-PCR and immunoblot. Oil red O staining of Dexras1 knockdown was performed at day 8. The siRNA sequences were as follows: si-Dexras1, #1, 5′-CAGGU UAUCA ACGAA ACUUU U-3′; #2, 5′-GGUCA UUUGC GGUAA CAAAU U-3′; #3, 5′-UCAAA CAGCA GAUCC UAGAU U-3′; and si-glucocorticoid receptor, 5′-GAUCC CCGAA AGCAU UGCAA CCTCA-3′.
Transient Transfection Assay.
Dexras1-overexpressing vector (pcDNA3-Dexras1-FLAG) was generated by inserting the whole ORF of mouse Dexras1 with an N-terminal FLAG tag into pcDNA3.0 (Invitrogen). To maximize transfection efficiency, we used microliter volume electroporation of 3T3-L1 preadipocytes with OneDrop MicroPorator MP-100 (Digital Bio). The cells were trypsinized, washed with 1× PBS, and resuspended in 10 μL of resuspension buffer R with 0.5 μg of plasmid at a concentration of 200,000 cells per pipette. The cells were then microporated at 1,300 V, with a 20-ms pulse width and two pulses. Following microporation, the cells were seeded in 35-mm cell culture dishes and placed at 37 °C in a 10% CO2-humidified atmosphere.
Animals.
We generated Dexras1 knockout mice by deleting whole Dexras1 exons flanked by loxp sites with Cre recombinase (28). Then, Dexras1 mice were backcrossed onto C57BL/6J background for eight generations. Mice were housed in a 12-h light, 12-h dark cycle. At 4 wk of age, mice were either fed normal chow diet or 60% HFD (Harlan Teklad). All mice used for studies were littermate males unless otherwise noted. Animal protocols were performed in accordance with National Institutes of Health guidelines and approved by the Johns Hopkins University Committee on Animal Care.
MEF Adipogenesis Assays.
Wild-type and knockout E14 embryos were isolated from a single heterozygous female that had been paired with a heterozygous male. The head and organs were removed, and the remaining carcasses were minced and incubated in trypsin to obtain single cells. MEFs were seeded in six-well plates and propagated to confluence. Forty-eight hours later, differentiation was initiated using DMEM containing 10% FBS, 0.5 mM IBMX, 1 μM dexamethasone, 10 μg/mL insulin, and 10 μM rosiglitazone for 2 d. Subsequently, cells were maintained in DMEM supplemented with 10% FBS, 10 μg/mL insulin, and 1 μM rosiglitazone. After 48 h, the medium was replaced with maintenance medium containing DMEM supplemented with 10% FBS. Cells were stained with oil red O as described.
Histology and Adipocyte Size Analysis.
Epididymal WAT specimens were fixed in 10% neutral-buffered formalin for 24 h before trimming for histology processing. Paraffin-embedded tissue sections were stained with hematoxylin/eosin according to standard protocols. Adipocyte size was determined by ImageJ.
Body Composition Analysis.
Whole-body lean mass and total fat of mice was measured with an EchoMRI whole-body composition analyzer. One-month-old mice were exposed to normal diet or HFD for 10 wk and were analyzed for lean and fat mass per body weight.
Statistical Analysis.
All results are expressed as means ± SD. Statistical comparisons of groups were made using an unpaired Student t test or two-way ANOVA.
Supplementary Material
Acknowledgments
We thank Prof. Su-Jae Lee (Hanyang University) for providing the H-Ras and K-Ras expression vectors. We thank Drs. Tetsuya Hosooka and Masato Kasuga (Kobe University) for providing a protocol for mouse embryonic fibroblast adipogenesis assays. This work was supported by US Public Health Service Grant MH18501 (to S.H.S.) and by National Research Foundation of Korea Grants 2011-0030711 and 2011-0015665, funded by the Korean government, Ministry of Science, ICT and Future Planning (MSIP).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320454110/-/DCSupplemental.
References
- 1.Friedman JM. Modern science versus the stigma of obesity. Nat Med. 2004;10(6):563–569. doi: 10.1038/nm0604-563. [DOI] [PubMed] [Google Scholar]
- 2.Kopelman PG. Obesity as a medical problem. Nature. 2000;404(6778):635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 3.Otto TC, Lane MD. Adipose development: From stem cell to adipocyte. Crit Rev Biochem Mol Biol. 2005;40(4):229–242. doi: 10.1080/10409230591008189. [DOI] [PubMed] [Google Scholar]
- 4.Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4(4):263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7(12):885–896. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 6.Cristancho AG, Lazar MA. Forming functional fat: A growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12(11):722–734. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacDougald OA, Lane MD. Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem. 1995;64:345–373. doi: 10.1146/annurev.bi.64.070195.002021. [DOI] [PubMed] [Google Scholar]
- 8.Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol. 2000;16:145–171. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 9.Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell. 1975;5(1):19–27. doi: 10.1016/0092-8674(75)90087-2. [DOI] [PubMed] [Google Scholar]
- 10.Kemppainen RJ, Behrend EN. Dexamethasone rapidly induces a novel ras superfamily member-related gene in AtT-20 cells. J Biol Chem. 1998;273(6):3129–3131. doi: 10.1074/jbc.273.6.3129. [DOI] [PubMed] [Google Scholar]
- 11.Fang M, et al. Dexras1: A G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28(1):183–193. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 12.Cheah JH, et al. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron. 2006;51(4):431–440. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng HY, et al. Dexras1 potentiates photic and suppresses nonphotic responses of the circadian clock. Neuron. 2004;43(5):715–728. doi: 10.1016/j.neuron.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 14.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330(6009):1349–1354. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc Natl Acad Sci USA. 2011;108(4):1657–1662. doi: 10.1073/pnas.1018375108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckel-Mahan KL, et al. Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci USA. 2012;109(14):5541–5546. doi: 10.1073/pnas.1118726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng D, Lazar MA. Clocks, metabolism, and the epigenome. Mol Cell. 2012;47(2):158–167. doi: 10.1016/j.molcel.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freytag SO, Paielli DL, Gilbert JD. Ectopic expression of the CCAAT/enhancer-binding protein alpha promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes Dev. 1994;8(14):1654–1663. doi: 10.1101/gad.8.14.1654. [DOI] [PubMed] [Google Scholar]
- 19.Rosen ED, et al. C/EBPalpha induces adipogenesis through PPARgamma: A unified pathway. Genes Dev. 2002;16(1):22–26. doi: 10.1101/gad.948702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 21.Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. 1993;73(4):725–734. doi: 10.1016/0092-8674(93)90252-l. [DOI] [PubMed] [Google Scholar]
- 22.Tong Q, et al. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290(5489):134–138. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- 23.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: Post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13(1):39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindroos J, et al. Human but not mouse adipogenesis is critically dependent on LMO3. Cell Metab. 2013;18(1):62–74. doi: 10.1016/j.cmet.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laplante M, et al. DEPTOR cell-autonomously promotes adipogenesis, and its expression is associated with obesity. Cell Metab. 2012;16(2):202–212. doi: 10.1016/j.cmet.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H, Lee YJ, Choi H, Ko EH, Kim JW. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J Biol Chem. 2009;284(16):10601–10609. doi: 10.1074/jbc.M808742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, et al. Dexras1, a small GTPase, is required for glutamate-NMDA neurotoxicity. J Neurosci. 2013;33(8):3582–3587. doi: 10.1523/JNEUROSCI.1497-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.