

Significance
The blood–brain barrier (BBB) prevents ingress of small molecules into the brain partly by expression of drug efflux transporters. Although several transporters are known to be involved, the role ABCG2 plays in this process is not clear. We demonstrate the ability to image ABCG2 function at the BBB by using luciferin in a luciferase-expressing mouse. This represents a direct measurement of ABCG2 function at the BBB, and demonstrates that ABCG2 does play a functional role at the BBB.
Abstract
ATP-binding cassette (ABC) transporters are a group of transmembrane proteins that maintain chemical homeostasis through efflux of compounds out of organelles and cells. Among other functions, ABC transporters play a key role in protecting the brain parenchyma by efflux of xenobiotics from capillary endothelial cells at the blood–brain barrier (BBB). They also prevent the entry of therapeutic drugs at the BBB, thereby limiting their efficacy. One of the key transporters playing this role is ABCG2. Although other ABC transporters can be studied through various imaging modalities, no specific probe exists for imaging ABCG2 function in vivo. Here we show that d-luciferin, the endogenous substrate of firefly luciferase, is a specific substrate for ABCG2. We hypothesized that ABCG2 function at the BBB could be evaluated by using bioluminescence imaging in transgenic mice expressing firefly luciferase in the brain. Bioluminescence signal in the brain of mice increased with coadministration of the ABCG2 inhibitors Ko143, gefitinib, and nilotinib, but not an ABCB1 inhibitor. This method for imaging ABCG2 function at the BBB will facilitate understanding of the function and pharmacokinetic inhibition of this transporter.
Provision of nutrients and maintenance of chemical homeostasis in the brain is performed by the endothelial cells of brain capillaries within a neurovascular unit termed the blood–brain barrier (BBB) (1). In contrast to endothelial cells of capillaries elsewhere in the body, those in the brain are joined by tight junctions forming a physiologic barrier. Drug delivery to the brain depends on physicochemical characteristics such as lipophilicity, molecular weight, and ionic state. For many compounds, brain entry is lower than other tissues/organs because of the presence of ATP-binding cassette (ABC) efflux transporters at the apical surface of endothelial cells at the BBB (2, 3). These transporters maintain chemical homeostasis in the brain, and prevent toxins from interfering with neural processes by regulating the compounds that can enter the brain.
ABC transporters contribute to the clinical challenge of drug delivery to the brain, and it has been estimated that only 2% of drug discovery compounds can cross the BBB to reach therapeutic targets (4). ABCG2 (also known as breast cancer resistance protein) and ABCB1 (also called P-glycoprotein) are the two most highly expressed efflux transporters at the BBB (5). Altered expression of ABC transporters at the BBB has been associated with a range of pathophysiological conditions (2, 6). ABC efflux transporters at the BBB also play a major role in limiting effective concentrations of chemotherapeutic agents to treat primary and metastatic tumors in the brain (7). ABCG2 has been shown to work in tandem with ABCB1 at the BBB (8, 9). However, its individual contribution is not understood.
Molecular imaging allows the measurement of the individual contribution and function of transporters in vivo (10). Efflux of a substrate by transporters at the BBB is reflected by little to no uptake in brain tissue, and when efflux transport is pharmacologically inhibited, increased accumulation occurs (11, 12). Although a number of radiolabeled specific substrates have been developed to study ABCB1 function by using positron emission tomography (PET), no specific probe exists for imaging ABCG2 function at the BBB (13, 14).
Whole-animal bioluminescent imaging (BLI) is increasingly used in mouse genetic studies to visualize cellular events (15). The primary reporters used for BLI are the light-generating luciferase enzymes and their substrates, such as firefly luciferase (fLuc) and d-luciferin. It has been reported that ABCG2 expression decreases bioluminescence in fLuc cells compared with control cells (16), and biodistribution studies have reported low distribution of d-luciferin in the brain (17). This suggests that ABCG2 may restrict the entry of d-luciferin at the BBB. We hypothesized that ABCG2 function at the BBB could be examined by using BLI in transgenic mice expressing fLuc in the brain.
In this study, we sought to answer two questions. First, is d-luciferin a specific substrate of human and murine ABCG2? To assess this directly, we measured the fluorescence levels of d-luciferin in human and mouse cells that overexpress select ABC transporters. Second, can d-luciferin be used in vivo as a probe to measure ABCG2 function at the BBB? To answer this question, we used BLI to measure the bioluminescence in the brain of fLuc-expressing transgenic mice administered d-luciferin with or without an inhibitor of ABCG2. Our goal was to develop time-course BLI of the mouse brain with a view to understanding the kinetics of ABCG2 activity at the BBB.
Results
d-Luciferin Is a Specific Substrate of Human ABCG2 and Not Human ABCB1 or ABCC1 (MRP1).
d-Luciferin’s (Fig. 1A) cellular accumulation was examined by measuring its fluorescence (excitation, 350 nm; emission, 530 nm) (18) with flow cytometry in human ABCG2-expressing cell lines (MCF7 FLV500, MCF7 MX100, and H460 MX20) and their parental cells (MCF7 and H460). Intracellular accumulation plateaued after 30 min and was pH-dependent (Fig. S1). The accumulation of d-luciferin (2 mM) was significantly lower in ABCG2-expressing sublines than parental cells (Fig. 1). d-Luciferin accumulation in MCF7 cells demonstrated high accumulation that was unaffected by the ABCG2 inhibitor Ko143 (19) (Fig. 1 B and C). The ABCG2-expressing subline MCF7 FLV500 demonstrated low accumulation, which was increased by Ko143 (Fig. 1D). Compared with parental cells, d-luciferin accumulation was significantly lower in the ABCG2-expressing sublines MCF7 MX100 (10.3 ± 1.5%), MCF7 FLV500 (8.7 ± 1.5%), and H460 MX20 (6.3 ± 0.6%) than their parental cells (Fig. 1 E and F). In the presence of Ko143 (5 µM), accumulation of d-luciferin was similar to that observed in parental cells. The parental cells express low-level functional ABCG2 that also results in slightly elevated d-luciferin accumulation in the presence of ABCG2 inhibitors (20). This inhibition of ABCG2-mediated efflux was also observed in the presence of another potent ABCG2 inhibitor, fumitremorgin C (FTC), at 5 µM (Fig. 1G). Confocal microscopy visually confirmed low-level fluorescence in H460 MX20 cells (Fig. 2A) that increased when treated with Ko143.
Fig. 1.

Flow cytometry analysis of d-luciferin (excitation, 350 nm; emission, 530/30 nm) accumulation in human ABC transporter-expressing cells and their parental cells demonstrates that d-luciferin is a specific substrate for human ABCG2. Structure of (A) d-luciferin and (B) Ko143. (C) MCF7 or (D) ABCG2-expressing MCF7 FLV500 cells incubated with d-luciferin alone (2 mM; gray) or with the ABCG2 inhibitor Ko143 (5 µM; black). For comparison, (E) MCF7 or (F) ABCG2-expressing MCF7 FLV500 cells incubated with the standard ABCG2 substrate pheophorbide A alone (10 μM; gray) or with the ABCG2 inhibitor Ko143 (5 µM; black). The average of the geometric mean for three experiments with d-luciferin in ABCG2-expressing (G) H460 MX20 and (H) MCF7 MX100 and MCF7 FLV500 cells compared with parental cells tested in the presence of ABCG2 inhibitors Ko143 (5 µM) or FTC (5 µM). (I) Incubation of H460 MX20 cells with d-luciferin was tested at 4 °C alone or in the presence of Ko143 and compared with incubation at 37 °C. d-Luciferin accumulation in human parental and sublines overexpressing (J) ABCB1 (KB-8-5-11) or (K) ABCC1 (HEK MRP1) alone, or with inhibitors of ABCB1 (verapamil 20 µM) or ABCC1 [indomethacin (INDO) 100 µM]. All accumulation values are normalized to the accumulation in the parental cell lines in the presence of inhibitor. Data represent means ± SD of three experiments (***P < 0.001 by one-way ANOVA; α = 0.01).
Fig. 2.

d-Luciferin is a substrate for ABCG2 (A) Confocal images of H460 MX20 cells incubated with d-luciferin alone or with Ko143 (5 µM) shown at a magnification of 63×. (Scale bar: 20 µm.) (B) Effect of d-luciferin and its derivatives on ATPase activity of ABCG2 and ABCB1. The ATPase activity in the presence of compound was divided by basal activity to calculate fold stimulation. Nilotinib (5 µM) was included as a positive control (+). Data represent means ± SD of three experiments (***P < 0.001 by one-way ANOVA; α = 0.05). (C) d-Luciferin (500 µM) accumulation was tested in BB19 immortalized human brain endothelial cells alone or with inhibitors of ABCB1 (verapamil 20 µM), ABCC1 [indomethacin (INDO) 100 µM], and ABCG2 (FTC 5 µM and Ko143 5 µM). Data represent means ± SD of three experiments (***P < 0.001 by one-way ANOVA; α = 0.01).
ABCG2-mediated efflux of substrates is driven by ATP-hydrolysis. Incubation at 4 °C (compared with 37 °C) elevated intracellular accumulation of d-luciferin (Fig. 1I), which was not affected by the presence of Ko143. To measure directly d-luciferin’s effect on the ATPase activity of ABCG2, ATPase activity was measured in the presence of d-luciferin (10, 50, and 100 µM). A significant increase in ABCG2 ATPase activity over basal activity was observed at 100 µM (1.40 ± 0.06 fold; P < 0.001; Fig. 2B). This absolute stimulation is low relative to positive control nilotinib, consistent with the majority of luciferin being ionized and not able to enter the lipid bilayer and interact with ABCG2, as observed for the organic acid substrate sulfasalazine (21). In contrast, the nonionizable ethyl ester of luciferin produced 2.3-fold stimulation of ABCG2 activity (Fig. S2). d-Luciferin can also act as an inhibitor of ABCG2 at high concentrations (Fig. S3).
Cell lines expressing ABCB1 (KB-8-5-11) and ABCC1 (HEK MRP1) were examined to assess the specificity of d-luciferin for ABCG2 among other ABC transporters at the BBB. The human parental and ABCB1-expressing or ABCC1-expressing sublines accumulated the same amounts of d-luciferin as parental cells. Furthermore, d-luciferin accumulation did not change when coincubated with ABCB1 inhibitor (verapamil 20 µM) or ABCC1 inhibitor (indomethacin 100 µM; Fig. 1 J and K). d-Luciferin (10, 50, and 100 µM) did not stimulate ABCB1 ATPase activity significantly beyond its basal activity (Fig. 2B). d-Luciferin was also shown to be a specific substrate of mouse Abcg2 (Fig. S4).
d-Luciferin Accumulation Is Highest in Cultured Human BBB Endothelial Cells When ABCG2 Is Inhibited.
d-Luciferin accumulation was assessed in BB19 immortalized human brain endothelial cells, which express the full complement of BBB transporters (22). BB19 cells were incubated with d-luciferin (500 µM) alone or in the presence of the inhibitors of ABCB1 (verapamil 20 µM), ABCC1 (indomethacin 100 µM), and ABCG2 (FTC, 5 µM and Ko143, 5 µM). Both ABCG2 inhibitors demonstrated a significant increase in d-luciferin accumulation (Ko143, 78 ± 2.30%, P < 0.0001; FTC, 43 ± 0.58%, P < 0.0001), but indomethacin [6 ± 3.46%; difference not significant (NS)] and verapamil (1 ± 0.57%; NS) did not demonstrate a significant effect.
Bioluminescence in Vitro Increases when Coincubated with ABCG2 Inhibitors.
To test the relative effectiveness of reported ABCG2 inhibitors in vitro, we measured the bioluminescence signal of HEK cells transfected with fLuc and ABCG2 (23) in the presence of the ABCG2 inhibitors Ko143 (19), tariquidar (24), elacridar (25), gefitinib, or nilotinib (26). The ABCB1 inhibitor (2R)-anti-5-f3-[4-(10,11-dichloromethanodibenzo-suber-5-yl)piperazin-1-yl]-2-hydroxypropoxygquinoline trihydrochloride(DCPQ) (27, 28) and HEK cells without ABCG2 (HEK/empty/fLuc) were used as controls. Bioluminescence was detected at lower concentrations of d-luciferin compared with direct fluorescence (Fig. S1C). Bioluminescence values were normalized to the highest signal achieved (Fig. 3A). Ko143 demonstrated the greatest increase in bioluminescence signal and the lowest effective concentration required to inhibit efflux activity by 50% (EC50 of 11 ± 7 nM). Gefitinib (EC50 of 666 ± 38 nM) and nilotinib (EC50 of 4 ± 0.7 µM) also demonstrated a significant increase in bioluminescence signal. Tariquidar and elacridar did not achieve a 50% increase in bioluminescent signal. The relative order of potency achieved by the inhibitors was Ko143, gefitinib, nilotinib, followed by elacridar and tariquidar. DCPQ did not alter the bioluminescent signal at any concentration tested. The inhibitors did not alter the bioluminescent signal in HEK/empty/fLuc (Fig. S5).
Fig. 3.

In vivo bioluminescent signal is increased by ABCG2 inhibitors. (A) HEK/ABCG2/fLuc cells were incubated with serial dilutions of the ABCG2 inhibitors Ko143, gefitinib, nilotinib, tariquidar, and elacridar. DCPQ, an ABCB1 inhibitor, was included as a control. Data points represent means ± SD of three observations. (B) Time–activity curve of total flux (photons per second) acquired from mice treated with 16 mg/kg of Ko143 or vehicle before d-luciferin administration. Data represent means ± SD of four experiments. (C) AUC calculated from each time–activity curve. Data represent means ± SD of four experiments (****P < 0.0001, ***P < 0.001, and **P < 0.01 by one-way ANOVA; α = 0.05).
Bioluminescence in the Mouse Brain Increases when Pretreated with ABCG2 Inhibitors.
To investigate the function of ABCG2 at the BBB in vivo, we used a transgenic mouse model that expresses fLuc controlled by the GFAP promoter (29). GFAP is an intermediate filament protein expressed mainly in astrocytes (30), and, as such, fLuc is expressed predominantly in the mouse brain (29, 31). For the production of bioluminescence in the brain, d-luciferin must diffuse across the endothelial cells in the BBB that contain ABCG2. We predicted that pharmacologic inhibition of ABCG2 function would increase d-luciferin entry into the brain parenchyma and result in increased bioluminescent signal.
Baseline bioluminescent signal in the brain of each animal was first determined following i.p. administration of d-luciferin (18 mg/kg). To subsequently test the relative efficacy of ABCG2 inhibitors in vivo, inhibitors (16 mg/kg) were administered i.v. before d-luciferin administration, and images of resulting light produced from mice were acquired for 60 min. The highest peak in total flux was observed in mice pretreated with Ko143 (Fig. 3B), and it produced the greatest increase in the area under the curve (AUC) of total flux (1.31 × 108 ± 1.14 × 107 p/s⋅min; P < 0.0001) relative to vehicle (Fig. 3C). A significant increase in the AUC of total flux was also observed for gefitinib (7.48 ± 2.15 × 107 p/s⋅min; P < 0.001) and nilotinib (7.02 ± 1.84 × 107 p/s⋅min; P < 0.005). Some increase in bioluminescence was observed with the administration of elacridar (4.60 × 107 ± 7.91 × 106 p/s⋅min; NS) and tariquidar (3.67 × 107 ± 1.61 × 107 p/s⋅min; NS), but it was not significant. The relative order of potency achieved by the inhibitors in vivo was similar to in vitro results for ABCG2 inhibition. No appreciable difference was observed with the administration of the ABCB1 inhibitor DCPQ (2.95 ± 1.73 × 107 p/s⋅min; P > 0.9999) relative to vehicle administration (2.56 1.19 × 107 p/s⋅min) or baseline signal (2.81 ± 0.13 × 107 p/s⋅min). The time to peak in photon flux with vehicle administration relative to the inhibitors was not significantly different (Fig. S6).
To confirm that the photons emitted were from the brain, mice injected with vehicle or Ko143 (16 mg/kg; Fig. 4 A and C) were removed for sampling 20 min after injection of d-luciferin, and the brains were immediately resected and imaged ex vivo (Fig. 4B). The ex vivo bioluminescence of the brain was significantly greater (P < 0.05) in the Ko143-treated mice (2.68 ± 0.68 × 107 p/s) than in those treated with vehicle (1.23 ± 0.15 × 107 p/s; Fig. 4D), and no detectable bioluminescence remained in the mouse following removal of the brain.
Fig. 4.

Ex vivo analysis confirms BLI signal from the brain. (A) Representative images of mice treated with vehicle or Ko143 (16 mg/kg). (B) Mice were euthanized 20 min after d-luciferin administration, and brains were resected for immediate ex vivo imaging. (C) In vivo analyses of the brain region for total flux at 20 min after d-luciferin administration. (D) Ex vivo analyses of resected brain tissue for total flux at 21 min after d-luciferin administration.
As Ko143 demonstrated the greatest efficacy among ABCG2 inhibitors in vitro and in vivo, it was selected for dose–response characterization at doses of 4, 8, 16, and 32 mg/kg. Ko143-treated mice produced increased signal from the brain region with increasing doses of Ko143, and a dose-dependent increase in the AUC of brain bioluminescent signal was observed (Fig. 5A). The AUC of total flux over 60 min duration showed a significant difference from vehicle control for every dose of Ko143 tested: 4 mg/kg (6.01 ± 0.42 × 107 p/s⋅min; P < 0.005), 8 mg/kg (8.31 ± 1.47 × 107 p/s⋅min; P < 0.0001), 16 mg/kg (1.31 ± 0.11 × 108 p/s⋅min; P < 0.0001), and 32 mg/kg (1.37 ± 0.15 × 108 p/s⋅min; Fig. 5B). The lack of a difference in the AUC of total flux between 32 mg/kg and 16 mg/kg suggested near-maximal inhibition of ABCG2 had been achieved at 16 mg/kg (Fig. 5B). A sigmoidal curve fit to the data produced a calculated ED50 of 6.3 ± 1.8 mg/kg (Fig. 5C).
Fig. 5.
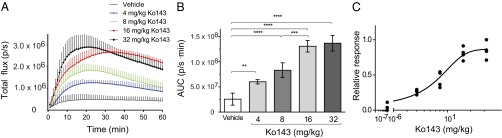
A Ko-143 dose-dependent increase in bioluminescence was observed in vivo. (A) Time-activity curve of total flux (photons per second) acquired from mice treated with Ko143 or vehicle before d-luciferin administration. Data represent means ± SD of four observations. (B) The AUC acquired from the time–activity curve for each dose of Ko143. (C) A sigmoidal curve fit to the AUC for each dose of Ko143. Data represent means ± SD of four observations.
It is possible that Ko143 inhibition of ABCG2 could alter peripheral luciferin pharmacokinetics, or that protein binding by Ko143 could elevate the free fraction of luciferin in blood, and subsequently increase brain accumulation by mass action. Plasma from mice injected with vehicle or 16 mg/kg Ko143 followed by luciferin was collected after 30 min, and immediately added to fLuc solution. No difference in bioluminescence was observed, indicating that plasma concentrations of luciferin were equivalent (Fig. S7A). Bioluminescent signal of luciferin-containing mouse plasma was also unaffected by addition of Ko143 ex vivo (150 and 300 µM), indicating that the free fraction of d-luciferin is not elevated by changes in protein binding in the presence of Ko143 (Fig. S7B).
Discussion
Bioluminescence Imaging of ABCG2 at the BBB.
Our results showed that d-luciferin is a specific substrate for mouse and human ABCG2 in vitro. Our in vitro findings are consistent with observations that bioluminescent signal is low in ABCG2-expressing cells (because of limited d-luciferin) but not ABCB1- or ABCC1-expressing cells. We observed relatively low bioluminescence in vivo from the brain when d-luciferin was administered to mice with luciferase expressed in the brain. The low brain signal observed is consistent with the reported low distribution to the brain when mice were injected with d-[14C]luciferin (<1% injected dose per gram) (17).
Two confounding processes could have occurred to artificially elevate brain BLI signal. First, ABCG2 inhibitors such as Ko143 could permeabilize tight junctions at the BBB, nonspecifically allowing d-luciferin uptake into the brain; several studies have shown that brain uptake of small molecules is not facilitated by Ko143 doses (8, 13), and toxicity and brain histology were reported to be unaffected by Ko143 (19). Second, Ko143 may elevate systemic luciferin levels, or the free fraction of luciferin; analysis of blood plasma showed that no systemic effect had occurred (Fig. S7).
Our imaging protocol may benefit from the irreversible enzymatic conversion of d-luciferin to oxyluciferin in the brain, driving further brain uptake of d-luciferin to amplify the signal. From an imaging perspective, a trapping mechanism is beneficial because it can amplify the signal. Berger et al. showed that fLuc-transfected cells trap >20-fold more d-[14C]luciferin than nontransfected cells, probably because of intracellular metabolic consumption of d-luciferin driving its uptake. This trapping increases bioluminescence. Signal amplification is also observed with [18F]fluorodeoxyglucose, which is irreversibly trapped via phosphorylation by the glucose-6-kinase (32) and [11C]desmethyl loperamide, which is trapped by lysosomal sequestration, again amplifying signal (11).
Studies investigating Abcg2 function at the BBB in mouse models have been extrapolated to human ABCG2 function (9, 29). The mouse ortholog of Abcg2 has 81% protein sequence homology with human ABCG2 (33), and a single amino acid mutation can alter the substrate and antagonist specificity in both species (34). We have demonstrated that the substrate and inhibitor specificity of human and mouse ABCG2 is very similar (20). The overlapping substrate and inhibitor specificity of human and mouse ABCG2 supports utilization of mouse models in understanding the clinical, physiological, and pharmacological roles of ABCG2.
Inhibition of ABCG2 at the BBB Using BLI.
Investigating ABCG2 function with BLI offers the ability to observe ABCG2 transport function in vitro and over time in vivo. Because d-luciferin emits a single photon on oxidation by luciferase (i.e., one photon per d-luciferin molecule), the signal represents d-luciferin levels entering the brain. ABCG2 modulates the kinetics of brain bioluminescent signal output by regulating the amount of d-luciferin crossing the BBB and thus controlling the availability of d-luciferin in the brain. By using GFAP-fLuc transgenic mice, we measured the ability of a number of ABCG2 inhibitors to prevent efflux of d-luciferin in mice. Of the drugs tested, Ko143 was the most effective inhibitor of ABCG2 function. It had previously been shown to inhibit ABCG2 at nanomolar concentrations by blocking ATPase activity of the transporter (35). We derived an ED50 for maximal inhibition of ABCG2 at the BBB of 6.3 ± 1.8 mg/kg. This compares well with that derived by Wanek et al., who assessed the brain uptake of the ABCB1/ABCG2 substrate [11C]tariquidar in double-KO mdr1a−/−/mdr1b−/− mice, and reported a Ko143 ED50 of 4.98 ± 1.4 mg/kg (36). Although not as potent as Ko143, gefitinib demonstrated a significant increase in brain bioluminescent signal.
Gefitinib has been reported to enhance the efficacy of photodynamic therapy by using 5-aminolevulinic acid in malignant brain tumor cells (37), and our results suggest that it may achieve this by blocking ABCG2-mediated efflux of 5-aminolevulinic acid at the BBB. We found that tariquidar and elacridar had little effect on ABCG2 transport of d-luciferin. Both molecules are avid substrates of ABCG2, and act as moderate inhibitors in vitro (20, 24). Elacridar has been reported to reach a saturable efflux in the brain at 10 mg/kg (38), and increases gefitinib brain uptake (which requires inhibition of ABCG2 and ABCB1) (25). The lack of efficacy of tariquidar and elacridar observed may result from their relatively poor inhibition of ABCG2 (Fig. 3A) and high plasma binding with consequent very low free fraction in blood plasma.
The Role of ABCG2 at the BBB.
Until now, it has not been possible to perform dynamic assessment of ABCG2’s function at the BBB in mice with WT ABC transporter function, and the two specific ABCG2 substrates assessed for PET imaging have not proved successful (14). Both [11C]dantrolene and the celecoxib metabolite [11C]SC-62807 have been assessed in mice (39, 40). Neither demonstrated appreciable brain uptake in Abcg2−/− mice. Both SC-62807 and dantrolene require transporter-mediated uptake processes, and these may be limiting at the BBB.
Few substrates are reported to be selectively transported by ABCG2 (8, 13, 41). As a consequence, ABCG2 has been proposed to play a minor role at the BBB (42). Zhao et al. assessed four ABCG2 substrates, three of which (alfuzosin, dipyridamole, and LY2228820) are also ABCB1 substrates, and one (cimetidine) did not display brain uptake under conditions tested. For each compound, ABCG2 KO mice did not exhibit elevated brain levels compared with WT mice, leading to the aforementioned conclusions. However, it is now well recognized that dual ABCG2/ABCB1 substrates require genetic KO of both transporter types in mice before brain levels are dramatically elevated (8). Conversely, Kodaira et al. did report that the specific ABCG2 substrates daidzein and genistein demonstrated elevated brain levels in ABCG2 KO mice (43). In our present study, pharmacologic inhibition of ABCG2 by Ko143 resulted in an appreciable increase in the brain accumulation of d-luciferin (fivefold increase) at levels comparable to those observed in studies examining ABCB1 substrates such as [11C]desmethyl loperamide (2.5-fold) (12) and [11C]verapamil (11-fold) (44).
It has been suggested that clinical inhibition of ABC transporter function at the BBB may not be achievable (45). We showed that ABCG2 plays a role in limiting the biodistribution of a specific substrate, and that saturable inhibition of ABCG2 at the BBB by using pharmacologic levels of inhibitor is possible. Nevertheless, in a therapeutic context, a dose of 16 mg/kg in the human with an inhibitor such as Ko143 could be achieved by direct intraarterial infusion, in a similar fashion to that used for nonspecific opening of the BBB by mannitol for drug delivery (46).
Important Considerations Concerning Brain Bioluminescence.
For in vivo BLI, limited d-luciferin biodistribution will limit bioluminescence intensity from that organ. It has been stated that d-luciferin can penetrate the BBB (47), but here we demonstrate that, by inhibiting ABCG2, the brain uptake of d-luciferin can be further enhanced. The GFAP-fLuc model used here is used primarily to assess astrogliosis associated with neuroinflammation, neurodegeneration, infection, and cancer progression (48–52). One implication of our finding is that these pathologic conditions are associated with BBB disruption (53, 54), which would increase the entry of d-luciferin and bioluminescence.
Comparison with Other Imaging Modalities to Measure ABC Transporter Function.
Noninvasive imaging of ABCB1 in living animals and humans has previously been reported with radiolabeled transport substrates for PET. Alternative techniques are available to assess whether a candidate small molecule can cross the BBB, but they are encumbered with limitations. Examples include confocal microscopy (by using a fluorescent substrate) or a long-t1/2 radiotracer (usually 3H or 14C), but both are necessarily end-point experiments whereby brain penetration is assessed ex vivo at a single time point. In contrast, BLI represents a relatively low-cost alternative for studying time-course transporter function in vivo compared with other strategies such as PET.
Conclusion
Drug delivery to the CNS still remains a problem because of the BBB (55). Patients with brain metastases are excluded from clinical trials because drugs are not BBB-permeable (56), and the efflux transporter ABCG2 affects the pharmacokinetics of many drugs. d-Luciferin is amenable to radiolabeling for dual PET and optical imaging (57) and therefore may be combined for multimodality imaging of ABCG2 function at the BBB, and development of radiolabeled d-luciferin for PET studies or pharmacokinetic microdosing studies would allow use of the molecule in animals not expressing fLuc, including humans. Methods to increase drug delivery to the brain, such as intranasal delivery (55), osmotic disruption of the BBB (46), and regulation of ABCG2 (58), can now be assessed in real time by using this model. Furthermore, the relationship between ABC transporter function and a range of pathophysiologies can also now be more easily investigated.
Materials and Methods
Details of cell culture, flow cytometry, fluorescence spectroscopy, confocal microscopy, ATPase assay, in vitro bioluminescence, animal protocol, and drug formulations are provided in SI Materials and Methods.
BLI.
The FVB/N-Tg(GFAP-fLuc)-Xen mice model was provided by Taconic Farms (29). Animals were injected i.v. with inhibitors followed by i.p. d-luciferin injection 10 min later. BLI signals from the ventral brain are higher (59); therefore, mice are imaged in supine position, which diminishes the signal from the cochlear spiral ganglion (60). A 60-min sequential acquisition with 1-min intervals was carried out to record the bioluminescent signals. To quantify the light output from mouse brains, we defined the region of interest (ROI) over the brain and examined all values in the same ROI in sequential images by using LivingImage software (PerkinElmer). Each data point was corrected for the host’s baseline signal level. The AUC value was calculated by numerical integration of light output (in photons per second) and time by using Prism 6.0 (GraphPad Software).
Statistical Analysis.
For in vivo BLI studies, data are expressed as mean ± SD from at least four mice. Time–activity curves were created for each mouse by plotting the total flux (in photons per second) vs. time (in minutes), and the area under the time–activity curve (i.e., photons per second·minutes) was calculated by the trapezoidal method by using Prism 6.0 (GraphPad software). After the data were tested for homogeneity of variance, differences in mean AUC (photons per second·minutes) were compared by using a one-way ANOVA, followed by Bonferroni posttest for multiple comparisons (α = 0.05).
Supplementary Material
Acknowledgments
We thank Mr. George Leiman for editorial assistance, Dr. Jeih-San Liow for training on tail vein injections, Dr. Michael Maurizi for fluorescence spectral analysis assistance, Ms. Karen Wolcott for FACS assistance, Ms. Susan Garfield for help with confocal microscopy, and Dr. Robert B. Innis and Dr. Victor W. Pike for helpful discussions. This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute. J.B. and K.-E.C. are NIH Medical Research Scholars. The Medical Research Scholars Program is a public–private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from Pfizer, the Leona M. and Harry B. Helmsley Charitable Trust, the Howard Hughes Medical Institute, as well as other private donors (listed at www.fnih.org/work/programs-development/medical-research-scholars-program).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312159110/-/DCSupplemental.
References
- 1.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kannan P, et al. Imaging the function of P-glycoprotein with radiotracers: Pharmacokinetics and in vivo applications. Clin Pharmacol Ther. 2009;86(4):368–377. doi: 10.1038/clpt.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Begley DJ. Delivery of therapeutic agents to the central nervous system: The problems and the possibilities. Pharmacol Ther. 2004;104(1):29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Pardridge WM. Crossing the blood-brain barrier: Are we getting it right? Drug Discov Today. 2001;6(1):1–2. doi: 10.1016/s1359-6446(00)01583-x. [DOI] [PubMed] [Google Scholar]
- 5.Uchida Y, et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem. 2011;117(2):333–345. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 6.Xiong H, et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J Neurosci. 2009;29(17):5463–5475. doi: 10.1523/JNEUROSCI.5103-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer. 2011;11(5):352–363. doi: 10.1038/nrc3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agarwal S, Hartz AM, Elmquist WF, Bauer B. Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des. 2011;17(26):2793–2802. doi: 10.2174/138161211797440186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vlaming ML, Lagas JS, Schinkel AH. Physiological and pharmacological roles of ABCG2 (BCRP): Recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev. 2009;61(1):14–25. doi: 10.1016/j.addr.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Mairinger S, Erker T, Muller M, Langer O. PET and SPECT radiotracers to assess function and expression of ABC transporters in vivo. Curr Drug Metab. 2011;12(8):774–792. doi: 10.2174/138920011798356980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kannan P, et al. Lysosomal trapping of a radiolabeled substrate of P-glycoprotein as a mechanism for signal amplification in PET. Proc Natl Acad Sci USA. 2011;108(6):2593–2598. doi: 10.1073/pnas.1014641108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannan P, et al. N-desmethyl-loperamide is selective for P-glycoprotein among three ATP-binding cassette transporters at the blood-brain barrier. Drug Metab Dispos. 2010;38(6):917–922. doi: 10.1124/dmd.109.031161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wanek T, et al. A novel PET protocol for visualization of breast cancer resistance protein function at the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):2002–2011. doi: 10.1038/jcbfm.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kusuhara H. Imaging in the study of membrane transporters. Clin Pharmacol Ther. 2013;94(1):33–36. doi: 10.1038/clpt.2013.85. [DOI] [PubMed] [Google Scholar]
- 15.Thorne N, Inglese J, Auld DS. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol. 2010;17(6):646–657. doi: 10.1016/j.chembiol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, et al. ABCG2/BCRP expression modulates D-luciferin based bioluminescence imaging. Cancer Res. 2007;67(19):9389–9397. doi: 10.1158/0008-5472.CAN-07-0944. [DOI] [PubMed] [Google Scholar]
- 17.Berger F, Paulmurugan R, Bhaumik S, Gambhir SS. Uptake kinetics and biodistribution of 14C-D-luciferin—a radiolabeled substrate for the firefly luciferase catalyzed bioluminescence reaction: Impact on bioluminescence based reporter gene imaging. Eur J Nucl Med Mol Imaging. 2008;35(12):2275–2285. doi: 10.1007/s00259-008-0870-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowie LJ. Synthesis of firefly luciferin and structural analogs. Methods Enzymol. 1978;57:15–28. [Google Scholar]
- 19.Allen JD, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1(6):417–425. [PubMed] [Google Scholar]
- 20.Bakhsheshian J, et al. Overlapping substrate and inhibitor specificity of human and murine ABCG2. Drug Metab Dispos. 2013;41(10):1805–1812. doi: 10.1124/dmd.113.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jani M, et al. Kinetic characterization of sulfasalazine transport by human ATP-binding cassette G2. Biol Pharm Bull. 2009;32(3):497–499. doi: 10.1248/bpb.32.497. [DOI] [PubMed] [Google Scholar]
- 22.Kusch-Poddar M, Drewe J, Fux I, Gutmann H. Evaluation of the immortalized human brain capillary endothelial cell line BB19 as a human cell culture model for the blood-brain barrier. Brain Res. 2005;1064(1-2):21–31. doi: 10.1016/j.brainres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, et al. Identification of inhibitors of ABCG2 by a bioluminescence imaging-based high-throughput assay. Cancer Res. 2009;69(14):5867–5875. doi: 10.1158/0008-5472.CAN-08-4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kannan P, et al. The “specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2) ACS Chem Neurosci. 2011;2(2):82–89. doi: 10.1021/cn100078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334(1):147–155. doi: 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozvegy-Laczka C, et al. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol. 2004;65(6):1485–1495. doi: 10.1124/mol.65.6.1485. [DOI] [PubMed] [Google Scholar]
- 27.Shepard RL, Cao J, Starling JJ, Dantzig AH. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int J Cancer. 2003;103(1):121–125. doi: 10.1002/ijc.10792. [DOI] [PubMed] [Google Scholar]
- 28.Bao X, et al. [¹¹C]Rhodamine-123: synthesis and biodistribution in rodents. Nucl Med Biol. 2012;39(8):1128–1136. doi: 10.1016/j.nucmedbio.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu L, et al. Non-invasive imaging of GFAP expression after neuronal damage in mice. Neurosci Lett. 2004;367(2):210–212. doi: 10.1016/j.neulet.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 30.Bignami A, Eng LF, Dahl D, Uyeda CT. Localization of the glial fibrillary acidic protein in astrocytes by immunofluorescence. Brain Res. 1972;43(2):429–435. doi: 10.1016/0006-8993(72)90398-8. [DOI] [PubMed] [Google Scholar]
- 31.Cho W, Hagemann TL, Johnson DA, Johnson JA, Messing A. Dual transgenic reporter mice as a tool for monitoring expression of glial fibrillary acidic protein. J Neurochem. 2009;110(1):343–351. doi: 10.1111/j.1471-4159.2009.06146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muzi M, et al. Quantitative assessment of dynamic PET imaging data in cancer imaging. Magn Reson Imaging. 2012;30(9):1203–1215. doi: 10.1016/j.mri.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen JD, Brinkhuis RF, Wijnholds J, Schinkel AH. The mouse Bcrp1/Mxr/Abcp gene: amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res. 1999;59(17):4237–4241. [PubMed] [Google Scholar]
- 34.Robey RW, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89(10):1971–1978. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glavinas H, et al. ABCG2 (breast cancer resistance protein/mitoxantrone resistance-associated protein) ATPase assay: A useful tool to detect drug-transporter interactions. Drug Metab Dispos. 2007;35(9):1533–1542. doi: 10.1124/dmd.106.014605. [DOI] [PubMed] [Google Scholar]
- 36.Wanek T, Mairinger S, Langer O. Radioligands targeting P‐glycoprotein and other drug efflux proteins at the blood–brain barrier. J Labelled Comp Radiopharm. 2013;56(3-4):68–77. doi: 10.1002/jlcr.2993. [DOI] [PubMed] [Google Scholar]
- 37.Sun W, et al. Gefitinib enhances the efficacy of photodynamic therapy using 5-aminolevulinic acid in malignant brain tumor cells. Photodiagn Photodyn Ther. 2013;10(1):42–50. doi: 10.1016/j.pdpdt.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 38.Sane R, Agarwal S, Mittapalli RK, Elmquist WF. Saturable active efflux by p-glycoprotein and breast cancer resistance protein at the blood-brain barrier leads to nonlinear distribution of elacridar to the central nervous system. J Pharmacol Exp Ther. 2013;345(1):111–124. doi: 10.1124/jpet.112.199786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Enokizono J, Kusuhara H, Ose A, Schinkel AH, Sugiyama Y. Quantitative investigation of the role of breast cancer resistance protein (Bcrp/Abcg2) in limiting brain and testis penetration of xenobiotic compounds. Drug Metab Dispos. 2008;36(6):995–1002. doi: 10.1124/dmd.107.019257. [DOI] [PubMed] [Google Scholar]
- 40.Takashima T, et al. Evaluation of breast cancer resistance protein function in hepatobiliary and renal excretion using PET with 11C-SC-62807. J Nucl Med. 2013;54(2):267–276. doi: 10.2967/jnumed.112.110254. [DOI] [PubMed] [Google Scholar]
- 41.Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333(3):788–796. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- 42.Zhao R, et al. Breast cancer resistance protein interacts with various compounds in vitro, but plays a minor role in substrate efflux at the blood-brain barrier. Drug Metab Dispos. 2009;37(6):1251–1258. doi: 10.1124/dmd.108.025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kodaira H, et al. Quantitative evaluation of the impact of active efflux by p-glycoprotein and breast cancer resistance protein at the blood-brain barrier on the predictability of the unbound concentrations of drugs in the brain using cerebrospinal fluid concentration as a surrogate. J Pharmacol Exp Ther. 2011;339(3):935–944. doi: 10.1124/jpet.111.180398. [DOI] [PubMed] [Google Scholar]
- 44.Bauer M, et al. Pgp-mediated interaction between (R)-[11C]verapamil and tariquidar at the human blood-brain barrier: A comparison with rat data. Clin Pharmacol Ther. 2012;91(2):227–233. doi: 10.1038/clpt.2011.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalvass JC, et al. International Transporter Consortium Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: The ITC evidence-based position. Clin Pharmacol Ther. 2013;94(1):80–94. doi: 10.1038/clpt.2013.34. [DOI] [PubMed] [Google Scholar]
- 46.Neuwelt EA, et al. Reversible osmotic blood-brain barrier disruption in humans: Implications for the chemotherapy of malignant brain tumors. Neurosurgery. 1980;7(1):44–52. doi: 10.1227/00006123-198007000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Aswendt M, Adamczak J, Couillard-Despres S, Hoehn M. Boosting bioluminescence neuroimaging: An optimized protocol for brain studies. PLoS ONE. 2013;8(2):e55662. doi: 10.1371/journal.pone.0055662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watts JC, et al. Bioluminescence imaging of Abeta deposition in bigenic mouse models of Alzheimer’s disease. Proc Natl Acad Sci USA. 2011;108(6):2528–2533. doi: 10.1073/pnas.1019034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamgüney G, et al. Measuring prions by bioluminescence imaging. Proc Natl Acad Sci USA. 2009;106(35):15002–15006. doi: 10.1073/pnas.0907339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo J, Ho P, Steinman L, Wyss-Coray T. Bioluminescence in vivo imaging of autoimmune encephalomyelitis predicts disease. J Neuroinflammation. 2008;5:6. doi: 10.1186/1742-2094-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee J, Borboa AK, Baird A, Eliceiri BP. Non-invasive quantification of brain tumor-induced astrogliosis. BMC Neurosci. 2011;12:9. doi: 10.1186/1471-2202-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tran DK, Jensen RL. Treatment-related brain tumor imaging changes: So-called “pseudoprogression” vs. tumor progression: Review and future research opportunities. Surg Neurol Int. 2013;4(suppl 3):S129–S135. doi: 10.4103/2152-7806.110661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Willis CL. Glia-induced reversible disruption of blood-brain barrier integrity and neuropathological response of the neurovascular unit. Toxicol Pathol. 2011;39(1):172–185. doi: 10.1177/0192623310385830. [DOI] [PubMed] [Google Scholar]
- 54.Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: Function and dysfunction. Semin Immunopathol. 2009;31(4):497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- 55.Pardridge WM. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx. 2005;2(1):3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirk R. Clinical trials: New options for melanoma? Nat Rev Clin Oncol. 2012;9(7):368. doi: 10.1038/nrclinonc.2012.95. [DOI] [PubMed] [Google Scholar]
- 57.Wang JQ, et al. PET imaging and optical imaging with D-luciferin [11C]methyl ester and D-luciferin [11C]methyl ether of luciferase gene expression in tumor xenografts of living mice. Bioorg Med Chem Lett. 2006;16(2):331–337. doi: 10.1016/j.bmcl.2005.09.082. [DOI] [PubMed] [Google Scholar]
- 58.Mahringer A, Fricker G. BCRP at the blood-brain barrier: Genomic regulation by 17β-estradiol. Mol Pharm. 2010;7(5):1835–1847. doi: 10.1021/mp1001729. [DOI] [PubMed] [Google Scholar]
- 59.Kadurugamuwa JL, et al. Reduction of astrogliosis by early treatment of pneumococcal meningitis measured by simultaneous imaging, in vivo, of the pathogen and host response. Infect Immun. 2005;73(12):7836–7843. doi: 10.1128/IAI.73.12.7836-7843.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanzaki S, et al. Novel in vivo imaging analysis of an inner ear drug delivery system in mice: comparison of inner ear drug concentrations over time after transtympanic and systemic injections. PLoS ONE. 2012;7(12):e48480. doi: 10.1371/journal.pone.0048480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.