

Significance
Multinucleated giant tumor cells are frequently observed in tissue sections of patients with cancer. In classical Hodgkin lymphoma (HL), these so-called Reed–Sternberg (RS) cells are pathognomonic for the disease. Despite the well-described disease-promoting functions of multinucleated RS cells, their development remains obscure. Long-term time-lapse microscopy and single-cell tracking of HL cell lines demonstrated that RS cells are generated by re-fusion of small mononuclear progenitors. Importantly, fusing cells share almost exclusively the same ancestor, and visualization of the microtubule network revealed a persistent connection between daughter cells in the majority of re-fusion events. Understanding the mechanisms involved in fusion-based RS cell development will further illuminate giant tumor cell formation and may lead to new therapeutic intervention strategies.
Abstract
Multinucleated Reed–Sternberg (RS) cells are pathognomonic for classical Hodgkin lymphoma (HL), and their presence is essential for diagnosis. How these giant tumor cells develop is controversial, however. It has been postulated that RS cells arise from mononucleated Hodgkin cells via endomitosis. Conversely, continuous single-cell tracking of HL cell lines by long-term time-lapse microscopy has identified cell fusion as the main route of RS cell formation. In contrast to growth-induced formation of giant Hodgkin cells, fusion of small mononuclear cells followed by a size increase gives rise to giant RS cells. Of note, fusion of cells originating from the same ancestor, termed re-fusion, is seen nearly exclusively. In the majority of cases, re-fusion of daughter cells is preceded by incomplete cytokinesis, as demonstrated by microtubule bonds among the cells. We confirm at the level of individual tracked cells that giant Hodgkin and RS cells have little proliferative capacity, further supporting small mononuclear Hodgkin cells as the proliferative compartment of the HL tumor clone. In addition, sister cells show a shared propensity for re-fusion, providing evidence of early RS cell fate commitment. Thus, RS cell generation is related neither to cell fusion of unrelated Hodgkin cells nor to endomitosis, but rather is mediated by re-fusion of daughter cells that underwent mitosis. This surprising finding supports the existence of a unique mechanism for the generation of multinuclear RS cells that may have implications beyond HL, given that RS-like cells are frequently observed in several other lymphoproliferative diseases as well.
Hodgkin lymphoma (HL) presents with a unique histological pattern compared with the numerous non-HL B-cell lymphomas (1). For instance, the involved tissue contains a high amount of activated immune cells surrounding the HL tumor cells, which usually account for less than 1% of the cellular infiltrate (2). The HL tumor cells are composed of Hodgkin and Reed–Sternberg (RS) cells, representing the mononucleated and multinucleated subtype, respectively, and collectively termed Hodgkin and Reed–Sternberg (HRS) cells (3–5). With a diameter of up to 100 µm, HRS cells are also referred to as giant cells (6). However, a fraction of mononuclear Hodgkin cells, prominent in HL cell lines, is considerably smaller, with a diameter of approximately 20–30 µm (6).
The cellular origin of HRS cells has long been controversial, until single-cell PCR of microdissected HRS cells revealed rearrangement of the Ig genes indicating a B-cell derivation (7, 8). Moreover, HRS cells carry mutations in the Ig variable region genes, which is a hallmark of B cells that have undergone or are undergoing a germinal center reaction, in which the process of somatic hypermutation is active (5, 9, 10). These findings identify germinal center B cells as precursors of HRS cells, even though they lost their distinct gene expression and cell surface marker profile characteristic for normal mature B cells (5, 11, 12).
Another fundamental question facing researchers is how giant HRS cells, especially the multinucleated RS subtype, evolve from mononucleated Hodgkin cells. Early experiments with HL cell lines revealed that giant RS cells have no proliferative and clonal growth potential (13–15); thus, RS cells were defined as a differentiated end-state of HL tumor cells, presumably playing a pivotal role in interaction with the tumor microenvironment in situ (16). The underlying mechanism of giant HRS cell development remained obscure, however.
Cell fusion of mononuclear Hodgkin cells has been explored as a mechanism for RS cell generation (15); however, a molecular analysis of primary HRS cells excluded the possibility that the HRS cell clone as such or the RS cells are derived from the fusion of different cells (e.g., a B cell and a non-B cell) (17). Moreover, a mixing experiment of dual fluorescent-labeled cells of the HL cell line L1236 provided evidence against cell fusion as the mechanism giving rise to RS cells (18). Thus, endomitosis instead of cell fusion has been proposed as the mechanism for RS cell formation in HL (17, 18). But endomitosis by definition means mitosis leading to polyploidy within a cell without nuclear division, and thus the proposed mechanism should have been called acytokinetic mitosis, which is defined as mitosis with nuclear division but without cellular division, causing binuclearity or multinuclearity of a cell. Remarkably, the first evidence of an abortive mitotic cycle in HRS cells was not reported until years later (19). Furthermore, the mechanism, as well as the aberrant expression of cell cycle-regulating proteins causing acytokinetic mitosis have not been identified to date (20).
In the present study, we tackled the question of giant HRS cell formation by applying a continuous cell observation and tracking method, allowing us to follow individual HRS cells and their progeny at high temporal and spatial resolution (21). We performed long-term time-lapse microscopy of HL cell lines KMH2, L428, and L1236 for more than 10 d under standard culture conditions. These HL patient-derived cell lines, covering two different subtypes of HL, nodular sclerosis (L428) and mixed cellularity (KMH2 and L1236), resemble long-standing adequate models for HRS cell physiology with tumorigenic potential (22, 23). Importantly, each of the three HL cell lines contains a rare giant HRS cell population. Combining time-lapse microscopy with lentiviral-based nuclear fluorescence labeling of HL cells also enabled us to monitor nuclear morphology in real time.
By tracking the cells and their progeny for multiple generations, it became obvious that the fusion of daughter cells, termed re-fusion, plays an essential role in the formation of RS cells. Our data show an alternative route to endomitosis-based giant HRS cell formation. Importantly, cell fusion events occur almost exclusively between cells sharing the same ancestor, explaining why it has been impossible to detect cell fusion in HL with traditional approaches. Visualization of the microtubule network in time-lapse microscopy experiments revealed a persistent connection between daughter cells in the majority of re-fusion events. Moreover, our analysis of giant HRS cell-containing pedigrees clearly shows an early commitment to fusion-induced RS cell development already in giant HRS cell progenitors.
Results
Giant HRS Cells Show a Tremendous Increase in Lifetime.
By continuous observation of individual HRS cells and their progeny from three HL cell lines and application of single-cell tracking, we aimed to gain insight into the cellular behavior of HRS cells, particularly rare giant HRS cells. In contrast to Burkitt lymphoma (BL) cell lines and probably most other non-HL B-cell lymphomas, in which cells either divide regularly or die, in HL cell lines we observed individual cells that sustained for many days without cell division (Fig. 1 A and B). Analysis of hundreds of individual cells of the HL cell lines KMH2, L428, and L1236 found that 4–9% of the cells persisted for longer than 50 h with no division or apoptosis (Fig. 1B and Fig. S1A). We termed this cell fate quiescence. In contrast, BL cell lines BL2 and BL41, which served as controls, did not contain any quiescent cells. Furthermore, the percentage of dying cells was up to 10-fold higher in the HL cell lines compared with the BL cell lines (Fig. 1B).
Fig. 1.

HL cell lines contain rare giant and long-lived HRS cells. Individual cells of HL cell lines KMH2, L428, and L1236 and their progeny were followed with time-lapse microscopy-based cell tracking for 10–12 d, and pedigrees were generated. (A and B) The cell fate of all seeded cells (n = 107–146; generation 0) within the first 50 h was analyzed. (A) Schematic diagram of the identified fates in HL cell lines. Quiescence is defined as cell survival of longer than 50 h without division. (B) Cell fate distribution in HL and BL cell lines (BL2 and BL41), which served as negative controls without a quiescent population. Results for subsequent generations 1 and 2 are shown in Fig. S1A. (C and D) Quiescent cells (n = 36–54, all generations) demonstrated a longer lifetime (C) and larger cell diameter (D) compared with the corresponding bulk population. (E and F) Example images showing representative HRS cell development in the L428 cell line. Shown are the lifespan of a long-lived, quiescent HRS cell (E; 8.5 d; Movie S1) and growth of a normal-sized cell becoming a giant HRS cell (F; 2.2-fold size increase). Stars in E and F designate cells tracked over time. Equivalent examples of KMH2 and L1236 cells are shown in Fig. S1 C and D.
We next focused on the quiescent HRS cell population, for which we found a lifetime of 89–112 h, although the average generation time of the cell lines ranged from 25 to 30 h (Fig. 1 C and E, Fig. S1C, and Movie S1). In addition, long-lived HRS cells showed a continuous increase in cell size (Fig. 1 D and F and Fig. S1D). They frequently reached a cell diameter of up to 100 µm, compared with the average size of 27–31 µm in the bulk population. The cell diameters of giant, long-lived HRS cells was closely correlated with their lifetime (Fig. S1B). The longevity and large cell diameters of the quiescent cell population suggest that these cells are equivalent to the giant cells detected in biopsy specimens from patients with HL. Of note, quiescent cells of the HL cell line KMH2 were markedly smaller than those of cell lines L428 and L1236; however, long-lived HRS cells were clearly identified in all HL cell lines (Fig. 1 C and D and Fig. S1B).
RS Cell Formation Is Based on Re-fusion of Daughter Cells.
We performed live-cell imaging to study the generation of giant HRS cells from small Hodgkin cells. Our time-lapse microscopy experiments identified re-fusion of daughter cells as the predominant event in giant HRS cell formation, with 60–70% of the cells evolving from this phenomenon (Fig. 2 A and B). These fused giant HRS (fHRS) cells were generated from average-sized Hodgkin cells in two steps: a division into two separate daughter cells, followed by re-fusion of these cells several hours after cytokinesis (Fig. 2D, Fig. S2A, and Movie S2). The fused cells increase in size to become giant, long-lived HRS cells. In addition, 21–43% of fHRS progenitor cells divide into three fully separate daughter cells, referred to as trichotomy, in which two cells fuse and give rise to a giant fHRS cell and the third cell usually dies (Fig. 2 A, C, and E; Fig. S2B; and Movie S3). Of note, we also observed multidaughter division followed by re-fusion of all daughter cells.
Fig. 2.

RS cells arise primarily from re-fusion of daughter cells. Development of giant HRS cells was corroborated by time-lapse microscopy-based cell tracking of HL cell lines KMH2, L428, and L1236. (A–C) Continuous HRS cell tracking (n = 36–54) identified re-fusion of daughter cells as a prominent route of giant cell formation. Occasionally, cell fusion was present as a so-called trichotomy, meaning a division into three cells with subsequent fusion of two of the three daughter cells. Most of the remaining nonfused daughter cells died. (A) Schematic diagram of observed routes of giant HRS cell formation. (B) Frequency of fHRS and gHRS cells. (C) Frequency of fHRS cells originating via trichotomy. (D and E) Examples of L1236 cell division and subsequent re-fusion (D; Movie S2) and trichotomy (E) demonstrating the division into three cells with fusion of two cells leading to two daughter cells of unequal size (Movie S3). Equivalent examples from HL cell lines KMH2 and L428 are shown in Fig. S2 A and B. (F) Tracking of the development of a KMH2 fHRS cell with nuclear fluorescence, focusing on cell fusion (Movie S5). An example of a gHRS cell is shown in Fig. S3A. (G) Comparison of the generation time of dividing cells with the division time of giant HRS cells before a cell fusion event. (H) Average cell size of the bulk population plotted against the cell diameter of giant HRS cells before cell fusion occurred. The tracked cells are denoted by a star in D–F.
While multidaughter divisions of HRS cells have so far been unknown, this phenomenon was recently described for polyploid cells as well (24). Furthermore, in 30–40% of cases, we observed generation of giant HRS cells with no previous cell fusion. We termed this process growth-induced giant HRS (gHRS) cell formation, because only growth of these cells could be observed (Fig. 2 A and B). Importantly, re-fusion events between nonrelated cells were not observed, and fusion of cousin cells instead of daughter cells was detected in only one instance, in L428 cells (Fig. S2C and Movie S4).
To better monitor the natural formation of giant HRS cells, especially multinuclear RS cells, in real time, we lentivirally transduced HL cell lines with a vector encoding a fluorescent reporter localized to the nuclear membrane (tdTOMATO-hIMPORTIN). We found that each of the fusing daughter cells contains an intact single nucleus before re-fusion, and that the generated fHRS cells contain both separated nuclei after cell fusion (Fig. 2F and Movie S5). Thus, larger fHRS cells resemble multinuclear RS cells. Conversely, gHRS cells exhibit an increase in nuclear mass over time, but different than fHRS cells, without becoming multinuclear. Rather, they display mononuclear giant Hodgkin cells that could have undergone endomitosis (Fig. S3A and Movie S6).
To further rule out the possibility of cell fusion between nonrelated Hodgkin cells, we prepared mixed cultures of HL cell lines nuclear-labeled with either VENUS- or tdTOMATO-hIMPORTIN over a 4-wk period. Flow cytometry or live-cell imaging did not reveal any double-positive HRS cells.
Interestingly, cell fusion events in cell lines L428 and L1236 occurred concurrently with division of the bulk cell population (Fig. 2G). In addition, the diameter of fHRS progenitor cells was only slightly increased compared with that of the proliferating cell population (Fig. 2H). On the other hand, the lifetime and size of giant HRS cells were significantly different than those of their progenitors and of the bulk cell population (dashed lines in Fig. 2 G and H). These findings indicate that re-fusion of daughter cells occurs as an initial step of RS cell formation.
To evaluate whether both daughter cells are completely separated before they re-fuse to initiate RS cell development, we genetically labeled HL cell lines with fluorescent α-tubulin (RFP-tubulin) and monitored the microtubule network during HRS cell division using time-lapse microscopy (Fig. 3). The midbody resembles the last connection of dividing cells and consists of microtubules derived from the central spindle (25). By introducing RFP-tubulin into HRS cells, we were able to detect midbody formation and disassembly in proliferating cells (Fig. 3A). With a focus on the fusion-based development of RS cells, it became obvious that 83.3% of re-fusion events (30 of 36) were accompanied by persistent microtubule bonds (Fig. 3B and Movie S7). Although both daughter cells individually formed their own nucleus with an assembled nuclear membrane after mitosis (Fig. 2F), suggesting complete cell division, visualization of the microtubule network revealed that both daughter cells remained connected until re-fusion. Furthermore, HRS cells undergoing multidaughter division established a multipart midbody that connected all cells with one another (Fig. 3C), resulting in one cell separating completely while the remaining cells re-fused. Interestingly, in 16.7% of re-fusion events (6 of 36), complete disassembly of the midbody was observed, raising the possibility that the fusing daughter cells were completely separated before re-fusion (Fig. 3D and Movie S8).
Fig. 3.

Incomplete cytokinesis precedes re-fusion of HRS daughter cells. (A) Proliferating HRS cells with fluorescent microtubules (RFP-tubulin) demonstrated midbody assembly during cytokinesis. Mitosis is completed by disassembly of the microtubule connection between the daughter cells. (B) In 83% of the 36 observed fHRS cell formations, daughter cells remained connected via the midbody until re-fusion occurred (Movie S7). (C) Multidaughter divisions displayed a three-part midbody connection. (D) In some cases (∼17%), cytokinesis was complete before cells re-fused, as demonstrated by disassembly of the connecting midbody (Movie S8). Tracked cells are denoted by a circle or cross in A and by a star in B, C, and D. The midbody is indicated by an arrowhead.
RS Cells Preserve a Residual Proliferation Capacity.
We next focused on the fate of the long-lived giant HRS cells. In contrast to previous studies (13–15), we found that giant HRS cells have a residual proliferation potential of 4–22% (Fig. 4A), significantly lower than the 61–75% dividing cells seen in the bulk population (Fig. 1B). However, the majority of giant HRS cells (77–95%) eventually died in our in vitro system (Fig. 4A). We compared fHRS and gHRS cells to investigate whether the route of giant HRS cell formation affects cell lifetime, size, and fate. Typical giant HRS cell characteristics, such as increased lifetime and diameter, did not differ between the two types of cells (Fig. S3 B and C). Interestingly, the fate of giant HRS cells was strongly influenced by the route of development (Fig. 4 B and C). The residual proliferation potential of giant HRS cells was provided solely by the fHRS cell subpopulation, whereas gHRS cells had no proliferation capacity (Fig. 4C). These findings indicate that RS cells, but not mononuclear giant Hodgkin cells, preserve the residual proliferation potential of giant HRS cells.
Fig. 4.
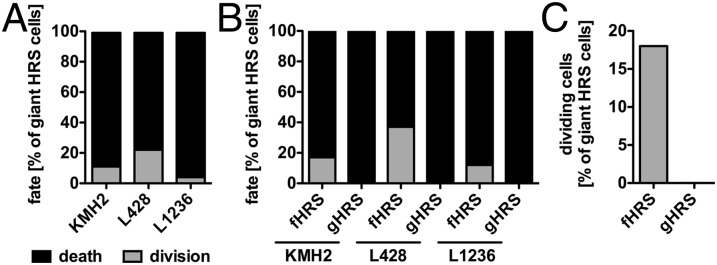
Re-fusion events predict the residual proliferation potential of RS cells. The fate of individual giant HRS cells (n = 36–54) was determined by continuous tracking of HL cell lines KMH2, L428, and L1236. (A) Only cells with a distinct cell fate until the end of the observation period (death or division) were subjected to analysis. (B) Giant HRS cells were grouped by dependency of their origin, and cell fate was reassessed. (C) Highlighted proliferation potentials of fHRS and gHRS cells.
Giant HRS Cell Development and Fate Are Already Committed in the Ancestor Generation.
To gain insight into the mechanism of giant HRS cell development and commitment, we analyzed the behavior of sister cells of giant HRS cells (Fig. 5A). These sister cells exhibited a markedly reduced proliferation potential, with only 25–52% dividing cells (Fig. 5B), compared with the bulk population (Fig. 1B), suggesting high functional similarity between giant HRS cells and their corresponding sister cells. Indeed, 40–44% of the sister cells developed into giant cells themselves (Fig. 5C). To illustrate this phenomenon of giant cell commitment, a selection of original generation pedigrees is shown in Fig. S4.
Fig. 5.

RS cell development is committed in ancestor generations. (A) Pedigree analyses revealed the cell behavior and fate of sister cells of giant HRS cells (n = 13–28) for the KMH2, L428, and L1236 cell lines. (B and C) Continuous single-cell tracking of HRS sister cells determined cell fate (B) and the incidence of giant cell development (C). (D) Likelihood of detecting fHRS cell development in fHRS or gHRS sister cells. (E) Likelihood of identifying giant HRS cell division when the corresponding sister cell is a dividing or dying giant cell.
We next sought to analyze whether the origin and fate of giant HRS cells are correlated to some extent with the behavior of the corresponding sister cells, analyzing only pairs of giant HRS cells (Fig. 5 D and E). We found that 87.5% of fHRS cells had sister cells also undergoing cell fusion, whereas only 22.2% of gHRS cells corresponded to sister fHRS cells (Fig. 5D). In line with this finding, the residual proliferation potential of RS cells was increased when the corresponding sister cells also divided (Fig. 5E).
Based on (i) the reduced proliferation potential of sister cells of giant HRS cells (Fig. 5B), (ii) the high propensity for sister cells to become giant HRS cells (Fig. 5C), and (iii) the commonly shared route of origin as well as cell fate between corresponding giant HRS cells (Fig. 5 D and E), it can be hypothesized that the commitment to giant HRS cell development appears in the ancestor generation, and that giant HRS cells demonstrate an imprinted cell behavior.
Discussion
In this study, we analyzed the cellular behavior of vital giant HRS cells by long-term time-lapse microscopy. Continuous observation of individual HRS cells demonstrated that HL cell lines contain a rare population of long-lived giant HRS cells with a mean lifetime of 89–112 h and a cell diameter of up to 100 µm (Fig. 1). These findings are in line with decades-old observations based on conventional microscopy showing that HL cell lines are heterogeneous with respect to cell size and number of nuclei per cell (6, 15, 26, 27). Apart from a few studies investigating the proliferation potential of RS cells (13, 14, 28), the giant cell population has not been characterized in detail up to now. Most previous functional studies used the bulk of HL cell lines, ignoring cellular heterogeneity (18, 29–31). Tracking of individual HRS cells and their progeny over time has allowed us to dissect giant cell dynamics within the heterogeneous cultures of three HL cell lines and to link HRS cell behavior, RS cell development, and the future fate of these cells. All of these processes require many days and are only observable with in vitro imaging technologies using adequate HL cell line models. In vivo imaging allows only short-term observations, and culturing of primary HRS cells is rather challenging (26, 32).
Interestingly, while exploring the origin of giant cells, we identified a previously unknown behavior of HRS cells, termed re-fusion (Fig. 2). Although cell fusion was ruled out as the origin of RS cells and endomitosis is the currently accepted mechanism for RS cell development (17, 33), we have clearly demonstrated that re-fusion of daughter cells is the initial step in RS cell formation (Fig. 2B), which occurs concurrently with the average division time point of the bulk population (Fig. 2G). Importantly, we found that cell fusion occurs preferentially between cells sharing the same ancestor, explaining why it has been impossible to detect this phenomenon with genetic approaches (17).
The finding that primarily daughter cells undergo re-fusion suggests an incomplete cytokinesis. However, the observation that daughter cells often remain visually separated for up to 1 d after putative cytokinesis, with cell distances of more than one-half a cell diameter before fusion, argues for a complete separation (Fig. 2D). Accordingly, in one case we observed cell fusion between cousin cells originating from different ancestors, supporting the fusion ability of HRS cells (Fig. S2C). To clarify whether the daughter cells were completely separated or still connected before re-fusion, we visualized the microtubule network in time-lapse microscopy experiments. Our observation of a persistent microtubule bond (midbody) connecting the divided daughter cells in the majority of re-fusion events suggests that an incomplete cytokinesis seems to provoke the formation of multinucleated RS cells (Fig. 3).
In line with this finding, a germ line mutation and a functionally relevant single nucleotide polymorphism for the gene of the midbody protein KLHDC8B in HL were reported recently (34, 35). Furthermore, down-regulation of this gene in HeLa cells caused increased frequencies of binucleated cells owing to incomplete cytokinesis (36). Thus, further studies to examine the functional role of KLHDC8B in HRS cells are warranted.
In this study, we assessed not only the origin of giant HRS cells, but also their fate (Fig. 4). RS cells have been characterized as postmitotic end-stage cells (13–15). This is also true for the majority of the tracked giant HRS cells in our study, although we can show that RS cells have a residual proliferation potential after elongated lifetime and enormous size increase, whereas mononuclear giant cells have completely lost their proliferation potential (Fig. 4C). It can be postulated that multinuclearity preserves a residual proliferation potential of giant HRS cells. Nonetheless, owing to the reduced proliferation potential of RS cells, only small Hodgkin cells are able to maintain the HRS cell clone in culture (Fig. 4A). This finding was already implied from previous population studies of HL cell lines (13, 14, 28), but here we have validated and extended these previous studies by tracking the behavior of single HRS cells.
By analyzing the sister cells of giant HRS cells, we have shown that development of giant cells is largely committed already in the ancestor generation (Fig. 5). The rare population of giant HRS cells consists mostly of giant HRS cell pairs, meaning that in 40–44% of cases, both sister cells simultaneously developed into giant cells (Fig. 5C). Interestingly, a fusion-based origin showed a high coincidence between giant HRS cell pairs, suggesting that the residual proliferation potential of RS cells is imprinted to some extent (Fig. 5 D and E). The early commitment of RS progenitors to give rise to fusion-competent progeny in a timed manner points to a distinct biological property of RS cells in HL pathogenesis.
In conclusion, we report the surprising observation that the main route for RS cell generation from Hodgkin cells is not via endomitosis, but rather through re-fusion of daughter cells. Considering that RS cells are the hallmark cells in classic HL, this finding is of major significance for our understanding of HL pathobiology. Visualization of the microtubule network revealed incomplete cytokinesis in the majority of investigated re-fusion events, and suggests an intrinsic mitotic failure in a fraction of the HRS cell clone. Identifying the underlying pathogenetic mechanism is essential. As mentioned above, an altered function of KLHDC8B might play a role (34–36). However, in a few cases we could not clearly discern whether cytokinesis was incomplete before re-fusion, because the connecting midbody had been disassembled. This additional scenario of a putative complete separation before re-fusion is intriguing and should likewise prompt further studies.
Independent of whether re-fusion is an active process of two separated daughter cells or a consequence of failed cytokinesis, one might wonder whether RS cells have a pivotal role in HL pathogenesis or are only “accidents.” As we confirm here, RS cell proliferation alone is insufficient for expansion or maintenance of the HRS cell clone. However, considering the longevity of RS cells, as shown here, as well as the important role of the microenvironment in HL (16, 37, 38), one may speculate that RS cells support tumor clone survival and expansion by producing cytokines and chemokines and promoting cellular interactions with other immune cells (39–42), shaping the HL microenvironment in a way that supports the proliferating Hodgkin cells (43–46). Based on this idea, understanding the mechanisms involved in fusion-based RS cell formation might lead to new therapeutic interventions making use of this process. Finally, our present findings have implications beyond HL, given that RS and RS-like cells are also regularly seen in other lymphoproliferative disorders, including infectious mononucleosis, B-cell chronic lymphocytic leukemia, and T-cell lymphomas (7, 47, 48). In these disorders, the RS-like cells may be generated in a similar way as we have described here.
Materials and Methods
Cell Culture.
HL cell lines KMH2, L428, and L1236 and BL cell lines BL2 and BL41 (obtained from the German Collection of Microorganisms and Cell Cultures) were cultured in RPMI 1640 medium (Life Sciences) supplemented with 10% FBS (PAA), 1% l-glutamine (Life Sciences), and 1% penicillin/streptomycin (Life Sciences) in a standard cell culture incubator with 5% CO2 at 37 °C.
Lentiviral Vectors and Transduction of HRS Cells.
Third-generation self-inactivating lentiviral vectors were used for fluorescent labeling of nuclear membranes and cell microtubules. For nuclear labeling, the ORF of a fusion protein consisting of either VENUS- or tdTOMATO-hIMPORTIN (subunit α2, AA2-67) was cloned into the lentiviral vector pRRL.PPT.SF.eGFP.wPRE by replacing the ORF of GFP (49). VSVG-pseudotyped lentiviral particles were produced by a split genome technique through calcium-phosphate-mediated transient transfection of human embryonic kidney 293T producer cells. After 60 h, supernatant was collected, filtered (45 µm), and enriched by ultracentrifugation (50,000 × g for 2 h). Viral titers were determined with the embryonic murine fibroblast cell line NIH 3T3. Labeling of microtubules was done using lentiviral particles encoding for a fusion protein of RFP and α-tubulin (RFP-tubulin; LentiBrite, Merck Millipore). HL cell lines were transduced with lentiviral particles at a multiplicity of infection of 5–10 and were spin-infected by centrifugation at 900 × g for 1 h at 31 °C.
Flow Cytometry.
Lentivirally transduced HL cell lines were analyzed for VENUS, tdTOMATO, or RFP expression by flow cytometry (MACSquant analyzer; Miltenyi Biotech). Cocultures of VENUS- and tdTOMATO-hIMPORTIN–expressing HL cells were assessed for double-positive cells by FACS analysis two to three times weekly for 4 wk, to detect cell fusion events between unrelated cells.
Time-Lapse Imaging and Single-Cell Tracking.
HL and BL cell lines were cultured in 24-well culture plates equipped with silicon stem cell inserts (ibidi). Time-lapse imaging was performed with a Zeiss CellObserver system as described previously (21). Phase-contrast images of each position were obtained every 1 min with a Zeiss 10× Neofluar objective and an AxioCamHRm camera (at 1,388 × 1,040 pixel resolution) with Zeiss AxioVision 4.8 software. Expression of VENUS (Zeiss filter set 46HE at 1,000 ms), tdTOMATO (Zeiss filter set 43HE at 200 ms), and RFP (Zeiss filter set 43HE at 500 ms) was detected with HXP-120 (Osram) illumination every 44–67 min. Videos were assembled using Apple QuickTime 7.7.3 software.
Tracking of individual cells and their progeny was performed using a self-written computer program as described previously (21, 50, 51). Cell size was determined by pixel measurement of the cell diameter and multiplication of the optical resolution, because most cells were almost round in shape. The generation time or the lifetime of an individual cell was defined as the time span from cytokinesis of its mother cell division to its own division or death, respectively. Dead cells can be readily identified by their shrunken, nonrefracting appearance with immobility. All cell tracking was done by scientists; the current analysis does not rely on data generated by an unsupervised computer algorithm for automated tracking. Only cells with unequivocal identity that could be clearly identified when evaluating the data were used for analysis, and all cells with questionable identity were excluded from relevant analyses.
Statistical Analyses.
Significance was calculated using either the Student t test (two-tailed, unpaired/unequal variances) or linear regression for correlation analysis.
Supplementary Material
Acknowledgments
We thank Claudia Jourdan, Robin Wistinghausen, and Tefik Merovci for their excellent technical assistance. B.R. is a doctoral candidate, and the data presented here are part of his PhD thesis. R.K. was supported by the Deutsche Forschungsgemeinschaft (Grant KU1315/7-1), and M.A.R. was supported by the LOEWE Center for Cell and Gene Therapy Frankfurt, Hessisches Ministerium für Wissenschaft und Kunst [Grant III L 4- 518/17.004 (2010)].
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312509110/-/DCSupplemental.
References
- 1.Hodgkin T. On some morbid appearances of the absorbent glands and spleen. Med Chir Trans. 1832;17:68–114. doi: 10.1177/095952873201700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drexler HG. Recent results on the biology of Hodgkin and Reed-Sternberg cells, I: Biopsy material. Leuk Lymphoma. 1992;8(4-5):283–313. doi: 10.3109/10428199209051008. [DOI] [PubMed] [Google Scholar]
- 3.Sternberg C. Über eine eigenartige unter dem bilde der pseudoleukämie verlaufende tuberkulose des lymphatischen apparates. Zeitschrift für Heilkunde. 1898;19:21–90. [Google Scholar]
- 4.Reed DM. On the pathological changes in Hodgkin’s disease, with special reference to its relation to tuberculosis. Johns Hopkins Hosp Rep. 1902;10:133–196. [Google Scholar]
- 5.Küppers R, Schwering I, Bräuninger A, Rajewsky K, Hansmann ML. Biology of Hodgkin’s lymphoma. Ann Oncol. 2002;13(Suppl 1):11–18. doi: 10.1093/annonc/13.s1.11. [DOI] [PubMed] [Google Scholar]
- 6.Drexler HG, Gaedicke G, Lok MS, Diehl V, Minowada J. Hodgkin’s disease-derived cell lines HDLM-2 and L-428: Comparison of morphology, immunological and isoenzyme profiles. Leuk Res. 1986;10(5):487–500. doi: 10.1016/0145-2126(86)90084-6. [DOI] [PubMed] [Google Scholar]
- 7.Kanzler H, et al. Hodgkin and Reed-Sternberg-like cells in B-cell chronic lymphocytic leukemia represent the outgrowth of single germinal-center B-cell–derived clones: Potential precursors of Hodgkin and Reed-Sternberg cells in Hodgkin’s disease. Blood. 2000;95(3):1023–1031. [PubMed] [Google Scholar]
- 8.Küppers R, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci USA. 1994;91(23):10962–10966. doi: 10.1073/pnas.91.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bräuninger A, et al. Identification of common germinal-center B-cell precursors in two patients with both Hodgkin’s disease and non-Hodgkin’s lymphoma. N Engl J Med. 1999;340(16):1239–1247. doi: 10.1056/NEJM199904223401604. [DOI] [PubMed] [Google Scholar]
- 10.Kanzler H, Küppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med. 1996;184(4):1495–1505. doi: 10.1084/jem.184.4.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Küppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9(1):15–27. doi: 10.1038/nrc2542. [DOI] [PubMed] [Google Scholar]
- 12.Schwering I, et al. Loss of the B-lineage–specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2003;101(4):1505–1512. doi: 10.1182/blood-2002-03-0839. [DOI] [PubMed] [Google Scholar]
- 13.Newcom SR, Kadin ME, Phillips C. L-428 Reed-Sternberg cells and mononuclear Hodgkin’s cells arise from a single cloned mononuclear cell. Int J Cell Cloning. 1988;6(6):417–431. doi: 10.1002/stem.5530060606. [DOI] [PubMed] [Google Scholar]
- 14.Hsu SM, et al. Reed-Sternberg cells in Hodgkin’s cell lines HDLM, L-428, and KM-H2 are not actively replicating: Lack of bromodeoxyuridine uptake by multinuclear cells in culture. Blood. 1988;71(5):1382–1389. [PubMed] [Google Scholar]
- 15.Drexler HG, Gignac SM, Hoffbrand AV, Minowada J. Formation of multinucleated cells in a Hodgkin’s-disease–derived cell line. Int J Cancer. 1989;43(6):1083–1090. doi: 10.1002/ijc.2910430622. [DOI] [PubMed] [Google Scholar]
- 16.Steidl C, Connors JM, Gascoyne RD. Molecular pathogenesis of Hodgkin’s lymphoma: Increasing evidence of the importance of the microenvironment. J Clin Oncol. 2011;29(14):1812–1826. doi: 10.1200/JCO.2010.32.8401. [DOI] [PubMed] [Google Scholar]
- 17.Küppers R, et al. Evidence that Hodgkin and Reed-Sternberg cells in Hodgkin disease do not represent cell fusions. Blood. 2001;97(3):818–821. doi: 10.1182/blood.v97.3.818. [DOI] [PubMed] [Google Scholar]
- 18.Re D, et al. Cell fusion is not involved in the generation of giant cells in the Hodgkin–Reed-Sternberg cell line L1236. Am J Hematol. 2001;67(1):6–9. doi: 10.1002/ajh.1068. [DOI] [PubMed] [Google Scholar]
- 19.Tzankov A, et al. Aberrant expression of cell cycle regulators in Hodgkin and Reed-Sternberg cells of classical Hodgkin’s lymphoma. Mod Pathol. 2005;18(1):90–96. doi: 10.1038/modpathol.3800276. [DOI] [PubMed] [Google Scholar]
- 20.Ullah Z, Lee CY, Depamphilis ML. Cip/Kip cyclin-dependent protein kinase inhibitors and the road to polyploidy. Cell Div. 2009;4:10. doi: 10.1186/1747-1028-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science. 2009;325(5937):217–218. doi: 10.1126/science.1171461. [DOI] [PubMed] [Google Scholar]
- 22.Steidl C, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471(7338):377–381. doi: 10.1038/nature09754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathas S, et al. Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol. 2006;7(2):207–215. doi: 10.1038/ni1285. [DOI] [PubMed] [Google Scholar]
- 24.Duncan AW, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467(7316):707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu CK, Coughlin M, Mitchison TJ. Midbody assembly and its regulation during cytokinesis. Mol Biol Cell. 2012;23(6):1024–1034. doi: 10.1091/mbc.E11-08-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drexler HG. Recent results on the biology of Hodgkin and Reed-Sternberg cells, II: Continuous cell lines. Leuk Lymphoma. 1993;9(1-2):1–25. doi: 10.3109/10428199309148499. [DOI] [PubMed] [Google Scholar]
- 27.Drexler HG, Minowada J. Morphological, immunophenotypical and isoenzymatic profiles of human leukemia cells and derived T-cell lines. Hematol Oncol. 1989;7(2):115–125. doi: 10.1002/hon.2900070203. [DOI] [PubMed] [Google Scholar]
- 28.Ikeda J, et al. Tumorigenic potential of mononucleated small cells of Hodgkin lymphoma cell lines. Am J Pathol. 2010;177(6):3081–3088. doi: 10.2353/ajpath.2010.100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tiacci E, et al. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood. 2012;120(23):4609–4620. doi: 10.1182/blood-2012-05-428896. [DOI] [PubMed] [Google Scholar]
- 30.Stanelle J, Döring C, Hansmann ML, Küppers R. Mechanisms of aberrant GATA3 expression in classical Hodgkin lymphoma and its consequences for the cytokine profile of Hodgkin and Reed/Sternberg cells. Blood. 2010;116(20):4202–4211. doi: 10.1182/blood-2010-01-265827. [DOI] [PubMed] [Google Scholar]
- 31.Hartmann S, et al. Spindle-shaped CD163+ rosetting macrophages replace CD4+ T-cells in HIV-related classical Hodgkin lymphoma. Mod Pathol. 2013;26(5):648–657. doi: 10.1038/modpathol.2012.217. [DOI] [PubMed] [Google Scholar]
- 32.Mader A, et al. U-HO1, a new cell line derived from a primary refractory classical Hodgkin lymphoma. Cytogenet Genome Res. 2007;119(3-4):204–210. doi: 10.1159/000112062. [DOI] [PubMed] [Google Scholar]
- 33.Küppers R. Molecular biology of Hodgkin’s lymphoma. Adv Cancer Res. 2002;84:277–312. doi: 10.1016/s0065-230x(02)84009-x. [DOI] [PubMed] [Google Scholar]
- 34.Krem MM, Luo P, Ing BI, Horwitz MS. The kelch protein KLHDC8B guards against mitotic errors, centrosomal amplification, and chromosomal instability. J Biol Chem. 2012;287(46):39083–39093. doi: 10.1074/jbc.M112.390088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krem MM, Salipante SJ, Horwitz MS. Mutations in a gene encoding a midbody protein in binucleated Reed-Sternberg cells of Hodgkin lymphoma. Cell Cycle. 2010;9(4):670–675. doi: 10.4161/cc.9.4.10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salipante SJ, et al. Mutations in a gene encoding a midbody kelch protein in familial and sporadic classical Hodgkin lymphoma lead to binucleated cells. Proc Natl Acad Sci USA. 2009;106(35):14920–14925. doi: 10.1073/pnas.0904231106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshall NA, et al. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood. 2004;103(5):1755–1762. doi: 10.1182/blood-2003-07-2594. [DOI] [PubMed] [Google Scholar]
- 38.Küppers R, Engert A, Hansmann ML. Hodgkin lymphoma. J Clin Invest. 2012;122(10):3439–3447. doi: 10.1172/JCI61245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skinnider BF, Mak TW. The role of cytokines in classical Hodgkin lymphoma. Blood. 2002;99(12):4283–4297. doi: 10.1182/blood-2002-01-0099. [DOI] [PubMed] [Google Scholar]
- 40.Tanijiri T, et al. Hodgkin’s Reed-Sternberg cell line (KM-H2) promotes a bidirectional differentiation of CD4+CD25+Foxp3+ T cells and CD4+ cytotoxic T lymphocytes from CD4+ naive T cells. J Leukoc Biol. 2007;82(3):576–584. doi: 10.1189/jlb.0906565. [DOI] [PubMed] [Google Scholar]
- 41.van den Berg A, Visser L, Poppema S. High expression of the CC chemokine TARC in Reed-Sternberg cells: A possible explanation for the characteristic T-cell infiltrate in Hodgkin’s lymphoma. Am J Pathol. 1999;154(6):1685–1691. doi: 10.1016/S0002-9440(10)65424-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto R, et al. PD-1–PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood. 2008;111(6):3220–3224. doi: 10.1182/blood-2007-05-085159. [DOI] [PubMed] [Google Scholar]
- 43.Aldinucci D, Celegato M, Borghese C, Colombatti A, Carbone A. IRF4 silencing inhibits Hodgkin lymphoma cell proliferation, survival and CCL5 secretion. Br J Haematol. 2011;152(2):182–190. doi: 10.1111/j.1365-2141.2010.08497.x. [DOI] [PubMed] [Google Scholar]
- 44.Aldinucci D, et al. Expression of CCR5 receptors on Reed-Sternberg cells and Hodgkin lymphoma cell lines: Involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int J Cancer. 2008;122(4):769–776. doi: 10.1002/ijc.23119. [DOI] [PubMed] [Google Scholar]
- 45.Aldinucci D, et al. IRF4 is modulated by CD40L and by apoptotic and anti-proliferative signals in Hodgkin lymphoma. Br J Haematol. 2010;148(1):115–118. doi: 10.1111/j.1365-2141.2009.07945.x. [DOI] [PubMed] [Google Scholar]
- 46.Cattaruzza L, et al. Functional coexpression of interleukin (IL)-7 and its receptor (IL-7R) on Hodgkin and Reed-Sternberg cells: Involvement of IL-7 in tumor cell growth and microenvironmental interactions of Hodgkin’s lymphoma. Int J Cancer. 2009;125(5):1092–1101. doi: 10.1002/ijc.24389. [DOI] [PubMed] [Google Scholar]
- 47.Quintanilla-Martinez L, et al. Peripheral T-cell lymphoma with Reed-Sternberg–like cells of B-cell phenotype and genotype associated with Epstein-Barr virus infection. Am J Surg Pathol. 1999;23(10):1233–1240. doi: 10.1097/00000478-199910000-00008. [DOI] [PubMed] [Google Scholar]
- 48.Moroch J, et al. Follicular peripheral T-cell lymphoma expands the spectrum of classical Hodgkin lymphoma mimics. Am J Surg Pathol. 2012;36(11):1636–1646. doi: 10.1097/PAS.0b013e318268d9ff. [DOI] [PubMed] [Google Scholar]
- 49.Schambach A, et al. Overcoming promoter competition in packaging cells improves production of self-inactivating retroviral vectors. Gene Ther. 2006;13(21):1524–1533. doi: 10.1038/sj.gt.3302807. [DOI] [PubMed] [Google Scholar]
- 50.Eilken HM, Nishikawa S, Schroeder T. Continuous single-cell imaging of blood generation from haemogenic endothelium. Nature. 2009;457(7231):896–900. doi: 10.1038/nature07760. [DOI] [PubMed] [Google Scholar]
- 51.Kimura A, et al. The transcription factors STAT5A/B regulate GM-CSF–mediated granulopoiesis. Blood. 2009;114(21):4721–4728. doi: 10.1182/blood-2009-04-216390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.