

Significance
We show that directly coupled evolutionary residue pairs provide a distinct footprint of conformational diversity in protein families. This is revealed as competing residue contacts unique to distinct configurations of proteins with multiple conformations. We demonstrate that combining this information with physical models of proteins is sufficient to uncover conformational diversity for several protein families. We discovered that such directly coupled residues not only allow us to accurately transition between apo and holo conformations but also help to uncover intermediate states that are not easily accessible to experimental or other computational methods. This enhanced sampling of the functional conformational space of proteins may have broad implications in protein structure determination and the design of ligands that can trap intermediate states.
Keywords: conformational plasticity, covariation, statistical inference, molecular dynamics
Abstract
A long-standing problem in molecular biology is the determination of a complete functional conformational landscape of proteins. This includes not only proteins’ native structures, but also all their respective functional states, including functionally important intermediates. Here, we reveal a signature of functionally important states in several protein families, using direct coupling analysis, which detects residue pair coevolution of protein sequence composition. This signature is exploited in a protein structure-based model to uncover conformational diversity, including hidden functional configurations. We uncovered, with high resolution (mean ∼1.9 Å rmsd for nonapo structures), different functional structural states for medium to large proteins (200–450 aa) belonging to several distinct families. The combination of direct coupling analysis and the structure-based model also predicts several intermediates or hidden states that are of functional importance. This enhanced sampling is broadly applicable and has direct implications in protein structure determination and the design of ligands or drugs to trap intermediate states.
As demonstrated by Anfinsen in 1973 (1) for small and intermediate-size proteins, amino acid sequences contain all of the necessary information to determine their native structure and function. In principle, a complete physical understanding of all molecular interactions should be sufficient to uncover not only the proteins’ native structures, but also all their respective functional states, including functionally important intermediates. This landscape is required for a complete knowledge of functional mechanisms and therefore it has implications for drug discovery. Advances in computational approaches have been promising in sampling such conformational intermediates (2, 3). However, in general, computational methods are limited by uncertainties in protein models as well as insufficient computational resources to achieve proper sampling. Experimental techniques such as crystallography or NMR spectroscopy have been successful in identifying functional protein structures but only for a fraction of the complete set of known protein sequences (4, 5). Additionally, the determination of functionally important intermediate states using such methods has been challenging due to their transient nature. One idea to confront this challenge is to search for clues in genomic data (6–10). Functional states under conformational selection should leave a trace in the evolutionary history of proteins. Recent results inspired by this hypothesis have led to the development of the powerful “direct coupling analysis” (DCA), which was able to predict a large number of direct structural contacts between residues from sequences alone (11). Other useful methods have been developed to define coupling among residue pairs (12, 13). Others have also looked into correlated electrostatic mutations to study the evolution of protein topology toward minimized interaction frustration (14). Integrating the DCA-predicted contacts into coarse-grained physical models of proteins such as structure-based models (SBMs) (15–19) led to predictions on protein–protein interactions (20–22) as well as tools to aid the prediction of native structures (19, 23–25). The idea of using predicted contacts to estimate native structures was also explored by other methodologies (26, 27). Here, we show that DCA predicts important structural interactions related not only to the native state but also to distinct functional conformational states of a protein, including intermediates. We develop a hybrid computational method to recover such important conformations, which derives the SBM energy function from a single experimental structure and incorporates DCA residue contacts into the energy function (Materials and Methods). We show that this model samples well beyond a single native structure to reveal conformational diversity, including hidden functional configurations, in proteins. We refer to this methodology as SBM+DCA.
Results
Conformational Diversity Is Embedded in Evolutionary Information.
In this study, we provide evidence that accurate information about conformational diversity can be extracted from evolutionarily related protein sequences. We focus on proteins that experience large conformational changes upon ligand binding (Table S1) (28, 29). Fig. 1 illustrates our first example, the l-leucine binding protein [Protein Data Bank (PDB) IDs 1usg and 1usi], which experiences large conformational changes upon binding to l-leucine (30). Fig. 1C displays a structural comparison between the ligand bound and unbound states. The residue–residue contact maps for open and closed conformations have very distinct signatures, as highlighted in Fig. 1A. This protein belongs to the domain family “periplasmic binding protein 6” (Pfam ID: PF13458). Applying DCA to 7,363 sequences in this domain, we obtained a large number of high-ranking pairings predicted to be physical contacts (69% true positives in the top 500 predictions). Interestingly, the predicted contacts belong to both open and closed conformations; compare highlighted regions in Fig. 1B. We use such predicted couplings in combination with a structure-based model (SBM+DCA) to study the conformational dynamics of the l-leucine binding protein. Using only structure parameters taken from the open-state topology (residue contacts and distances and dihedral angles and bond distances), the DCA-predicted contacts led the model to identify an ensemble of closed conformations in addition to the open state (Fig. 1D). We used the gromos clustering algorithm (31) to obtain representative structures (cluster centroids) in the ensemble. Both centroids are within 2-Å rmsd accuracy to the experimental structures (Movie S1). For comparison, replacing those DCA contacts by the same number of random contacts led to fluctuations only around the open conformation (Fig. S1). A dual-basin SBM (32) is also able to recover both the open and the closed conformations. However, it requires, for both bound and unbound conformations, the complete knowledge of the contacts and their experimentally determined distances (Fig. S2).
Fig. 1.

A hybrid SBM+DCA model of the l-leucine binding protein is able to uncover its two-state (apo/holo) conformational landscape. A compares the native open and closed contact maps and B compares a DCA contact map with the native closed state. In A, comparing the native contact map of the open conformation (PDB ID 1usg; lower triangular map) and the closed conformation (PDB ID 1usi; upper triangular map) shows a clear set of contacts (shaded box) that are exclusive to the closed state. In B a predicted contact map using highly ranked DCA residue pairs (lower triangular map) shows a very accurate reconstruction of the complete map that includes the extra contacts in the closed conformation (upper triangular map). (C) Structural comparison between the apo and the holo states of the l-leucine binding protein, showing domain closure. (D) Integrating a SBM of the open-state topology with DCA contacts produces a distinct bimodal landscape, as opposed to the single-basin distribution observed when we use the same number of extra contacts but randomly distributed.
Multiple States with Functionally Important Intermediates Are Found by SBM+DCA Methodology.
We next studied the glutamate receptor (GluR2), which belongs to the family of “bacterial extracellular solute-binding proteins” (PF00497). Armstrong and Gouaux (33) provided structural evidence suggesting that GluR2 uses an agonist-induced domain closure mechanism to gate the transmembrane channel and that its activation is dependent on the degree of domain closure.
We analyzed the 20,059 protein sequences in this family using DCA, and the predicted contacts are again used to study the dynamics of the structure. The 2D rmsd frequency distribution in Fig. 2A illustrates how DCA-predicted couplings can be used to sample the conformational space of the open and closed states to an accuracy of 1 Å. However, the conformational space is more complex in this case. Fig. 2B shows the rmsd measures of the top three clusters from the SBM trajectory. One cluster centroid (6%) has an rmsd of 1.1 Å with respect to the closed glutamate-bound state (PDB ID: 1ftm); a second cluster centroid (10%) has an accuracy of 0.9 Å with respect to the apo state (PDB ID: 1fto). Interestingly, the centroid of the most populated cluster (82%) is far away from both the open and the closed state. However, this centroid structure is only 0.8 Å apart from a Kainate-bound structure of the receptor (PDB ID 1fw0). Kainate was shown to be a partial agonist that induced an intermediate semiclosed state in the glutamate receptor (33). Fig. 2C compares the cluster centroids with the experimentally determined structures and shows the sequential closure of the domains. As a control, we show that a dual-basin SBM can sample a similar conformational space but it requires the complete knowledge of open and closed structures (Fig. S3). These results suggest that coevolutionary information can sample multiple functionally relevant states with high accuracy. We analyzed another protein of the same family, glutamine binding protein (GBP) (PDB IDs 1ggg and 1wdn) (34). We predict an intermediate state (Fig. S4), for which there is only indirect experimental evidence of its existence (35). We propose that this intermediate state for the GBP can be trapped experimentally by designing an appropriate ligand, in the same manner that kainate was used to crystallize the intermediate for GluR2.
Fig. 2.

The glutamate receptor has a ligand-dependent domain closure. (A) The conformational landscape observed after combining an open-state topology with coevolutionary restraints obtained from the family of bacterial extracellular solute-binding proteins (PF00497). The landscape includes conformations with an rmsd of less than 2 Å from the crystal structures of open and closed states. An intermediate state is also present that is between the closed and the open conformations. (B) After using the gromos clustering algorithm (31) for the molecular dynamics trajectory, the top three cluster centroids cover 98% of the conformations. The centroid of the most populated cluster is in fact structurally very similar to the kainite-bound structure of the glutamate receptor (rmsd 0.8 Å). Kainate is a partial agonist that brings the protein to a semiclosed state. (C) Structural comparison between the centroids and experimental structures of antagonist (open; PDB ID 1fto), partial agonist (semiclosed; PDB ID 1fw0), and agonist (closed; PDB ID 1ftm) states. The predictions for these three states have an ∼1-Å rmsd with respect to the crystal structures.
We next examined sugar-binding proteins that also experience large conformational changes. The d-Ribose binding protein is a protein of the “family of periplasmic protein binding domains” (PF13407) with more than 8,000 members. Fig. 3A shows a comparison between the open (PDB ID: 1urp) and closed (PDB ID: 2dri) residue contact maps (upper triangular map) as well as the contacts obtained via DCA (lower triangular map). We identified contacts common for both open and closed structures but also some unique to the closed state (red dashed boxes). We also found a series of contacts (black dashed box) that belong to neither the open nor the closed state. These couplings gave rise to a conformational landscape that includes a very distinct third state (Fig. 3B). For comparison, a dual-basin model of the open and closed conformations combined with the same number of random extra contacts did not yield the intermediate-state basin (Fig. S5), but instead a broadening is observed compared with the dual-basin landscape (Fig. S6). This finding suggests that this third state is a feature uniquely captured by the extra DCA couplings. Fig. 3B shows a closed-state ensemble with a tighter domain closure with respect to the experimental closed state but conserving the same topological features (rmsd open >5 Å). We attribute this to the difference between the ligand-bound and the ligand-free closed states (3). We model the presence of the ligand by using the exact experimental contact distances for only a few contacts (landscape in Fig. 3C). Now we observe the ligand-bound closed ensemble instead of the ligand-free state present in Fig. 3B, while still accessing the intermediate state. Fig. 3D shows the contact maps of the cluster centroids from the distributions observed in Fig. 3B. The open-state cluster centroid has an rmsd of 2.2 Å whereas the closed-state cluster has an rmsd of 2.6 Å. The intermediate-state cluster centroid has a twisted semiclosed state that is equidistant to the open and closed conformations and has unique contacts that are also present in the DCA estimated map (compare black dashed boxes in Fig. 3 A and D). A comparison between the three cluster centroids is available in Movie S2. Using umbrella sampling molecular dynamics simulations, Ravindranathan et al. provided computational evidence supporting the existence of such a twisted state (3). Such an intermediate state seems to be functionally relevant because it has been hypothesized to facilitate ribose transfer in the permease complex—a partially closed conformation with a more weakly bound ribose might help in providing an easier release of ribose into the membrane-bound permease (3).
Fig. 3.

d-Ribose binding protein (PDB IDs 1urp and 2dri) goes through large conformational changes upon ribose binding. (A) Comparison between the open and the closed native contact maps. Blue marks illustrate open-state contacts and red contacts are uniquely found in the closed state. The lower triangular part of the map shows the contacts estimated using DCA for which we can identify the global structure as well as the closed-state contacts (red dashed box). We can also see in the map an additional set of contacts (black dashed box) that may lead to a third state that has not been observed experimentally. (B) The conformational landscape shows a very distinct third state, which is not observed in the dual-basin control simulations, even if random extra contacts are added to increase the sampling of the conformational space (control in Fig. S5). (C) If we represent ligand constraints with few contacts with experimental distance parameters, then the landscape shows the ligand-bound closed state while the population of the intermediate state is still present. (D) The contact maps and structures for the cluster centroids are shown for the open, intermediate, and closed states. There is a distinct set of competing contacts that induce the observed landscape only when we integrate our model with DCA information. The comparison of the centroid cluster structures shows how two helices from the two domains are aligned for the open and closed states, as in their native structures, whereas a twisted alignment was found for the intermediate state. Ravindranathan et al. have provided evidence for this intermediate state (3). Similar evidence exists for other sugar-bound proteins like d-Glucose and Maltose binding protein (36, 38, 39).
A similar twisted intermediate state was suggested for the d-Glucose binding protein, another member of the same family, based on experimental studies using disulfite-trapping and fluorescence spectroscopy (36, 37). Very recently, accelerated molecular dynamics were used to suggest the existence of a semiclosed state for the Maltose binding protein, a member of a closely related family (38, 39). Our SBM+DCA-based model predicts another twisted intermediate for the d-Allose binding protein (PDB IDs 1gud and 1rpj) of the same family (Fig. S7). The existence of such a semiclosed twisted state seems to be a general feature of the sugar-bound periplasmic proteins.
We have investigated additional systems using this methodology with consistent results. For example, in the case of the 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase (PDB IDs 1rf5 and 1rf4), we have also found a hidden intermediate state (Fig. S8). Experimental evidence supporting this claim exists in the form of an ortholog (PDB ID 3roi) with similar topological features. The structure of this ortholog suggests that the state we found may be of functional relevance for EPSP. As a matter of negative control, we have also studied the case of the D class β-lactamase (PDB ID 1h8y) that is a member of a largely populated family of transpeptidases (PF00905). This protein is known to have only one state. When adding the same amount of DCA pairs to the open-state topology, we did not observe any spurious additional state (Fig. S9).
Discussion
In this study, we combined physical models of proteins, for instance SBM, with coevolutionary constraints obtained using direct coupling analysis. Our results provide support that information about conformational plasticity can be retrieved from a collection of evolutionary related protein sequences. This is a consequence of the fact that diverse states (intermediates/closed), which are of functional importance, are selected by evolution, because DCA captures evolutionarily significant residue–residue correlations, regardless of whether the interaction stabilizes the final or intermediate state. To clearly observe these types of transitions, the conformational states need to have unique subsets of contacts. Subtle conformational changes where only contact distances are changed, e.g., conformation differences between ATP and ADP binding in the active site, are harder to capture by SBM+DCA. Nonetheless, this enhanced sampling of the functional conformational space of proteins might have broader implications in protein structure determination as well as in the design of ligands that can trap intermediate states. Such ligands could be used to crystallize states that were previously difficult to access and also be used in the process of rational drug design. Our observations and theoretical framework are general enough to be applied to many protein families with enough sequence information, in principle even for those families without any experimental structural state available.
Materials and Methods
Directly Coupled Residue Pairs.
We use DCA to estimate directly coupled coevolving residue–residue physical contacts. DCA models the joint probability distribution of amino acid sequences with an exponential function that depends on single-site amino acid frequencies and pairwise interactions. An approximation of pairwise energies is calculated by inverting the connected correlation matrix computed from multiple sequence alignments. These pairwise terms are used to compute probabilities of “direct couplings” among amino acid pairs. When applying DCA to a set of sequences of a given family [e.g., Pfam domains (40)], then residue pairs that show the largest amount of direct coupling or direct information (DI) tend to be a proxy of residue–residue contacts in the 3D fold of a protein that is part of such family. For more details about DCA and an evaluation of its performance using a mean field formulation, refer to Morcos et al. (11). For each of the families analyzed, we used the top ranked pairs based on the DI metric. We used a pseudocount value of  . The value of
. The value of  in turn was computed using a correction of sampling bias for proteins with sequence identity of 80%. The number of
in turn was computed using a correction of sampling bias for proteins with sequence identity of 80%. The number of  is shown as “effective sequences” in Table S1. The number of DCA contacts used was proportional to the total number of native contacts in the open state of a protein, using shadow contact maps. For all of the systems studied, the number of DCA contacts used was 1.75 times the number of native contacts. The results are robust for a range of 1.5–2 times the number native shadow contacts. We used a cutoff value for shadow contacts of 6 Å + 1 Å atom “shadowing” radius; this is the standard value used in the smog web server (16).
is shown as “effective sequences” in Table S1. The number of DCA contacts used was proportional to the total number of native contacts in the open state of a protein, using shadow contact maps. For all of the systems studied, the number of DCA contacts used was 1.75 times the number of native contacts. The results are robust for a range of 1.5–2 times the number native shadow contacts. We used a cutoff value for shadow contacts of 6 Å + 1 Å atom “shadowing” radius; this is the standard value used in the smog web server (16).
Single-Basin SBM.
We built our single-basin SBM from a single native structure (open state of the binding protein) by placing a single bead of unit mass for each amino acid at the location of the Cα atom (10, 11). The energy function used for the SBM is given as
Here, the superscript O refers to the open state and 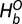 represents the local bonded component of the Hamiltonian,
represents the local bonded component of the Hamiltonian,
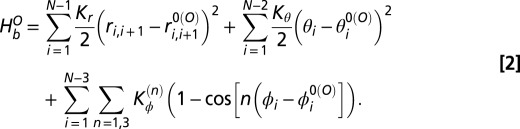 |
The first term in  ensures that the bond distance
ensures that the bond distance  between the neighboring residues i and i + 1 is constrained harmonically with respect to its native bond distance
between the neighboring residues i and i + 1 is constrained harmonically with respect to its native bond distance 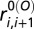 by a spring constant
by a spring constant  . The second term constrains the angle
. The second term constrains the angle  among the residues i, i + 1, and i + 2 with respect to its native value
among the residues i, i + 1, and i + 2 with respect to its native value  by a harmonic spring constant
by a harmonic spring constant  . The third term represents the dihedral angle potential with
. The third term represents the dihedral angle potential with  that describes the rotation of the backbone involving successive residues from i to i + 3. The native values
that describes the rotation of the backbone involving successive residues from i to i + 3. The native values  ,
, 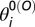 , and
, and  are taken from the open conformation crystal structure. The value of
are taken from the open conformation crystal structure. The value of  is chosen carefully to ensure better sampling of the conformational space while ensuring sufficient stabilization of the open state. The nonlocal part of the Hamiltonian,
is chosen carefully to ensure better sampling of the conformational space while ensuring sufficient stabilization of the open state. The nonlocal part of the Hamiltonian,  is given by
is given by
 |
The 6–12 Lennard–Jones (LJ) potential is used in  to describe the interactions that stabilize the nonbonded native contacts. Native contact pairs (i and j) are obtained using the shadow contact map that is implemented in SMOG (16). If i and j residues are in contact in the native state,
to describe the interactions that stabilize the nonbonded native contacts. Native contact pairs (i and j) are obtained using the shadow contact map that is implemented in SMOG (16). If i and j residues are in contact in the native state,  ; otherwise
; otherwise 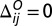 . Native contact pair distance
. Native contact pair distance  is obtained from open-state structure. Nonnative pairs with
is obtained from open-state structure. Nonnative pairs with  are under repulsive potential with a distance parameter
are under repulsive potential with a distance parameter  Å. The strength of repulsive potential
Å. The strength of repulsive potential  is 1 kJ/mol. However, the value of
is 1 kJ/mol. However, the value of  is chosen carefully to ensure better sampling of the conformational space and sufficient stabilization of the open state. These parameters are sampled until we observe the existence of new basins with a sufficiently large population.
is chosen carefully to ensure better sampling of the conformational space and sufficient stabilization of the open state. These parameters are sampled until we observe the existence of new basins with a sufficiently large population.
Dual-Basin Structure-Based Model.
As the name suggests, the dual-basin structure-based model (dSBM) is built using two experimental structures of a single protein, namely open and ligand-bound closed states. The energy function used for this model is given as
Here,  represents the local part of the Hamiltonian as in the single SBM.
represents the local part of the Hamiltonian as in the single SBM.  describes the nonlocal part of the dual-SBM Hamiltonian and has the form
describes the nonlocal part of the dual-SBM Hamiltonian and has the form
 |
The first term in the summation is same as the first term in  and derived from open structure. The second term is derived from the closed-state structure and
and derived from open structure. The second term is derived from the closed-state structure and  if residues i and j are in contact in closed state but not in open state. For these unique closed-state contacts, we also use the native contact distances
if residues i and j are in contact in closed state but not in open state. For these unique closed-state contacts, we also use the native contact distances  derived from the closed-state crystal structure. The last term accounts for the repulsion between the nonbonded pairs that are not in contact either in open state or in closed state and the logic or operator ∨ is used for that purpose. Here, the values for
derived from the closed-state crystal structure. The last term accounts for the repulsion between the nonbonded pairs that are not in contact either in open state or in closed state and the logic or operator ∨ is used for that purpose. Here, the values for  and
and  are chosen carefully to sample both the states.
are chosen carefully to sample both the states.
SBM+DCA Model.
For our hybrid SBM+DCA model, we combine the Hamiltonian of the open state and supplement with DCA-predicted contact pairs. The energy function for this hybrid method is given by
Here,  represents the local part of the Hamiltonian as in the single SBM and derived from open-state crystal structure.
represents the local part of the Hamiltonian as in the single SBM and derived from open-state crystal structure. 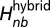 has the form
has the form
 |
The first term in the summation is the same as the first term in 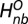 and derived from open structure. The second term is derived from the DCA pair and
and derived from open structure. The second term is derived from the DCA pair and  if the residue pair i and j appear as a top-ranked DCA contact but not in open state. For these unique DCA contacts, we use the native contact distance
if the residue pair i and j appear as a top-ranked DCA contact but not in open state. For these unique DCA contacts, we use the native contact distance  Å and LJ well-depth
Å and LJ well-depth  . The last term accounts for the repulsion between the nonbonded pairs that are not in contact either in open state or in DCA pairs and the logic or operator ∨ is used for that purpose. Here, the value of
. The last term accounts for the repulsion between the nonbonded pairs that are not in contact either in open state or in DCA pairs and the logic or operator ∨ is used for that purpose. Here, the value of  is chosen carefully to sample the phase space efficiently. The values for
is chosen carefully to sample the phase space efficiently. The values for  and
and  range between 0.4 and 0.6.
range between 0.4 and 0.6.
Supplementary Material
Acknowledgments
This work was supported by the Center for Theoretical Biological Physics sponsored by the Welch Foundation Grant C-1792, by the National Science Foundation (NSF) (Grant PHY-1308264), and by Grant NSF-MCB-1214457. J.N.O. is a CPRIT Scholar in Cancer Research sponsored by the Cancer Prevention and Research Institute of Texas.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315625110/-/DCSupplemental.
References
- 1.Anfinsen CB. Principles that govern folding of protein chains. Science. 1973;18(4096):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Kim MK, Chirikjian GS, Jernigan RL. Elastic models of conformational transitions in macromolecules. J Mol Graph Model. 2002;21(2):151–160. doi: 10.1016/s1093-3263(02)00143-2. [DOI] [PubMed] [Google Scholar]
- 3.Ravindranathan KP, Gallicchio E, Levy RM. Conformational equilibria and free energy profiles for the allosteric transition of the ribose-binding protein. J Mol Biol. 2005;353(1):196–210. doi: 10.1016/j.jmb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.UniProt Consortium Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40(Database issue):D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Juan D, Pazos F, Valencia A. Emerging methods in protein co-evolution. Nat Rev Genet. 2013;14(4):249–261. doi: 10.1038/nrg3414. [DOI] [PubMed] [Google Scholar]
- 7.Göbel U, Sander C, Schneider R, Valencia A. Correlated mutations and residue contacts in proteins. Proteins. 1994;18(4):309–317. doi: 10.1002/prot.340180402. [DOI] [PubMed] [Google Scholar]
- 8.Park S, Kono H, Wang W, Boder ET, Saven JG. Progress in the development and application of computational methods for probabilistic protein design. Comput Chem Eng. 2005;29(3):407–421. [Google Scholar]
- 9.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286(5438):295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 10.Halabi N, Rivoire O, Leibler S, Ranganathan R. Protein sectors: Evolutionary units of three-dimensional structure. Cell. 2009;138(4):774–786. doi: 10.1016/j.cell.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morcos F, et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci USA. 2011;108(49):E1293–E1301. doi: 10.1073/pnas.1111471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor WR, Sadowski MI. Structural constraints on the covariance matrix derived from multiple aligned protein sequences. PLoS ONE. 2011;6(12):e28265. doi: 10.1371/journal.pone.0028265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones DT, Buchan DW, Cozzetto D, Pontil M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28(2):184–190. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- 14.Haq O, Andrec M, Morozov AV, Levy RM. Correlated electrostatic mutations provide a reservoir of stability in HIV protease. PLoS Comput Biol. 2012;8(9):e1002675. doi: 10.1371/journal.pcbi.1002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: What determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J Mol Biol. 2000;298(5):937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 16.Noel JK, Whitford PC, Sanbonmatsu KY, Onuchic JN. SMOG@ctbp: Simplified deployment of structure-based models in GROMACS. Nucleic Acids Res. 2010;38(Web Server issue):W657-W661. doi: 10.1093/nar/gkq498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oklejas V, Zong C, Papoian GA, Wolynes PG. Protein structure prediction: Do hydrogen bonding and water-mediated interactions suffice? Methods. 2010;52(1):84–90. doi: 10.1016/j.ymeth.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davtyan A, et al. AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B. 2012;116(29):8494–8503. doi: 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sułkowska JI, Morcos F, Weigt M, Hwa T, Onuchic JN. Genomics-aided structure prediction. Proc Natl Acad Sci USA. 2012;109(26):10340–10345. doi: 10.1073/pnas.1207864109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weigt M, White RA, Szurmant H, Hoch JA, Hwa T. Identification of direct residue contacts in protein-protein interaction by message passing. Proc Natl Acad Sci USA. 2009;106(1):67–72. doi: 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schug A, et al. Computational modeling of phosphotransfer complexes in two-component signaling. Methods Enzymol. 2010;471:43–58. doi: 10.1016/S0076-6879(10)71003-X. [DOI] [PubMed] [Google Scholar]
- 22.Dago AE, et al. Structural basis of histidine kinase autophosphorylation deduced by integrating genomics, molecular dynamics, and mutagenesis. Proc Natl Acad Sci USA. 2012;109(26):E1733–E1742. doi: 10.1073/pnas.1201301109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marks DS, et al. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE. 2011;6(12):e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopf TA, et al. Three-dimensional structures of membrane proteins from genomic sequencing. Cell. 2012;149(7):1607–1621. doi: 10.1016/j.cell.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dill KA, MacCallum JL. The protein-folding problem, 50 years on. Science. 2012;338(6110):1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- 26.Nugent T, Jones DT. Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc Natl Acad Sci USA. 2012;109(24):E1540–E1547. doi: 10.1073/pnas.1120036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor WR, Jones DT, Sadowski MI. Protein topology from predicted residue contacts. Protein Sci. 2012;21(2):299–305. doi: 10.1002/pro.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brylinski M, Skolnick J. What is the relationship between the global structures of apo and holo proteins? Proteins. 2008;70(2):363–377. doi: 10.1002/prot.21510. [DOI] [PubMed] [Google Scholar]
- 29.Seeliger D, de Groot BL. Conformational transitions upon ligand binding: Holo-structure prediction from apo conformations. PLoS Comput Biol. 2010;6(1):e1000634. doi: 10.1371/journal.pcbi.1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magnusson U, Salopek-Sondi B, Luck LA, Mowbray SL. X-ray structures of the leucine-binding protein illustrate conformational changes and the basis of ligand specificity. J Biol Chem. 2004;279(10):8747–8752. doi: 10.1074/jbc.M311890200. [DOI] [PubMed] [Google Scholar]
- 31.Daura X, et al. Peptide folding: When simulation meets experiment. Angew Chem Int Ed. 1999;38(1):236–240. [Google Scholar]
- 32.Okazaki K, Koga N, Takada S, Onuchic JN, Wolynes PG. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc Natl Acad Sci USA. 2006;103(32):11844–11849. doi: 10.1073/pnas.0604375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Neuron. 2000;28(1):165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 34.Hsiao CD, Sun YJ, Rose J, Wang BC. The crystal structure of glutamine-binding protein from Escherichia coli. J Mol Biol. 1996;262(2):225–242. doi: 10.1006/jmbi.1996.0509. [DOI] [PubMed] [Google Scholar]
- 35.Hsiao CD, et al. Crystals of glutamine-binding protein in various conformational states. J Mol Biol. 1994;240(1):87–91. doi: 10.1006/jmbi.1994.1420. [DOI] [PubMed] [Google Scholar]
- 36.Careaga CL, Sutherland J, Sabeti J, Falke JJ. Large amplitude twisting motions of an interdomain hinge: A disulfide trapping study of the galactose-glucose binding protein. Biochemistry. 1995;34(9):3048–3055. doi: 10.1021/bi00009a036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Messina TC, Talaga DS. Protein free energy landscapes remodeled by ligand binding. Biophys J. 2007;93(2):579–585. doi: 10.1529/biophysj.107.103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bucher D, Grant BJ, Markwick PR, McCammon JA. Accessing a hidden conformation of the maltose binding protein using accelerated molecular dynamics. PLoS Comput Biol. 2011;7(4):e1002034. doi: 10.1371/journal.pcbi.1002034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bucher D, Grant BJ, McCammon JA. Induced fit or conformational selection? The role of the semi-closed state in the maltose binding protein. Biochemistry. 2011;50(48):10530–10539. doi: 10.1021/bi201481a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finn RD, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38(Database issue):D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.