

Abstract
A key step in the cyclooxygenase reaction cycle of cyclooxygenase 1 (COX-1) is abstraction of the pro-S hydrogen atom of the arachidonic acid by a radical that is formed at the protein residue Tyr-385. Here we investigate this reaction step by a quantum-mechanics/molecular-mechanics approach in combination with molecular-dynamics simulations. The simulations identify the hydrogen abstraction angle as a crucial geometric determinant of the reaction, thus revealing the importance of the cyclooxygenase active site for calculating the potential energy surface of the reaction.
Cyclooxygenases (COXs) are monotopic membrane enzymes that catalyze the conversion of arachidonic acid (ACD) to prostaglandin H2 (PGH2), which is a common biosynthetic precursor to prostaglandins and thromboxanes (1). The two most important isoenzymes, COX-1 and COX-2, exhibit distinct properties. Whereas the inhibition of COX-1 may lead to ulcerogenic effects in the gastrointestinal system, inhibition of COX-2 is considered a target for therapy to control inflammation and pain (2). In addition, modeling studies have indicated that COX-1 and COX-2 may interact differently with the membrane (3). COX enzymes contain two active sites: a cyclooxygenase site, where ACD is converted into hydroperoxy endoperoxide prostaglandin G2 (PGG2), and a heme-dependent peroxidase site, where PGG2 is reduced to PGH2 (4). The rate-limiting step of the cyclooxygenase reaction cycle is believed to be the reaction of abstraction of the 13-pro-S hydrogen atom from the ACD chain by the Tyr-385 tyrosyl radical (Scheme 1) (5,6). This hydrogen abstraction step leads to formation of the planar pentadienyl radical, which subsequently reacts with dioxygen to form the endoperoxide PGG2.
Scheme 1.
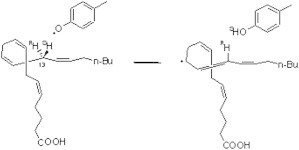
Hydrogen abstraction by the Tyr-385 radical in COX-1.
Previous computational investigations (5,6) were based on small cluster models of COX active sites. Although such calculations provide mechanistic insights into COX-1-catalyzed reactions, they can have important limitations (7). Cluster-based calculations do not include the effects of the whole protein structure, nor do they account directly for the influence of flexibility and relaxation of the enzyme-substrate (ES) complex on the potential energy of the reaction.
Enzymes are subject to conformational flexibility, which can influence the potential energy surface along a reaction coordinate (8). Distinct enzyme conformations are often linked to different potential energy barriers, as shown by simulations on chorismate mutase (9), fatty acid amide hydrolase (10), and enzymatic reactions involving quantum tunneling (11).
In this study, we included both the COX-1 structure and the effect of molecular flexibility in calculating the potential energy barrier for the rate-limiting H-abstraction reaction in COX-1. It is important to clarify that we analyzed the influence of enzyme structural variation on the potential energy surface but not on catalysis. Our strategy is based on two modeling steps: 1), generation of multiple structures of the ES complex with molecular-dynamics (MD) simulations; and 2), exploration of the reaction mechanism for five ES structures by a hybrid quantum-mechanics/molecular-mechanics (QM/MM) method. We selected four initial structures from periodic boundary MD simulations in the presence of the natural membrane environment (3) (Fig. 1 A), and one from stochastic boundary MD (Fig. 1 B). Details of stochastic- and periodic-boundary MD methods are given in the Supporting Material. The system was divided into QM and MM regions, and the whole ACD molecule and side chain of the catalytically active Tyr-385 radical were included in the QM region.
Figure 1.
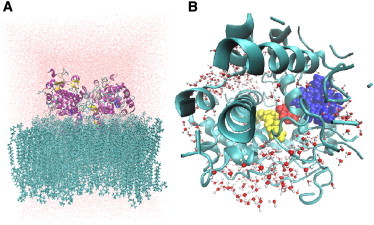
(A and B) Representative structures of COX-1 from periodic-boundary (A) and stochastic-boundary (B) MD simulations, employed for QM/MM reaction modeling.
It is important to include the whole ACD in the QM region so that one can evaluate whether the flexibility of the substrate and its orientation with respect to the tyrosine radical influence the potential energy surface of the reaction. The reaction coordinate is selected as the difference between two distances (d1[C13-Hs] − d2[Hs-O]) to account for the bond-creating and bond-breaking processes. More details about the QM/MM method are presented in the Supporting Material.
The five B3LYP/6-31G(d)-CHARMM27 reaction path profiles are shown in Fig. 2. They have similar shapes, and the transition states (TSs) have comparable positions along the reaction coordinate (close to zero). Nevertheless, differences in the calculated barriers were observed.
Figure 2.
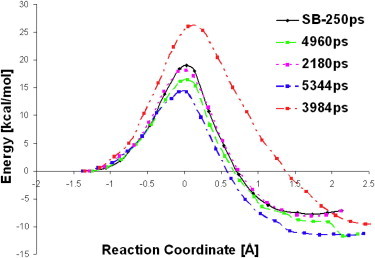
Calculated reaction paths starting from five different initial structures.
The reaction path originating from the stochastic-boundary MD structure gave a potential energy barrier of 18.4 kcal mol−1. The calculations performed using structures from periodic-boundary MD gave the following potential energy barriers: 18.2 kcal mol−1 (for the structure obtained after 2.2 ns of simulation), 16.4 kcal mol−1 after 5.0 ns; 25.9 kcal mol−1 after 4.0 ns, and 14.3 kcal mol−1 from 5.3 ns (Table 1). With two of the starting structures (e.g., after 5.0 and 5.3 ns of periodic-boundary MD simulations), we achieved very good agreement with the experimental barrier (15.0 kcal/mol) (12). A more comprehensive picture, however, is provided by averaged values. Although the arithmetically averaged energy barrier (18.6 kcal mol−1) is in fair agreement with the experimentally deduced value of 15.0 kcal/mol (10), the Boltzmann-averaged one (15.2 kcal mol−1) is in excellent agreement with experimentally derived barriers. Boltzmann averaging tends to eliminate the contributions of unreasonably high barriers and weigh the lower and more-representative barriers (13). Despite the apparent agreement near the chemical accuracy between the Boltzmann-averaged barrier and the experimental one, an absolute variation of 11.6 kcal mol−1 in the calculated barrier (measured as the highest Ea minus the lowest Ea) was found. The reaction energy varies within 3.7 kcal mol−1 and is not correlated to the activation barrier. The results therefore suggest that any single calculation would not necessarily be close to the experimental value or even to the average one. With this in mind, we performed a careful analysis of the geometry of the starting structures, in a manner similar to that described by Lodola et al. (14). Visual inspection of five reactant complexes (RCs) revealed that in all of the RCs, the pro-S hydrogen atom and phenoxyl radical appear to be ideally oriented for initiation of the hydrogen transfer. The distances between the pro-S hydrogen atom and the phenoxyl radical oxygen are reasonably consistent in all of the considered Michaelis complexes (Table 1).
Table 1.
Geometrical descriptors and energy barriers (kcal mol−1) of the reaction paths
| Structure | [O,HS]RC | [O,HS]TS | α1RC | α1TS | Ea |
|---|---|---|---|---|---|
| SB250 | 2.37 | 1.37 | 109.0 | 110.2 | 18.4 |
| 2180 | 2.38 | 1.30 | 95.2 | 108.0 | 18.2 |
| 3984 | 2.47 | 1.41 | 111.0 | 114.7 | 25.9 |
| 4960 | 2.36 | 1.29 | 89.3 | 108.4 | 16.4 |
| 5344 | 2.48 | 1.37 | 83.4 | 106.7 | 14.3 |
The TS structures identified for the five reaction paths show the hydrogen atom HS partially transferred to Tyr-385, and partial formation of the pentadienyl radical. The O–HS distance was found to vary from 1.29 to 1.41 Å, which is consistent with the gas-phase calculations (1.37 Å) (5,6).
To look for a qualitative model that is capable of explaining the difference in the calculated Ea, we analyzed a variety of geometric descriptors (including distances and angles) that account for the main interactions between the enzyme and substrate in RCs, TSs, and products (PC) (Supporting Material, Fig. S2, and Tables S1, S2, and S3). Here, we focus on the importance of the hydrogen abstraction angle α1, defined by the HS atom of ACD, and the phenol O of Tyr-385 and its directly attached aromatic C atom. Fig. 3 suggests a negative correlation between the hydrogen abstraction angle α1 in the TS (α1TS) and the barrier height, at least within the domain of the starting structures explored in this study.
Figure 3.
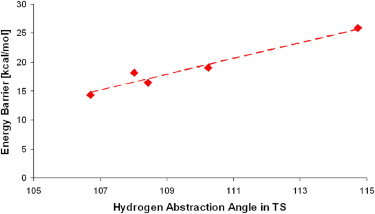
Calculated energy barriers versus hydrogen abstraction angles as found in the TSs.
Furthermore, our analysis indicates a correlation between the hydrogen abstraction angle, α1, in the TS and the same angle in the Michaelis complex (Fig. 3 and Fig. S3), suggesting that the geometry of the TS may be determined by the geometry of the reactants.
Our study demonstrates that the hydrogen abstraction angle is an important geometric determinant for COX reactivity that is correlated to the energy barrier for the reaction. In addition, the results indicate that it is important to include enzyme flexibility (i.e., by considering multiple structures of the ES complex and then averaging the resulting barrier heights (e.g., by Boltzmann averaging)) to obtain an accurate estimate of the potential energy barrier. Finally, our findings indicate the need to move toward free-energy simulations of enzyme mechanisms that intrinsically account for enzyme motion and relaxation (15).
Acknowledgments
The authors thank all of our collaborators from the IntBioSim project (in particular Prof. Mark Sansom and Prof. Jon Essex) and Dr. Kia Balali-Mood for useful discussions.
This work was supported by grant BBSB16011 from the Biotechnology and Biological Sciences Research Council. C.C. and T.K. received International Marie Curie fellowships. A.J.M. is an Engineering and Physical Sciences Research Council Leadership Fellow (grant number EP/G007705/1).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Contributor Information
Christo Z. Christov, Email: Christo.Christov@northumbria.ac.uk.
Adrian J. Mulholland, Email: Adrian.Mulholland@bristol.ac.uk.
Supporting Material
References and Footnotes
- 1.Garavito R.M., Mulichak A.M. The structure of mammalian cyclooxygenases. Annu. Rev. Biophys. Biomol. Struct. 2003;32:183–206. doi: 10.1146/annurev.biophys.32.110601.141906. [DOI] [PubMed] [Google Scholar]
- 2.Rao P., Knaus E.E. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci. 2008;11:81s–110s. doi: 10.18433/j3t886. [DOI] [PubMed] [Google Scholar]
- 3.Wan S., Coveney P.V. A comparative study of the COX-1 and COX-2 isozymes bound to lipid membranes. J. Comput. Chem. 2009;30:1038–1050. doi: 10.1002/jcc.21130. [DOI] [PubMed] [Google Scholar]
- 4.Hamberg M., Samuelsson B. On the mechanism of the biosynthesis of prostaglandins E-1 and F-1-alpha. J. Biol. Chem. 1967;242:5336–5343. [PubMed] [Google Scholar]
- 5.Blomberg L.M., Blomberg M.R.A., Tsai A.L. A quantum chemical study of synthesis of prostaglandin G(2) by the cyclooxygenase active site in prostaglandin endoperoxide H synthase 1. J. Phys. Chem. B. 2003;107:3297–3308. [Google Scholar]
- 6.Silva P.J., Fernandes P.A., Ramos M.J. A theoretical study of radical-only and combined radical/carbocationic mechanisms of arachidonic acid cyclooxygenation by prostaglandin H synthase. Theor. Chem. Acc. 2003;110:345–351. [Google Scholar]
- 7.Prasad B.R., Warshel A. Prechemistry versus preorganization in DNA replication fidelity. Proteins. 2011;79:2900–2919. doi: 10.1002/prot.23128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamerlin S.C.L., Warshel A. At the dawn of the 21st century: is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roca M., Messer B., Warshel A. On the relationship between folding and chemical landscapes in enzyme catalysis. Proc. Natl. Acad. Sci. USA. 2008;105:13877–13882. doi: 10.1073/pnas.0803405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lodola A., Mor M., Mulholland A.J. Conformational effects in enzyme catalysis: reaction via a high energy conformation in fatty acid amide hydrolase. Biophys. J. 2007;92:L20–L22. doi: 10.1529/biophysj.106.098434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glowacki D.R., Harvey J.N., Mulholland A.J. Taking Ockham’s razor to enzyme dynamics and catalysis. Nat. Chem. 2012;4:169–176. doi: 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- 12.Kulmacz R.J., Pendleton R.B., Lands W.E.M. Interaction between peroxidase and cyclooxygenase activities in prostaglandin-endoperoxide synthase. Interpretation of reaction kinetics. J. Biol. Chem. 1994;269:5527–5536. [PubMed] [Google Scholar]
- 13.Lonsdale R., Harvey J.N., Mulholland A.J. Compound I reactivity defines alkene oxidation selectivity in cytochrome P450cam. J. Phys. Chem. B. 2010;114:1156–1162. doi: 10.1021/jp910127j. [DOI] [PubMed] [Google Scholar]
- 14.Lodola A., Sirirak J., Mulholland A.J. Structural fluctuations in enzyme-catalyzed reactions: determinants of reactivity fatty acid amide hydrolase from multivariate statistical analysis of quantum mechanics/molecular mechanics paths. J. Chem. Theory Comput. 2010;6:2948–2960. doi: 10.1021/ct100264j. [DOI] [PubMed] [Google Scholar]
- 15.Plotnikov N.V., Kamerlin S.C.L., Warshel A. Paradynamics: an effective and reliable model for ab initio QM/MM free-energy calculations and related tasks. J. Phys. Chem. B. 2011;115:7950–7962. doi: 10.1021/jp201217b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.