

Abstract
Background
Tumor irradiation blocks local angiogenesis, forcing any recurrent tumor to form new vessels from circulating cells. We have previously demonstrated that the post-irradiation recurrence of human glioblastomas in the brains of nude mice can be delayed or prevented by inhibiting circulating blood vessel–forming cells by blocking the interaction of CXCR4 with its ligand stromal cell-derived factor (SDF)–1 (CXCL12). In the present study we test this strategy by directly neutralizing SDF-1 in a clinically relevant model using autochthonous brain tumors in immune competent hosts.
Methods
We used NOX-A12, an l-enantiomeric RNA oligonucleotide that binds and inhibits SDF-1 with high affinity. We tested the effect of this inhibitor on the response to irradiation of brain tumors in rat induced by n-ethyl-N-nitrosourea.
Results
Rats treated in utero with N-ethyl-N-nitrosourea began to die of brain tumors from approximately 120 days of age. We delivered a single dose of whole brain irradiation (20 Gy) on day 115 of age, began treatment with NOX-A12 immediately following irradiation, and continued with either 5 or 20 mg/kg for 4 or 8 weeks, doses and times equivalent to well-tolerated human exposures. We found a marked prolongation of rat life span that was dependent on both drug dose and duration of treatment. In addition we treated tumors only when they were visible by MRI and demonstrated complete regression of the tumors that was not achieved by irradiation alone or with the addition of temozolomide.
Conclusions
Inhibition of SDF-1 following tumor irradiation is a powerful way of improving tumor response of glioblastoma multiforme.
Keywords: angiogenesis, CXCL12, CXCR4, CXCR7, ENU-induced tumors, glioblastoma, irradiation, NOX-A12, SDF-1, vasculogenesis
Radiotherapy is an important component of the treatment of glioblastoma multiforme (GBM), but despite the high radiation doses used and the combination of anticancer agents and targeted therapies, the tumors invariably recur, leading to the demise of more than 75% of the patients by 2 years. Importantly, most of the recurrences occur within the radiation field.1–4 Thus any method of improving local control of the primary tumor would improve the curability of GBM patients.
We recently proposed a novel paradigm for the treatment of solid tumors with radiotherapy with a particular emphasis on GBM.5 This was based on our previous finding that doses of radiation in the therapeutic range abrogate local angiogenesis,6 forcing the tumor to rely on the vasculogenesis pathway. Vasculogenesis involves de novo growth of blood vessels from circulating cells for the restoration of the tumor vasculature, thereby allowing tumor recurrence. We demonstrated both with the subcutaneously transplanted FaDu human head and neck tumor7 and with the intracranially implanted U251 human GBM5 that tumor recurrence could be markedly reduced or even prevented by inhibition of irradiation-induced recruitment of the pro-angiogenic CD11b+ myelomonocytes. This could be achieved either with anti-CD11b antibodies7 or by inhibiting the interaction of stromal cell derived factor–1 (SDF-1, CXCL12) with its chemokine receptor CXCR4,5 which is highly expressed on CD11b+ monocytes. Kozin and colleagues8 have also shown that tumor irradiation induces uptake of CD11b+ monocytes into the tumors and that the SDF-1/CXCR4 inhibitor AMD3100 delivered for 21 days after irradiation enhances the antitumor efficacy of radiation.
Except in the case of the highly radiosensitive U251, post-irradiation treatment with AMD3100, however, only prolonged the radiation-induced growth delay, and the tumors eventually recurred in the 2 studies mentioned. Although CD11b+ monocytes facilitate vasculogenesis by tissue remodeling requiring matrix metalloproteinase–9,6 they do not form blood vessels themselves.5,8 Therefore, we hypothesized that other circulating cells, such as endothelial cells (ECs) or endothelial progenitor cells (EPCs),9 as well as possibly pericytes, may be needed to restore the irradiated vasculature. Since ECs in solid tumors, including GBM, have been shown to express high levels of CXCR7,10–12 the more recently discovered second receptor for SDF-1,13,14 we hypothesized that the most effective strategy for preventing post-irradiation vasculogenesis in GBM would be to block both SDF-1 receptors, CXCR4 and CXCR7. A logical way to achieve concomitant inhibition would be to target the shared ligand SDF-1.15
A modality fulfilling these requirements is the PEGylated mirror-image RNA oligonucleotide (a so-called Spiegelmer) NOX-A12 that binds with high affinity to SDF-1. NOX-A12 consists of 45 l-enantiomeric RNA nucleotides and carries a 40-kDa polyethylene glycol (PEG) modification at its 5′-end in order to increase plasma residence time. The 45 l-nucleotides of the oligonucleotide form a structural scaffold that recognizes SDF-1 with high affinity and thus efficiently blocks the chemokine's interaction with CXCR4 and CXCR7. The nonnatural l-nucleotides confer oligonucleotide biostability because naturally abundant nucleases are not able to recognize l-oligonucleotides. Also, their mirror-image nature renders Spiegelmers immunologically passive, with a low risk of neutralizing antibodies and no Toll-like receptor activation.16 NOX-A12 is currently in phase II studies with chronic lymphocytic leukemia and multiple myeloma.
To test our strategy to block SDF-1 interaction with both its receptors CXCR4 and CXCR7, we used N-ethyl-N-nitrosourea (ENU)–induced brain tumors in the Sprague-Dawley rat, a model that has proved to be extremely resistant to anticancer therapy in prior studies by a variety of investigators17,18 (L. Recht, unpublished). After in utero exposure to ENU on days 17–18 of gestation, the pups appeared healthy for over 100 days, at which time they began to demonstrate neurological distress and die progressively from brain tumors from day 120 after birth. The current data demonstrate that NOX-A12–mediated SDF-1 blockade was effective in inhibiting or delaying recurrences following irradiation in this tumor model that mimics the genetic diversity and histology of human brain tumors.19 As NOX-A12 was delivered following brain irradiation at doses and time periods similar to exposures that have been described to be safe and well tolerated in humans, we believe that these results justify a human trial in first-line glioblastoma patients.
Materials and Methods
In vitro Characterization of NOX-A12
THP-1 cells were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ) and cultured overnight at a cell density of 0.3 × 106/mL at 37°C and 5% CO2 in Roswell Park Memorial Institute 1640 medium with GlutaMAX (Invitrogen), 10% fetal bovine serum, and 50 µM β-mercaptoethanol. Five nanomolar human recombinant SDF-1α (R&D Systems) was preincubated with NOX-A12 in various concentrations in Hank’'s Balanced Salt Solution (Invitrogen) containing 1 mg/mL bovine serum albumin (BSA) and 20 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) (HBH) in the lower compartments of a 96-transwell plate with 5-μm pores (Costar Corning, #3388) at 37°C for 20 min. Added to the upper compartments were 1 × 105 THP-1 cells in HBH buffer, which were allowed to migrate at 37°C for 3 h. Migrated cells were quantified by resazurin (Sigma) fluorescence. Relative fluorescence intensity was normalized and plotted against NOX-A12 concentrations for determination of half-maximal inhibitory concentration (IC50) using GraphPad Prism.
PathHunter eXpress CXCR7 activated G protein coupled receptor (GPCR) internalization cells (DiscoveRX) were incubated with 10 nM human recombinant SDF-1α (R&D Systems) and various concentrations of NOX-A12 at 37°C for 3 h. CXCR7 internalization was quantified according to the instructions of the supplier, and relative luminescence intensity was normalized and plotted against NOX-A12 concentrations for IC50 determination using GraphPad Prism software.
Human umbilical vein endothelial cells (HUVECs) were obtained from Lonza and cultured in endothelial cell growth medium–2 (Lonza) at 37°C and 5% CO2. One day before the experiment, HUVECs were seeded in a 10-cm culture plate at 40% confluency and incubated overnight. Cells were washed once with phosphate buffered saline (PBS) and incubated with DiIC12(3) fluorescent dye (BD Biosciences) at a concentration of 5 µg/mL in endothelial cell basal medium–2 (Lonza) containing 0.5% BSA and 0.5% fetal bovine serum for 15 min at 37°C. Cells were washed twice with PBS, mildly trypsinized, washed, and resuspended in Dulbecco's modified Eagle's medium (DMEM) without phenol red containing 20 mM HEPES and 0.1% BSA at a concentration of 3.2 × 105 cells/mL. Cells were incubated at 37°C and 5% CO2 at least 60 min before adding to the FluoroBlok Transwell inserts with fluorescent blocking PET membrane and 3-μm pores (BD Biosciences, Falcon, # 351151). The lower side of the inserts was coated with 5 µg/mL human fibronectin (R&D Systems) at room temperature for 1 h, washed once with PBS, and then incubated with 1 µg/mL human SDF-1α (R&D Systems) and NOX-A12 in DMEM without phenol red and 20 mM HEPES at room temperature for 2 h. The filters were washed once and inserted in a BD Falcon 24-well companion plate containing 750 µL DMEM without phenol red, 20 mM HEPES, and 0.1% BSA. Two hundred fifty microliters of stained and equilibrated HUVECs as we have described were added to the upper compartment. Cell migration was quantified using a bottom fluorescence reading plate reader (Synergy2, Biotek) and plotted against incubation time.
Experimental Animals
Brain tumors were induced by i.p. injection of 50 mg/kg of the carcinogen ENU (Sigma) into Sprague-Dawley rats (Taconic Farms) at gestational day 18. During each experiment, at least one control pregnant rat received saline vehicle under identical conditions. Pups were born and weaned at normal age, after which they were monitored at least weekly for signs of illness, which usually developed after 120 days of age, at which time the pups started to show signs of brain lesions (loss of weight, hind limb paralysis, and lethargy). In the first study, designed only to evaluate survival, the rats were assigned randomly to the various groups on day 115 of age, just before any rats started to die. In the second study, designed to evaluate tumor response by MRI as well as survival, tumor size was estimated by MRI starting on day 130 of age, and the rats were assigned to groups so as to have approximately the same total tumor volume in each group. All animal procedures were approved by Stanford University's Administrative Panel on Laboratory Animal Care.
Drug Treatment and Irradiations
Whole brain irradiation was performed with a Phillips X-ray unit operated at 200 kVp with a dose rate of 1.21 Gy/min (20 mA with added filtration of 0.5 mm copper, distance from X-ray source to the target of 31 cm, and a half value layer of 1.3 mm copper). The rats were anesthetized and placed in individual lead boxes with a cutout that allowed the brain to be irradiated tangentially with full shielding of the buccal cavity as well as the rest of the body. To ensure the maximum uniformity of the dose delivered, the animals were turned 180 degrees halfway through each irradiation (giving the equivalent of parallel opposed fields).
Animals were treated with the anti–SDF-1 Spiegelmer NOX-A12 (primary sequence: 5′-GCGUGGUGUGAUCUAGAUGUAUUGGCUGAUCCUAGUCAGGUACGC-3′, modified with 40 kDa PEG at the 5′-terminus and synthesized at NOXXON Pharma, Berlin, Germany); adequate exposure of the drug was confirmed in a separate pharmacokinetics study (Supplemental Fig. S1). The compound was dissolved at a concentration of 10 mg/mL in sterile 5% glucose solution with occasional shaking for up to 30 min at room temperature. This solution was then stored frozen. For the 5 mg/kg group, the frozen solution was thawed on the day of injection and diluted to 5 mg/mL, and 1 mL/kg was subcutaneously administered every 2 days for 8 weeks; for the 20 mg/kg group, the 10 mg/mL solution was used directly and 2 mL/kg was administered for 4 or 8 weeks; for the 10 mg/kg group, the 10 mg/kg solution was used directly and 1 mL/kg was administered for 10 weeks. In the study in which NOX-A12 was combined with temozolomide (TMZ), the TMZ (10 mg/kg i.p; Sigma-Aldrich) was given 5 days/week for 3 weeks.
MRI Measurements of Rat Brain Tumors
MRI measurements (T2-weighted images) were performed on a 1 T machine (Bruker) commencing on day 130 after birth and then repeated every 2 weeks until death. Rats were distributed into the various treatment groups so as to have approximately equal total tumor volumes in each group. Only rats with identifiable tumors were sorted into the groups.
Rats were securely fixed to an MRI head holder (Stanford Center of Biomedical Imaging), and T2-weighted coronal images of 1.5 mm thickness were obtained.
Images were read by a reviewer who was blinded to treatment group using an OsiriX computer program. Each imaging slice that contained tumor was outlined freehand and the volume calculated; the sum of all the slice volumes was considered the tumor volume for that subject.
Histological Examination of the Brains
Rats were perfused with 4% paraformaldehyde at the end of the imaging experiment. Brains were removed and positioned for cutting using the same stereotactic readings from the bregma and lambda, so that they were at the precise angle in which they underwent MRI and brains removed. Fifty-micrometer sections were cut from ∼2.2 mm anterior to ∼7.5 mm posterior to the bregma; all sections were stained with standard hematoxylin and eosin. Using the serial imaging for verification, we then identified each tumor using standard microscopy.
Statistical Analysis
Statistical analyses were performed by the 2-tailed Student's t-test or 1-way ANOVA to determine statistical significance. P values (exact significance) of <.05 were considered statistically significant. Kaplan–Meier curves and the log-rank test were used to compare survival times among the groups. All calculations were performed using Prism5 (GraphPad).
Results
NOX-A12 Is a Specific Inhibitor of SDF-1 Blocking Interaction With CXCR4 and CXCR7
THP-1 myelomonocytes (which highly express CXCR4 but do not express CXCR7 as demonstrated by flow cytometry) show SDF-1–mediated chemotaxis. NOX-A12 is able to inhibit a stimulus of 5 nM SDF-1 in a dose-dependent manner with an IC50 of 3.9 ± 0.2 nM (Fig. 1A). PathHunter eXpress CXCR7 activated GPCR internalization cells (DiscoveRX) were used to demonstrate SDF-1–dependent CXCR7 activation. NOX-A12 shows inhibition of SDF-1 mediated CXCR7 internalization dose dependently with an IC50 of 3.0 ± 0.9 nM (Fig. 1B). HUVECs, which are known to express both SDF-1 receptors, CXCR420 and CXCR7,21 migrate toward immobilized SDF-1 on fibronectin. NOX-A12 inhibits SDF-1–stimulated migration of HUVECs (Fig. 1C). In summary, we demonstrated that NOX-A12 blocks SDF-1–dependent activation of both receptors, CXCR4 and CXCR7, with high potency. Furthermore, NOX-A12 inhibits SDF-1–dependent chemotaxis of monocytes and endothelial cells, which both play an important role in vasculogenesis of irradiated solid tumors.
Fig. 1.

(a) NOX-A12 inhibits SDF-1–mediated and CXCR4-dependent chemotaxis of THP-1 myelomonocytes. THP-1 cells are attracted by 5 nM of human SDF-1 (set to 100%). SDF-1 was preincubated with NOX-A12 at various concentrations. One representative dose-response curve (mean ± SD of triplicates) of 3 independent experiments is shown. NOX-A12 inhibits SDF-1–mediated chemotaxis of CXCR4-expressing THP-1 cells with an IC50 of 3.9 ± 0.2 nM. (b) NOX-A12 inhibits SDF-1–mediated internalization of CXCR7. PathHunter eXpress CXCR7 activated GPCR internalization cells show SDF-1–mediated internalization of CXCR7. Internalization of CXCR7 by incubation with 10 nM SDF-1 was set to 100%. SDF-1 was preincubated with various concentrations of NOX-A12. One representative dose-response curve (mean ± SD of triplicates) of 4 independent experiments is shown. NOX-A12 inhibits SDF-1–mediated CXCR7 internalization with an IC50 of 3.0 ± 0.9 nM. (c) NOX-A12 inhibits SDF-1–mediated migration of human ECs. HUVECs were stained with the fluorescent dye DiIC12(3), and migration through the FluoroBlok membrane was quantified in a bottom reading plate reader; 1 µg/mL of human SDF-1 was preincubated either with or without an equimolar concentration of NOX-A12 and then added to the bottom of the transwell inserts, which were coated with fibronectin. SDF-1 immobilized on fibronectin increases migration of HUVECs, which is completely inhibited by NOX-A12. Each curve reflects the means of duplicates ± SD from a single experiment and is representative of 5 independent experiments.
Blockade of SDF-1 Post-irradiation With NOX-A12 Prolongs Survival of Rats With ENU-induced Brain Cancer
To ensure that the rats entering the first set of studies would have brain tumors of a size close to those producing neurological symptoms and death, we randomized rats born to mothers treated with ENU (50 mg/kg) on day 18 of gestation at day 115 of age. This was just before any of the rats began to die from their brain tumors (Fig 2). In this study we included a group of rats given NOX-A12 alone and controls given vehicle alone for 28 days. We also tested 2 different concentrations of NOX-A12 and 2 different periods of drug administration after irradiation. In order to increase the power of the comparisons, we pooled the data from 2 prior experiments in which identically treated rats were either not irradiated or given 20 Gy whole brain irradiation (WBI). As can be seen (Fig. 2 and Table 1), the dose of 20 Gy WBI extended the median life span of the rats by a small and not quite significant amount of approximately 7 days (P = .07 vs non-irradiated). However, the addition of NOX-A12 prolonged the median life span of the irradiated rats by a significant amount: by 95 days for the rats given the 8-week infusion at the low dose (5 mg/kg) and by 153 days for the rats given the higher dose (20 mg/kg) in comparison with the rats receiving 20 Gy WBI alone. As can be seen, there was no significant effect of NOX-A12 alone. The experiment was terminated after 418 days. We also gave 20 Gy WBI with or without 8 weeks of NOX-A12 to rats that had not been given ENU in utero (so did not have brain tumors). We saw no deaths in these rats up to the observation period of 418 days (not shown in Fig. 2). The median life spans of the different groups and their significance levels are shown in Table 1.
Fig. 2.
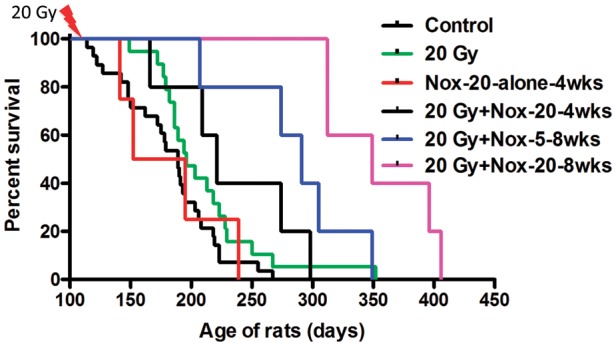
SDF-1 inhibition after irradiation prolongs the survival of the brain tumor–bearing rats. Rats born to mothers treated with a single injection of the carcinogen ENU on day 18 of gestation were sham irradiated or given a dose of 20 Gy to the whole brain with shielding of the buccal cavity. The rats receiving NOX-A12 were injected subcutaneously every 2 days with either 5 or 20 mg/kg starting soon after irradiation and continued for either 4 or 8 weeks. Survival times are shown in Table 1.
Table 1.
Analysis of survival of ENU-treated rats at day 384
| P value | Control | NOX-20 Alone | 20 Gy Alone | 20 Gy + NOX-5, 4 wk | 20 Gy + NOX-20, 4 wk | 20 Gy + NOX-5, 8 wk | 20 Gy + NOX-20, 8 wk | 20 Gy + No ENU | No ENU + NOX-20, 8 wk |
|---|---|---|---|---|---|---|---|---|---|
| Med. Surv | 189 | 173.5 | 196 | 244.5 | 221 | 291 | 349 | >384 | >418 |
| Vs 20 Gy | 0.07 | NS | – | 0.10 | NS | 0.05 | 0.003 | <0.0001 | 0.01 |
| Vs Control | – | NS | 0.07 | 0.05 | 0.02 | 0.0006 | <0.0001 | <0.0001 | 0.008 |
Controls and 20 Gy alone were pooled data from 3 studies.
Assessment of Rat Brain Tumor Response by MRI
In a second set of experiments, our aim was to take the study to a more clinically realistic setting by assessing the effect of the treatments on the brain tumors in real time using MRI and to compare the efficacy of SDF-1 blockade with that of the clinically used drug TMZ, in potentiating the efficacy of irradiation. To perform the study, we began MRI of the rat brains on day 130 after birth and treated only animals that had brain tumors identified by MRI. This protocol therefore had a key difference versus the first study in that treatment was started from 2 to 7 weeks later than day 115 used previously. Thus the average initial tumor sizes were larger in this study, making it more of a challenge to affect survival.
Prior to commencing the study, we established the sensitivity of MRI to detect brain tumors of various sizes. We imaged rats at various times after day 130 of birth and sacrificed them to relate the MRIs to tumor brain histology. We performed serial sections on the rats sacrificed and attempted to correlate the histological images (hematoxylin and eosin staining) with serial section MRI. As shown in Fig. 3, we saw an excellent correlation between the 2 methods except for the smallest tumors, barely detectable in the histological sections. We therefore concluded that MRI was measuring all the significant brain tumors induced by ENU.
Fig. 3.
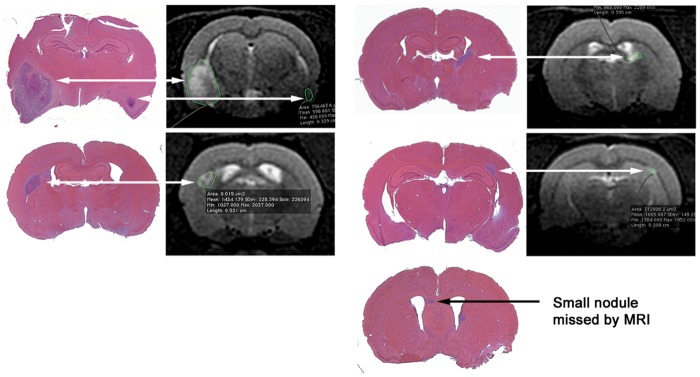
MRI is effective in detecting the ENU-induced brain tumors. Rats born to mothers given a single injection of ENU on day 18 of gestation were imaged by MRI, and when 1 or more tumors per rat were detected, the rats were sacrificed and the brains serially sectioned and stained with hematoxylin and eosin to identify the same lesions as seen on the MRI scans. MRI also detected all but the smallest lesions detected from the histology. Shown are representative images of 3 rats from a total of 11 analyzed.
Figure 4 shows the geometric means of the tumors in all the groups as a function of time from the beginning of treatment (initiated days 132–165). All the estimations of tumor size from the MRI measurements were made by one individual (T.J.) without any knowledge of the treatment that the rat had received. A second individual sorted the data into the treatment groups and calculated the group means. The salient features of the data are as follows:
The tumors in the rats treated with NOX-A12 alone continued to grow as expected (Fig. 4).
The tumors in the rats treated with 20 Gy + NOX-A12 (blue line) disappeared by 28 days after the start of treatment. The tumors in this group continued to be undetectable until the appearance of 2 recurrences 105 days after the initiation of treatment.
The tumors in the rats given 20 Gy alone or 20 Gy + TMZ behaved similarly with an initial decrease in volume to day 45 followed by a regrowth.
Fig. 4.
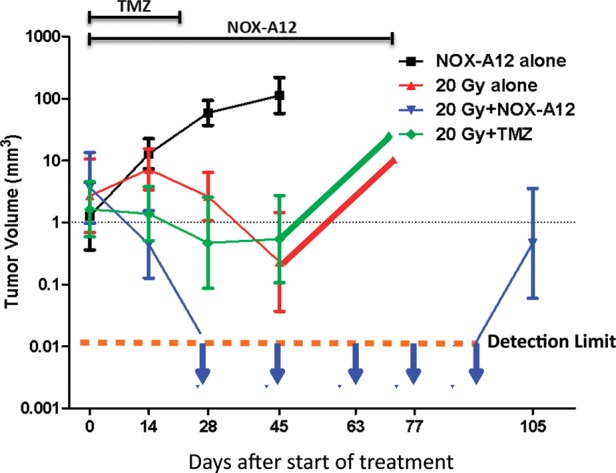
Addition of NOX-A12 following irradiation of the ENU-induced brain tumors produces complete responses by MRI. In utero ENU-treated rats were imaged by MR starting on day 130 of age, repeated every 2 weeks until death. Rats were distributed into the various treatment groups so as to have approximately equal total tumor volumes in each group at the start of treatment.
Though our primary purpose in this study was to measure tumor size following treatment, we also observed that the survival of the rats given NOX-A12 following irradiation was significantly extended compared with those rats given irradiation alone or irradiation + TMZ. We also in the same experiment assessed the effect of adding TMZ to the irradiation + NOX-A12 protocol and found that the addition of TMZ did not further extend the life span of the rats given irradiation + NOX-A12, further supporting our conclusion that the addition of NOX-A12 is superior to the addition of TMZ.
In order to be sure that the efficacy of NOX-A12 was not just confined to this rat model, we performed an experiment with U251 human GBM implanted intracranially into immune deficient nude mice, measuring tumor response using bioluminescent imaging. The results obtained showed that the addition of NOX-A12 (10 mg/kg 3× per wk) following 12 Gy WBI produced substantial additional tumor shrinkage and growth delay compared with irradiation only (Supplemental Fig. S2).
Discussion
In earlier preclinical studies, we have shown that we could inhibit or delay the regrowth of subcutaneously implanted FaDu human head and neck cancer7 or intracranially implanted U251 human GBM5 following irradiation using a variety of strategies to prevent the radiation-induced influx of CD11b+ myelomonocytes. These included inhibition of the transcription factor hypoxia inducible factor–1, inhibition of CXCR4 (by the small molecule plerixafor as well as by anti-CXCR4 antibodies), depletion of macrophages using carrageenan,5 and use of CD11b blocking antibodies.7 Although we obtained total abrogation of regrowth in the U251 tumor, we achieved only a modest increase in growth inhibition with the U87 human GBM.5 This result suggests that some tumors might be more, or could become more, dependent on alternative pathways of vasculogenesis after irradiation. Such pathways may also include CXCR7, the more recently discovered second receptor for SDF-1. This assumption seems an attractive possibility because CXCR7 is highly expressed on endothelial cells of many tumors, including GBM,10–12 and CXCR7 has also been shown to facilitate transendothelial cell migration and vascular tube formation by EPCs in vitro.22
The present study was performed with 2 main objectives: first, to investigate the efficacy of an inhibitor to the chemokine SDF-1, which blocks the signaling of both known SDF-1 receptors, CXCR4 and CXCR7 (Fig. 1A–C); second, to evaluate the strategy of employing a more refractory and naturally occurring tumor model as close as possible to clinically appearing brain tumors. Therefore, we selected the ENU-induced brain tumor model in rats. The key advantages of this model are that the tumors arise autochthonously in immune competent hosts and have a genetic diversity and aggressiveness comparable to human brain tumors.19 Furthermore, macroscopic tumors frequently contain high levels of vascular endothelial growth factor, hemorrhage, and focal necrosis, all general characteristics of the most malignant glioblastomas. Most importantly, these tumors, similar to human glioblastomas, are very resistant to treatment: maximum tolerated repeated doses of TMZ do not affect survival, and high doses of vascular endothelial growth factor inhibitors have only a slight effect on tumor progression (Recht et al, unpublished). As is evident from Fig. 2, we confirmed the resistance of these tumors by the fact that a dose of 20 Gy WBI resulted in a small and not significant (P = .07) increased survival of the rats.
As can be deduced from Fig. 2 as well, we showed that inhibition of the SDF-1 pathway in combination with a single dose of 20 Gy markedly enhanced the survival of the rats. This finding of an enhanced response on tumors was also confirmed by real-time MRI measurements (Fig. 4). In this second study we also showed that the effect of blocking SDF-1 with NOX-A12 in combination with irradiation was significantly greater than the effect of addition of TMZ to tumor irradiation.
To address the question of whether the in vivo effects of NOX-A12 on tumor regrowth are the result of a direct reduction of tumor cell growth by SDF-1 inhibition, we irradiated log phase U251 cells in vitro and maintained similar levels of NOX-A12 in the medium following irradiation. We saw no effect of the drug on the survival of U251 human GBM cells to irradiation, nor to the growth of the cells with or without prior treatment with irradiation (Supplemental Fig. S3). We therefore conclude that the effect of NOX-A12 is not a direct one on the tumor cells but is consistent with inhibition of the post-irradiation recovery of tumor blood flow in line with the observations published earlier with U251 GBM using the CXCR4 inhibitor AMD-3100 following irradiation.5
These data lead to the question on the mechanism by which SDF-1 inhibition prevents the eventual restoration of the tumor vasculature after irradiation. To address this question we first have to ask, how does the tumor restore its vasculature following irradiation? There are 3 main possibilities:
Angiogenesis sprouting from nearby unirradiated blood vessels eventually reaches the irradiated tumors. However, we consider this possibility unlikely because the whole brains of the rodents were irradiated and not just the tumors themselves. We note, however, that this is a possibility in the more normal situation in radiotherapy in which the radiation dose is more directly focused on the tumor itself. Despite this, there are situations such as with multiple brain metastases in which WBI is delivered.23
The tumor vasculature regrows from surviving ECs in and around the tumors. This is a scenario favored in a recent comment published by Kozin and colleagues.24 They argue that several investigators (including ourselves5) have failed to find bone marrow derived (BMD) ECs in tumors after irradiation and that the radiation-induced influx of pro-inflammatory and pro-angiogenic monocytes/macrophages would protect the ECs from radiation killing. Although this is a possibility, the protective effect of the BMD monocytes on the irradiated endothelial cells would have to be beyond what has ever been observed for radiation cell killing to account for the magnitude of the effects we see with inhibition of the SDF-1 pathway. Also, increased levels of SDF-1 and BMD monocytes in the tumor occur days to weeks after irradiation5,8 and therefore after the completion of cellular DNA repair.
Circulating ECs or EPCs and pro-angiogenic monocytes colonize the tumor after irradiation, thereby restoring the vasculature through the process of vasculogenesis. We and others have demonstrated the influx of pro-angiogenic CD11b+ monocytes into solid tumors following irradiation5,6,8 but, as noted earlier, failed to detect BMD ECs in the tumors. However, it was reported that the circulation of ECs or EPCs derives from other tissues, including vessel walls, the gastrointestinal tract, and liver.25–27 Furthermore, several authors, including ourselves, have shown an increase in circulating ECs or EPCs after treatment of tumors with vascular disruptive agents, chemotherapy,28,29 and irradiation.30
Based on these considerations, we believe that the evidence supports the post-irradiation colonization of the tumors with myelomonocytes and ECs and/or EPCs; these cells seem to play a major role in restoring tumor vasculature after irradiation. Several investigators have shown that the recruitment and retention of pro-angiogenic cells from the bone marrow to sites of ischemic tissue damage or to tumors are mediated by the interaction of SDF-1 with CXCR4.31–33 In support of this, our in vitro data (Fig. 1) show that NOX-A12 potently inhibits SDF-1–dependent chemotaxis of both myelomonocytes and ECs. Interestingly, we have previously demonstrated that SDF-1 is induced by hypoxia in vitro in a manner dependent on hypoxia inducible factor–1, that U251 tumors become hypoxic following irradiation, and that SDF-1 is produced in the tumors as they become hypoxic.5 As noted earlier, we demonstrated with the U251 intracranial model that the post-irradiation recruitment of BMD cells can be prevented using AMD3100 (plerixafor), a drug that blocks the interaction of SDF-1 with CXCR4, and that this delays tumor recurrence following both single and fractionated doses of irradiation. Of note, besides its activity to block CXCR4, AMD3100 is an allosteric agonist of CXCR734 that can signal through β-arrestin and may activate the endothelium, leading to increased invasiveness of cancer cells.35
One possible extension of our work is with the use of anti-angiogenic therapy that does not involve irradiation. Xu and colleagues36 have demonstrated that anti-angiogenic therapy of patients with rectal cancer with bevacizumab upregulates both SDF-1 and CXCR4 in the tumors. These data raise the possibility that anti-angiogenic therapy may increase vasculogenesis and that blockage of SDF-1 or its receptors may enhance the efficacy of this therapy. In preliminary studies we have in fact shown this to be the case with the C6 tumor implanted intracranially in rats (Liu and Brown, unpublished).
In summary, the present data confirm and extend our earlier findings with CXCR4 blockade with the U251 tumor using a more clinically realistic glioma model in which the tumors develop in an autochthonous manner in immune competent hosts. We further show that inhibition of SDF-1 post-irradiation is superior in shrinking brain tumors after irradiation compared with TMZ at doses equivalent to those used with concomitant radiotherapy in treating GBM. A summary of the proposed mechanism of tumor vasculogenesis and the various successful inhibitors of the process is presented in Fig 5.
Fig. 5.
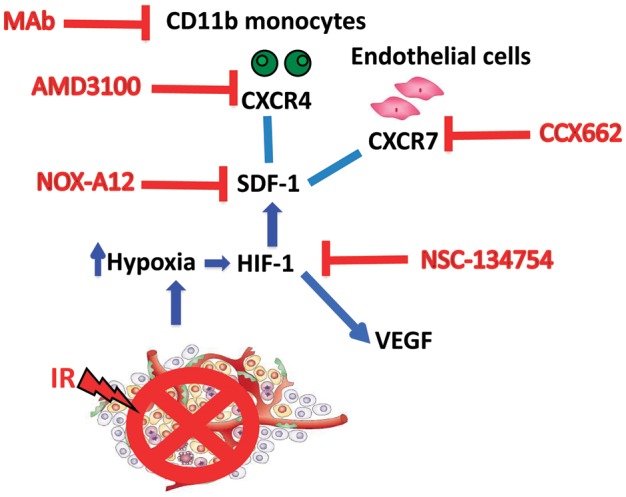
Model showing the vasculogenesis pathway that restores tumor vasculature after irradiation and the various inhibitors of this pathway that can improve the radiation response of the tumor. In addition to summarizing the data from the present study, the figure draws data from prior publications.5,7 Abbreviations: MAb, monoclonal antibody; HIF-1, hypoxia-inducible factor 1; VEGF, vascular endothelial growth factor; IR, irradiation.
Based on the results obtained from this study, we believe that a clinical trial of NOX-A12 in combination with standard therapy in first-line glioblastoma patients would be justified. NOX-A12 doses and treatment times in these preclinical studies were chosen based on equivalents found to be safe and well tolerated in humans, and the drug is currently in phase II studies for the treatment of chronic lymphocytic leukemia and multiple myeloma.
Supplementary Material
Funding
This work was supported by grants from the National Institutes of Health (nos. R01CA128873 and R01 CA149318) and from NOXXON Pharma AG.
Conflict of interest statement. D.Z., S.Z., A.K., and S.K. are employees of NOXXON Pharma AG. J.M.B. is the recipient of a research grant from NOXXON Pharma AG.
Supplementary Material
References
- 1.Hochberg FH, Pruitt A. Assumptions in the radiotherapy of glioblastoma. Neurology. 1980;30(9):907–911. doi: 10.1212/wnl.30.9.907. [DOI] [PubMed] [Google Scholar]
- 2.Liang BC, Thornton AF, Jr., Sandler HM, Greenberg HS. Malignant astrocytomas: focal tumor recurrence after focal external beam radiation therapy. J Neurosurg. 1991;75(4):559–563. doi: 10.3171/jns.1991.75.4.0559. [DOI] [PubMed] [Google Scholar]
- 3.Sneed PK, Gutin PH, Larson DA, et al. Patterns of recurrence of glioblastoma multiforme after external irradiation followed by implant boost. Int J Radiat Oncol Biol Phys. 1994;29(4):719–727. doi: 10.1016/0360-3016(94)90559-2. [DOI] [PubMed] [Google Scholar]
- 4.McDonald MW, Shu HK, Curran WJ, Jr., Crocker IR. Pattern of failure after limited margin radiotherapy and temozolomide for glioblastoma. Int J Radiat Oncol Biol Phys. 2011;79(1):130–136. doi: 10.1016/j.ijrobp.2009.10.048. [DOI] [PubMed] [Google Scholar]
- 5.Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120(3):694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahn GO, Brown JM. Matrix metalloproteinase–9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow–derived myelomonocytic cells. Cancer Cell. 2008;13(3):193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107(18):8363–8368. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70(14):5679–5685. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Carson-Walter EB, Cooper A, Winans BN, Johnson MD, Walter KA. Vascular gene expression patterns are conserved in primary and metastatic brain tumors. J Neurooncol. 2010;99(1):13–24. doi: 10.1007/s11060-009-0105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madden SL, Cook BP, Nacht M, et al. Vascular gene expression in nonneoplastic and malignant brain. Am J Pathol. 2004;165(2):601–608. doi: 10.1016/s0002-9440(10)63324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miao Z, Luker KE, Summers BC, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104(40):15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balabanian K, Lagane B, Infantino S, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280(42):35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- 14.Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203(9):2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinrich EL, Lee W, Lu J, Lowy AM, Kim J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J Transl Med. 2012;10:68. doi: 10.1186/1479-5876-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eulberg D, Klussmann S. Spiegelmers: biostable aptamers. Chem Bio Chem. 2003;4(10):979–983. doi: 10.1002/cbic.200300663. [DOI] [PubMed] [Google Scholar]
- 17.Kish PE, Blaivas M, Strawderman M, et al. Magnetic resonance imaging of ethyl-nitrosourea-induced rat gliomas: a model for experimental therapeutics of low-grade gliomas. J Neurooncol. 2001;53(3):243–257. doi: 10.1023/a:1012222522359. [DOI] [PubMed] [Google Scholar]
- 18.Yabuno T, Konishi N, Nakamura M, et al. Drug resistance and apoptosis in ENU-induced rat brain tumors treated with anti-cancer drugs. J Neurooncol. 1998;36(2):105–112. doi: 10.1023/a:1005878402133. [DOI] [PubMed] [Google Scholar]
- 19.Jang T, Savarese T, Low HP, et al. Osteopontin expression in intratumoral astrocytes marks tumor progression in gliomas induced by prenatal exposure to N-ethyl-N-nitrosourea. Am J Pathol. 2006;168(5):1676–1685. doi: 10.2353/ajpath.2006.050400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salcedo R, Wasserman K, Young HA, et al. Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: in vivo neovascularization induced by stromal-derived factor–1alpha. Am J Pathol. 1999;154(4):1125–1135. doi: 10.1016/s0002-9440(10)65365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe K, Penfold ME, Matsuda A, et al. Pathogenic role of CXCR7 in rheumatoid arthritis. Arthritis Rheum. 2010;62(11):3211–3220. doi: 10.1002/art.27650. [DOI] [PubMed] [Google Scholar]
- 22.Dai X, Tan Y, Cai S, et al. The role of CXCR7 on the adhesion, proliferation and angiogenesis of endothelial progenitor cells. J Cell Mol Med. 2011;15(6):1299–1309. doi: 10.1111/j.1582-4934.2011.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kienast Y, Winkler F. Therapy and prophylaxis of brain metastases. Expert Rev Anticancer Ther. 2010;10(11):1763–1777. doi: 10.1586/era.10.165. [DOI] [PubMed] [Google Scholar]
- 24.Kozin SV, Duda DG, Munn LL, Jain RK. Neovascularization after irradiation: what is the source of newly formed vessels in recurring tumors? J Natl Cancer Inst. 2012;104(12):899–905. doi: 10.1093/jnci/djs239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin-Padura I, Bertolini F. Circulating endothelial cells as biomarkers for angiogenesis in tumor progression. Front Biosci (Schol Ed) 2009;1:304–318. doi: 10.2741/s28. [DOI] [PubMed] [Google Scholar]
- 26.Aicher A, Rentsch M, Sasaki K, et al. Nonbone marrow–derived circulating progenitor cells contribute to postnatal neovascularization following tissue ischemia. Circ Res. 2007;100(4):581–589. doi: 10.1161/01.RES.0000259562.63718.35. [DOI] [PubMed] [Google Scholar]
- 27.Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105(1):71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaked Y, Ciarrocchi A, Franco M, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313(5794):1785–1787. doi: 10.1126/science.1127592. [DOI] [PubMed] [Google Scholar]
- 29.Bertolini F. Chemotherapy and the tumor microenvironment: the contribution of circulating endothelial cells. Cancer Metastasis Rev. 2008;27(1):95–101. doi: 10.1007/s10555-007-9110-y. [DOI] [PubMed] [Google Scholar]
- 30.Russell JS, Brown JM. Investigation of endothelial progenitor cells as mediators of radiation resistance. Paper presented at: Proceedings of the 104th Annual Meeting of the American Association for Cancer Research 2013; Washington, DC. [Google Scholar]
- 31.Aghi M, Cohen KS, Klein RJ, Scadden DT, Chiocca EA. Tumor stromal-derived factor–1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res. 2006;66(18):9054–9064. doi: 10.1158/0008-5472.CAN-05-3759. [DOI] [PubMed] [Google Scholar]
- 32.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10(8):858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 33.Jin DK, Shido K, Kopp HG, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12(5):557–567. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol Pharmacol. 2009;75(5):1240–1247. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 35.Rajagopal S, Kim J, Ahn S, et al. Beta-arrestin– but not G protein–mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107(2):628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Duda DG, di Tomaso E, et al. Direct evidence that bevacizumab, an anti-VEGF antibody, up-regulates SDF1alpha, CXCR4, CXCL6, and neuropilin 1 in tumors from patients with rectal cancer. Cancer Res. 2009;69(20):7905–7910. doi: 10.1158/0008-5472.CAN-09-2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.