

Abstract
Intracellular fat droplets are large and have a distinct morphology, which makes their imaging at the light level simple and informative. We detail how to image the fat droplet core by metabolic labeling with fluorescent fatty acids or lipophilic fluorochromes. Further, we describe the use of indirect immunostaining to image fat droplet proteins and fat cores in the same field. We also address the use of appropriate controls for determining signal specificity and other practical considerations for optimizing image quality.
I. Introduction and Rationale
The fat droplet is the main energy depot in many cells, and is composed of neutral lipid surrounded by a protein and phospholipid monolayer. Fat droplets have been observed in liver (Mottram, 1909) and in muscle fibers (Bullard, 1912) for over a century. Further, imaging of fat droplets has revealed their close association with mitochondria (Craig et al., 1963) the endoplasmic reticulum (Ashworth et al., 1959), and peroxisomes (Blanchette-Mackie et al., 1995).
Before the discovery of perilipin 1 as a fat droplet specific protein 20 years ago (Greenberg et al., 1991), the existence of specific fat coat proteins was not appreciated. This discovery sparked investigations that revealed that fat droplets in mammalian cells have a dynamic protein coat that controls access to the fat stores. This coat includes structural proteins, enzymes, and transport proteins (Brasaemle et al., 2004; Liu et al., 2004). Due to the large size and distinct morphology of fat droplets, fluorescence microscopy has played a key role in these studies. With increased availability of antibodies, fluorescence stains and fluorescing biologic molecules, fluorescence microscopy will continue to be an important tool.
Here we discuss strategies using light microscopy to image fat droplets and the proteins that package fat. In addition to staining protocols, we discuss considerations for choosing cell lines and optimal physical formats in which to grow and stain cells. We detail strategies to eliminate ambiguous signals and accomplish the simultaneous imaging of multiple molecules. Finally, we discuss image collection strategies, image presentation and the importance of microscope choice for the imaging outcome.
II. Materials
Twenty-two millimeter square glass coverslips (no 1.5 thickness preferred), glass slides, six-well tissue culture plates, finely pointed biological forceps (tweezers) (See Fig 2).
Phosphate-buffered saline (PBS) at pH 7.2 to 7.4.
Cell fixation solution: add 13.5 ml methanol stabilized 37 % formaldehyde to 236.5 ml PBS to make a 2% solution.
Microscopy buffer for cell permeabilization and antibody dilution: To about 400 ml PBS, add 5 g BSA, 0.5 g saponin, and 0.5 ml of a 20 % w/v solution of Sodium azide, NaN3. Mix until dissolved and bring to 500 ml with PBS. Final solution is 1 % BSA, 0.1 % saponin, and 0.02 % Sodium azide. Pass solution through 0.2 μm filter. Microscopy buffer is stable for two months at room temperature. If solution becomes cloudy from precipitation, replace.
Antibodies against proteins of interest (primary antibodies) and antibody-fluorochrome conjugates recognizing primary antibody species (secondary antibodies). Aliquot antibodies and store aliquots at −20 or −80 °C and when thawed do not refreeze.
DAPI (4′, 6-diamidino-2-phenylindole) stock solution is 10 mg/ml dissolved in water (store at 4°C) and BODIPY 493/503 stock is 1 mg/ml in DMSO (store at −20 °C).
Mounting media for adhering the cell bearing coverslip to the glass slide. We routinely make and use a polyvinyl alcohol based solution commonly called Elvanol (Wessel et al., 2004). Similar solutions can be purchased and some include DAPI.
An epifluorescence equipped microscope with a camera system and optic filters.
Figure 2. Staining cells on coverslips.

A) After cells are fixed and washed, the coverslips are removed from the plate in which the cells were grown. Keeping the cell-bearing side up, the coverslip is placed on a parafilm lined tray. B) Antibody is applied to the cell bearing side of the coverslip. C) Before mounting the coverslip, the non cell-bearing side is dried. D) The coverslip is carefully lowered onto a drop of mounting medium, with the cell-bearing side facing the slide.
III Methods
General considerations
We describe a simple staining protocol, because time and manipulation degrade cellular morphology. We recommend formalin fixation, because solvent fixation degrades fat droplet morphology (DiDonato and Brasaemle, 2003). Since coverslips are easily transferred using forceps, it is most convenient and economical to grow cells on coverslips. We find that 22 mm square coverslips in 6 well plates are useful for most applications; this allows independent treatment for the cells in each well. Using both glass coverslips and slides yields the best images. To maximize the information gathered, we routinely co-stain with two antibodies made in different species and DAPI (4′, 6-diamidino-2-phenylindole), or a single antibody together with the lipophilic fluorochrome BODIPY 493/503, and DAPI. Minimize light exposure because fluorochromes are subject to photobleaching. Finally, do not allow the cell bearing-side of the coverslip to dry out.
A. Cell lines
The cell-line chosen is often the difference between obtaining a clear, easily imaged answer or an ambiguous result. For many imaging applications, cells having broad flat processes lessen ambiguities caused by overlapping structures. For example, the perilipin 3 puncta falling along the endoplasmic reticulum (ER) in Figure 1 (red arrowheads) are evident, and it is likely that this ER-perilipin 3 association continues into the thick perinuclear region. However, overlapping signal obscures this relationship. Cells with large flat processes include primary fibroblasts, 3T3 cells, COS-7, and undifferentiated 3T3-L1 or OP9. Round cells include HEK293 cells and well differentiated 3T3-L1 adipocytes. In addition to morphology, antibody availability and transfectability are considerations in choosing cell lines.
Figure 1. Perilipin 3 falls along the ER at the cell periphery.

OP9 cells were incubated for 1 h with 1.8 mM of the fatty acid oleate bound to albumin at a ratio of 5:1. Cells were fixed and stained for perilipin 3 at 1.6 μg/ml, and the ER was visualized by immunostaining for transfected FLAG-DGAT1 as described previously (Skinner et al., 2009). Bar = 10 micrometers in all figures.
Cell Growth and Density
Cell density is also important. Cells with fibroblastic morphology are typically plated at between 30% to 60% confluency (surface area covered), since they spread out and form large flat processes that abut neighboring cells (Figs. 1 and 3). As cells proliferate, the cell layer thickens and overlapping structures make images difficult to interpret. At very low density (less than 10 % confluent) it is difficult to inspect large numbers of cells and to capture images containing multiple cells. In addition, we have observed that maintaining most fibroblastic cell-types at between 20% and 90% confluency results in the growth of broad, flat, easily imaged processes. Maintaining cells at very low density results in spindly processes and maintaining cells at very high density results in cells that remain thick after replating to a lower density.
Figure 3. BODIPY 493/503 stains fat droplets in living cells.
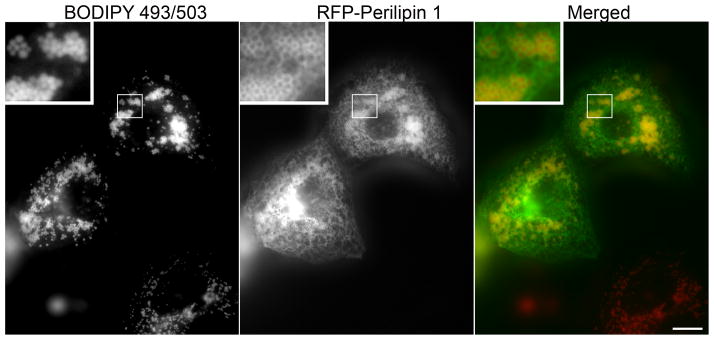
COS7 cells were transfected to express a red fluorescent protein/perilipin 1 fusion as described previously (Skinner et al., 2013) 18 hours later, 2 ug/ml of BODIPY 493/503 and 1.8 mM fatty acid were added to the media. After a 4 h incubation, the coverslip was rinsed in PBS and imaged. For clearer presentation, in the merged image the green fluorescing BODIPY 493/503 is shown as red, and the red fluorescent protein/perilipin 1 fusion is shown as green.
B. Microscopy
The figures shown were acquired using common wide-field immunofluorescence microscopy, rather than confocal microscopy. We find that, when imaging flat cells, a standard research grade microscope with appropriate filter sets and a sensitive digital camera is sufficient. Further, wide field microscopes are less expensive and generally less difficult to operate than confocal microscopes.
The independent imaging of multiple molecules in a single sample requires optic wavelength filters. Both the light exciting fluorochromes and the emitted light are filtered and detected by a monochromatic camera. The use of appropriate fluorochromes and filters enables the serial imaging of different molecules in the same field of view. The green and red light emitting fluorochromes are the most convenient and most commonly used. The human eye is sensitive to these wavelengths and there are many antibody-fluorochrome conjugates, dyes and proteins engineered to fluoresce at these wavelengths. We routinely use these channels to detect antibody-fluorochrome conjugates and for most unconjugated dyes and use the blue channel to image nuclei using blue-fluorescing DNA-binding dye the DAPI (4′, 6-diamidino-2-phenylindole).
Optimizing antibody concentration
Antibody binding increases with concentration. However, specific binding saturates faster than nonspecific binding. Thus, it is best to determine the optimal antibody concentrations by titration from 0.1 to 5 μg/ml for monoclonal or affinity purified antibodies and from 1:100 to 10,000 for serum. For antibodies conjugated to bright and photostable fluorochromes (usually secondary antibodies), such as the red and green ALEXA Fluor®, 2 μg/ml antibody is usually sufficient, while for blue and far red channels 5 μg/ml maybe required. Finally, when imaging multiple signals, similar signal intensity minimizes cross reactivity and bleed-through problems.
C. Labeling droplets with lipophilic fluorochromes
Lipophilic fluorochromes partition into the neutral lipid cores of fat droplets (Figs. 3 and 4). In addition to being conceptually simple, their use is straightforward. These lipophilic fluorochromes stain both living and fixed cells. We routinely add BODIPY 493/503 from a 1 mg/ml stock to fixed or living cells at a final concentration of 1 μg–10 μg/ml; followed by at least a 1-h incubation at 37°C. The final concentration of BODIPY 493/503 depends on fat droplet size, the larger the fat droplets, the less stain is needed. For fixed cells it is convenient to add BODIPY with the primary or secondary antibodies. BODIPY 493/503 is widely utilized, because it is inexpensive, has a narrow excitation/emission spectrum that is compatible with most green channels, is very bright, and more photostable than many other lipophilic fluorochromes. However, if channel availability precludes using BODIPY 493/503, other lipophilic fluorochromes with different excitation/emission spectra are available (Liu et al., 2009). Finally, like DAPI, these dyes are potent and care must be taken to avoid the unintentional staining of other specimen-containing coverslips.
Figure 4. Fat droplets and associated mitochondria imaged in a live muscle fiber.
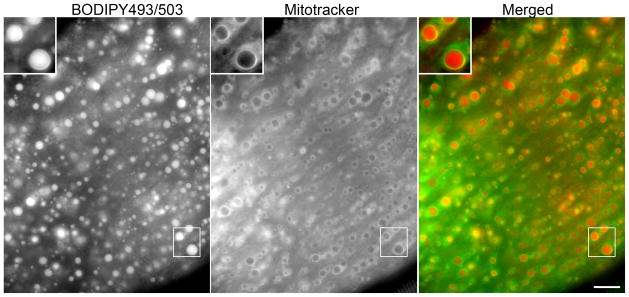
Muscle fibers from mice overexpressing perilipin 5 were collagenase dispersed for 1 h in 10 mg/ml collagenase in complete cell growth media supplemented with 25 mM HEPES, then exposed to BODIPY 493/503 and mitotracker for 3 h after which dyes were washed out. Labeled fibers were spread on a glass slide and imaged using standard wide field fluorescence microscopy. For clearer presentation, the green fluorescing BODIPY 493/503 is show as red.
D. Fluorescent fatty acids
Cells incorporate fatty acids into TAG (triacylglycerol) therefore TAG-containing droplets can be labeled with incorporated fluorescent fatty acids. Adding 5–50 μg/ml of labeled fatty acids from a 20 mg/ml stock dissolved in DMSO to culture media and incubating for one or more hours usually results in bright fat droplet staining. When using serum-containing media, thorough mixing sufficiently solubilizes low concentrations of fatty acids. BODIPY® 12 carbon fatty acids are useful because: they are incorporated into TAG–containing droplets, are available with emission spectra in multiple wavelengths, and are bright and reasonably photostable. In our experience, these molecules can give a higher signal to noise ratio than lipophilic fluorochromes. Further, lipophilic fluorochrome labeling intensity (see above) increases dramatically with fat droplet size, while BODIPY® fatty acid labeling is less size dependent, which can be advantageous when imaging adipocytes or other cells with both large and small fat droplets. Care must be taken in the interpretation of images produced when using these fluorescent fatty acids, since fatty acids have multiple fates; we have observed the labeling of both the ER and mitochondria with various dye-conjugated fatty acids. In addition, we have observed changes in emission spectra when these fatty acids are used at high concentrations. Finally, BODIPY fatty acids are not naturally occurring and hence, the fluorophore may affect both the rate of processing and metabolic fate of the labeled fatty acid. Thus, their utility for kinetic and partitioning studies is limited. In addition, fluorescing cholesteryl BODIPY has been used to distinguish cholesteryl ester droplets from TAG droplets (Hsieh et al., 2012).
E. Testing for optical bleed through and cross reactivity
Optic filter bleed-through can occur when using simple stains such as BODIPY and DAPI.. Using the BODIPY 493/503 stain as an example, bleed-through can be tested for as follows: 1) obtain the maximal signal by staining the cells with the highest concentration of BODIPY 493/503 that will be used then 2) confirm the presence of a strong signal in the green channel and the absence of appreciable signal in the red and blue channels.
Signal not dependant on the primary antibody for a given channel can result light bleeding though from another channel, or from antibody cross-reactivity. Cross-reactivity can include a fluorochrome-labeled secondary antibody binding: an unintended primary antibody, another fluorochrome labeled secondary antibody, or directly to fixed cells. To test for such unintended signals (as controls for Fig. 1), we stained two coverslips, omitting one of the primary antibodies in each case—the anti-FLAG from the first coverslip and the anti-perilipin 3 antibody from the second coverslip. Both coverslips were exposed to both secondary antibodies. The first coverslip showed no appreciable signal in the green channel with imageable signal in the red channel, and the second coverslip showed no appreciable signal in the red channel with imageable signal in the green channel. This demonstrated that the channels are independently imageable. However, these controls do not address the specificity of the primary antibodies.
F. Antibody staining protocol
Grow and treat cells on coverslips in 6-well plates.
Aspirate media. Do not wash cells.
Add 2 ml of fix solution to each well (cells will be facing up)*.
Incubate 10–20 min at room temperature then rinse coverslips 4 times with 2 ml PBS.
Replace PBS with 2 ml microscopy buffer. (After fixation cells can be stored at 4°C in microscopy buffer for up to four weeks).
Place coverslips cell-bearing side up** on a parafilm lined tray with a cover (Fig 2A). To avoid drying place a small water saturated paper towel inside the tray.
Add primary antibodies diluted in 150 μl microscopy buffer to the cell-bearing side of coverslips (Fig. 2B) and cover with lid.
After 1 h, rinse coverslips 4 times with PBS and apply fluorochrome-antibody conjugates (secondary antibodies) in microscopy buffer and cover with lid.
After 1 h incubation in the dark, rinse coverslips 5 times with PBS.
Pipette a bead of elvanol (or similar mounting solution) about 2 mm from the top edge of a microscope slide.
Dip coverslip in distilled water to wash off PBS.
Wipe water off the non-cell-bearing side of the coverslip (Fig. 2C).
Mount coverslip on slide so that cells are sandwiched between the two pieces of glass. Place the edge of the coverslip between the top edge of the slide and the bead of elvanol. Then lower slowly until the coverslip is flat (Fig. 2D).
Do not get elvanol on non cell-side of the coverslip.
Aspirate excess elvanol from edge of coverslip.
Position coverslips on slide so that they are not touching. Dyes such as DAPI and BODIPY diffuse in the mounting media and will stain abutting coverslips.
Let dry for at least 1 h.
Add DAPI along with the primary antibodies, the secondary fluorochrome-antibody conjugates or in the mounting media at 0.5 μg/ml.
-
*
In some cases formaldehyde fixation abolishes antibody binding. In this case, fix cells by adding 2 ml 100% methanol at −20 °C, and incubate at that temperature for 4 min. Wash 5 times in PBS, then proceed as per protocol (step 5). Fat droplet morphology will be dramatically compromised (DiDonato and Brasaemle, 2003), but may still be informative.
-
**
Cells on 22 mm square coverslips can be stained with less than 50 μl of antibody by placing the coverslip cell-bearing side down on a drop of antibody. The volume of antibody used can be further reduced by using smaller coverslips if necessary.
Fat droplets in dispersed muscle fibers
We have adapted this protocol for collagenase dispersed skeletal- and cardiomyocytes. We fix and stain these myocytes in suspension. Excess dye and antibodies are washed away by low speed centrifugation of cells (200 × g for 5 min), removal of supernatant and resuspension of myocytes in PBS, 5 times after fixation, and 5 times between addition of primary and fluorochrome-conjugated antibodies. After the final wash we resuspend the myocytes in elvanol, pipette the elvanol-myocyte suspension onto a slide and cover the suspension with a coverslip. If appropriate, vital staining of live fibers can be imaged without fixation (Fig 4).
IV Discussion
Validation of signals
The microscopy techniques for imaging fat droplets and associated cellular structures described herein, employ antibody-based labeling of fat droplet protein coats and the direct fluorophore staining of the fat droplet core. Immunofluorescence based protein detection generally utilizes a two-antibody detection system. Cells are incubated with an antibody against the protein of interest (the primary antibody). Then species specific antibody-fluorochrome conjugates are used to recognize the primary antibody. Both primary and secondary antibodies can contribute signal unrelated to the antigen--that is, background. For example, if there is no protein of interest present, all of the signal is background.
Unlike immunoblotting, where the predicted antigen migration in electrophoresis provides an indication whether a signal is specific or spurious. Immunostaining does not provide this information. Thus, immunofluorescence studies require rigorous controls to show signals are specificity to the protein of interest. Demonstrating an increased signal in cells with overexpressed protein of interest (e.g., by transfection) can provide convincing evidence that the signal detected is from the protein of interest. However, it does not guarantee that the endogenous protein will be discernable from background. Using a variety of antibodies raised against the protein of interest is another strategy to authenticate signal. Ideally, antibodies raised in different species can be used to detect different epitopes of the same protein. If these controls are not possible, imaging data can be supported by immunoblotting of subcellular fractions. Specifically, if imaging and cell fractionation show a specific signal around fat droplets and in fat droplet fractions, respectively, this is good evidence that both signals are indeed against the putative antigen (Wolins et al., 2001). Further, if conditions that perturb the subcellular localization of a protein reveal the same changes by both immunofluorescence microscopy and subcellular fractionation procedures, then this provides strong evidence that antibody labeling is specific. In this way other techniques are a valuable complement to cell imaging.
Placing fat droplets in intracellular context
In addition to authentic, well-defined signals, useful images of fat droplets require subcellular context. For example, perilipin 3 staining in fat storing OP9 cells reveals only puncta (Fig. 1) and round hollow appearing structures (Fig. 3). This staining pattern by itself is uninformative, because the cell boundaries are indiscernible, as is the intracellular distribution. However, imaging the ER reveals that the nuclear envelope and the ER extend to the cell periphery, and delineates the cell boundaries. In this context, we see that the perilipin 3 puncta at the periphery fall along the smooth ER (Fig. 1). We also see that the majority of the perilipin 3 puncta are in the perinuclear region. Several factors contribute to interpreting these observations. First, the signal from this anti perilipin 3 antibody is unambiguous, because its authenticity has been demonstrated in several different contexts and systems (Skinner et al., 2009; Wolins et al., 2005; Wolins et al., 2006). Second, the identity of the ER marker signal is also unambiguous. We showed that the signal for the ER marker requires both transfection with an expression plasmid and the primary antibody. Also, this transfection protocol results in varying levels of antigen in different cells, and this is reflected in ER marker signal levels. Finally, DAPI staining makes the expression difference evident by identifying individual cells. The blue arrow points to the DAPI labeled nucleus with no surrounding signal in the ER marker channel, thus showing that the ER signal is transfection dependent. Note that the ER marker is a fat synthesis enzyme (DGAT1) which increases TAG storage in lipid droplets and the untransfected cell is almost devoid of perinuclear perilipin 3 coated fat droplets. Also notable, the extra-nuclear DAPI staining represents small particles of condensed DNA from the transfection. Revealing the cytoskeleton by staining cellular filaments brings context to the signal of interest. Staining actin filaments with phalloidin-fluorochrome conjugates defines the cell boundary, is simple and is not antibody based.
Imaging is often used to localize proteins to compartments by comparing the signal of the protein of interest to specific organellar markers. Digital imaging allows the precise overlap of images by showing each image as a different color. The merged image in Figure 1 shows perilipin 3 puncta along the DGAT1 labeled ER as previously reported (Skinner et al., 2009). Signal overlap is shown as a third color. For example, red and green in an RBG image is shown in yellow (Figure 1). In Figure 1, the separate images for perilipin 3 and DGAT1 are patently dissimilar. Nonetheless, copious yellow is seen in the merged view. This is due to the ER and fat droplets overlapping in the thick perinuclear region of the cell, but not due to co-localization of the signals. This example illustrates that caution must be used when interpreting overlapping signals.
The goal of imaging is to present data as pictures. When visually scanning and positioning cells for photography, it is important to observe the more stable fluorochromes, usually the antibody-fluorochrome conjugates. This avoids photobleaching of less stable fluorochromes. Once the field is selected, images of the more easily bleached fluorochrome should be collected first—in this case the BODIPY lipophilic dye. Once a field is chosen, it is usually best to collect light for a duration that saturates a few pixels in the brightest area. Larger regions of saturation may be necessary; for example, in Figure 1 (ER-marker panel), clear depiction of the “lacy” ER at the cell periphery required exposure times that resulted in a large area of saturation in the perinuclear region. To identify representative data, image multiple fields. When assembling a figure comparing multiple conditions, chose fields where the cells are similar in size and shape.
Humans perceive green structures as sharper and brighter than red structures; for this reason we suggest presenting the individual channels as grayscale images. Merged color images are more easily interrupted when the most sharply defined structure is depicted in green. For example, the perilipin 1 signal in Figure 3 sharply outlines the more nebulous BODIPY 493/503-labeled neutral fat. This image is more understandable when the perilipin 1 signal is shown as green and the BODIPY 493/503 signal as red. Note that this presentation reverses the wavelengths at which the separate channel images were collected, and hence requires explanation in the text of reports or manuscripts. Finally, to assure images are published at a size large enough to show the relevant features, use a font size that is only legible when the relevant features are apparent. It is also helpful to submit images in a size that is compatible with journal specifications to avoid arbitrary resizing by the typesetter.
Summary
Proteins forming a coat around intracellular fat droplets have been identified, and antibodies against fat droplet proteins can be conveniently imaged using standard epifluorescence microscopy. In addition, the lipid core of fat droplets in living and fixed cells can be directly labeled by lipophilic incorporation of BODIPY dyes, or in living cells by the metabolic incorporation of BODIPY conjugated fatty acids. In this way both lipid cores and the surrounding protein coats of fat droplets can be imaged simultaneously. Placing fat droplets in subcellular context, especially with respect to the endoplasmic reticulum and mitochondria, is also possible with these methods. Cross linking fixation is superior because methanol fixation disrupts fat droplet morphology. Fat droplets are most conveniently imaged in cells grown directly on glass coverslips in individual wells of tissue culture plates. This format makes multiple experimental conditions feasible and analysis by imaging practical.
Acknowledgments
This work is supported in whole or in part by the National Institutes of Health Grant R01 DK088206–01A1 (N.E.W). Postgraduate support (to J.R.S.) was from the Training Program in Cardiovascular Biology NHLBI T32HL007275-33. We also thank the Washington University Nutrition Obesity Research Center supported by grant number P30DK056341 NORC.
References
- Ashworth CT, Race GJ, Mollen Hauer HH. Study of functional activit of adrenocortical cells with electron microscopy. Am J Pathol. 1959;35:425–37. [PMC free article] [PubMed] [Google Scholar]
- Blanchette-Mackie EJ, Dwyer NK, Barber T, Coxey RA, Takeda T, Rondinone CM, Theodorakis JL, Greenberg AS, Londos C. Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J Lipid Res. 1995;36:1211–26. [PubMed] [Google Scholar]
- Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279:46835–42. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- Bullard HH. On the interstitial granules and fat droplets of striated muscle. American Journal of Anatomy. 1912;14:1–46. [Google Scholar]
- Craig EL, Eglitis JA, McConnell DG. Observations on the Oil of Droplets of the Principal Cone Cells of the Frog Retina. Exp Eye Res. 1963;2:268–71. doi: 10.1016/s0014-4835(63)80046-9. [DOI] [PubMed] [Google Scholar]
- DiDonato D, Brasaemle DL. Fixation methods for the study of lipid droplets by immunofluorescence microscopy. J Histochem Cytochem. 2003;51:773–80. doi: 10.1177/002215540305100608. [DOI] [PubMed] [Google Scholar]
- Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266:11341–6. [PubMed] [Google Scholar]
- Hsieh K, Lee YK, Londos C, Raaka BM, Dalen KT, Kimmel AR. Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. J Cell Sci 2012. 2012 Sep 1;125(Pt 17):4067–76. doi: 10.1242/jcs.104943. Epub 2012 Jun 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Zhou S, Kim JY, Tillison K, Majors D, Rearick D, Lee JH, Fernandez-Boyanapalli RF, Barricklow K, Houston MS, Smas CM. Functional analysis of FSP27 protein regions for lipid droplet localization, caspase-dependent apoptosis, and dimerization with CIDEA. Am J Physiol Endocrinol Metab. 2009;297:E1395–413. doi: 10.1152/ajpendo.00188.2009. [DOI] [PubMed] [Google Scholar]
- Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem. 2004;279:3787–92. doi: 10.1074/jbc.M311945200. [DOI] [PubMed] [Google Scholar]
- Mottram VH. Fatty infiltration of the liver in hunger. J Physiol. 1909;38:281–352. 3. doi: 10.1113/jphysiol.1909.sp001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner JR, Harris LALS, Shew TM, Abumrad NA, Wolins NE. Perilipin 1 moves between the fat droplet and the endoplasmic reticulum. Adipocyte. 2013;2:6–13. doi: 10.4161/adip.22864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner JR, Shew TM, Schwartz DM, Tzekov A, Lepus CM, Abumrad NA, Wolins NE. Diacylglycerol enrichment of endoplasmic reticulum or lipid droplets recruits perilipin 3/TIP47 during lipid storage and mobilization. J Biol Chem. 2009;284:30941–8. doi: 10.1074/jbc.M109.013995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel GM, Voronina E, Brooks JM. Obtaining and handling echinoderm oocytes. Methods Cell Biol. 2004;74:87–114. doi: 10.1016/s0091-679x(04)74005-4. [DOI] [PubMed] [Google Scholar]
- Wolins NE, Quaynor BK, Skinner JR, Schoenfish MJ, Tzekov A, Bickel PE. S3–12, Adipophilin, and TIP47 package lipid in adipocytes. J Biol Chem. 2005;280:19146–55. doi: 10.1074/jbc.M500978200. [DOI] [PubMed] [Google Scholar]
- Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N, Kern PA, Finck BN, Bickel PE. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes. 2006;55:3418–28. doi: 10.2337/db06-0399. [DOI] [PubMed] [Google Scholar]
- Wolins NE, Rubin B, Brasaemle DL. TIP47 associates with lipid droplets. J Biol Chem. 2001;276:5101–8. doi: 10.1074/jbc.M006775200. [DOI] [PubMed] [Google Scholar]