

Abstract
The cytochromes P450 (CYPs) play a central role in many biologically important oxidation reactions, including the metabolism of drugs and other xenobiotic compounds. Because they are often assayed as both drug targets and anti-targets, any tools that provide: (a) confirmation of active site binding and (b) structural data, would be of great utility, especially if data could be obtained in reasonably high throughput. To this end, we have developed an analog of the promiscuous heme ligand, cyanide, with a 13CH3-reporter attached. This 13C-methyl isocyanide ligand binds to bacterial (P450cam) and membrane-bound mammalian (CYP2B4) CYPs. It can be used in a rapid 1D experiment to identify binders, and provides a qualitative measure of structural changes in the active site.
Keywords: NMR, Heme, P450cam, CYP2B4, Screening
Introduction
The cytochromes P450 (CYPs) are a family of heme-containing monooxygenase enzymes that use molecular oxygen to hydroxylate specific substrates, often with high stereoselectivity (Poulos and Johnson 2005). While CYPs are present in both eukaryotes and prokaryotes, the prokaryotic enzymes are soluble and therefore easier to study. A prototypical and well-studied bacterial CYP is P450cam (CYP101), from Pseudomonas putida (Katagiri et al. 1968; Poulos et al. 1986). P450cam is a 46 kDa soluble enzyme that catalyzes the 5-exo-hydroxylation of camphor (Gelb et al. 1982). A prototypical and well-studied mammalian CYP is CYP2B4. CYP2B4 is a 55 kDa membrane-bound protein from rabbit liver that either hydroxylates or demethylates a variety of foreign compounds, such as the drug benzphetamine (Kanaeva et al. 1992). The mammalian CYPs, being the enzymes predominantly responsible for drug and xenobiotic metabolism in humans, are of particular interest in the pharmaceutical industry. They are relatively large, membrane-bound proteins with a typical molecular weight of ~65 kDa (Furge and Guengerich 2006). Consequently, it is difficult to obtain structural information on the mammalian CYPs, either via X-ray or current multidimensional NMR techniques. Although, there have been some recent successes in crystallizing mammalian CYPs that lack the membrane-binding N-terminal region, and have other solubilizing mutations (Williams et al. 2000; Williams et al. 2003; Williams et al. 2004; Scott et al. 2003). But, the mammalian CYPs are too large to structurally characterize with NMR, and membrane-bound CYPs have yet to be crystallized. Hence, there is a need to develop new techniques that rapidly probe at least part of the active site structure, to better define binding interactions with substrates or inhibitors.
With the above goal in mind, we have decided to capitalize on the fact that cyanide is a promiscuous ligand for heme iron (binding through its carbon), and that alkyl isocyanides (alkyl-NC) are known to be ligands for a number of CYPs (Tomita et al. 2001; Lee et al. 2001). Their absorption spectra are typical of Type II binding spectra, where the Soret band is shifted upon binding of the alkyl isocyanide, with absorption maxima occurring at either or both 430 and 455 nm, depending upon the extent of back-bonding from iron into the isonitrile triple bond. Indeed, resonance Raman experiments (Tomita et al. 2001; Lee et al. 2001) have established that the heme-bound isocyanide binds in two distinct orientations, linear or bent depending on C–Fe bond order; these forms are in slow exchange on the resonance Raman timescale, but would normally be in fast exchange on the NMR timescale. These two orientations are illustrated in Fig. 1, for the methyl isocyanide ligand that is the topic of this paper. The linear and bent forms are related to the two resonance forms of methyl isocyanide (Scheme 1), although the imine form is highly disfavored unless there is electron donation to stabilize it via formation of a dative bond (as in Fig. 1; in this case, electron donation is via backbonding from the iron).
Fig. 1.

Methyl isocyanide bound to the heme iron. Schematic representation of methyl isocyanide bound to heme with (a) an iron-carbon bond order of two (“bent”) and (b) an iron-carbon bond order of one (“linear”). The heme pyrrole rings, along with the CN triple (or double) bond, produce a highly anisotropic environment that can produce large chemical shift perturbations for the labeled methyl group
Scheme 1.
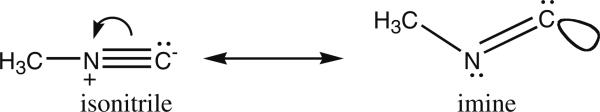
Resonance structures for methyl isocyanide
Alkyl isocyanides bind particularly strongly when the iron of the heme is in its reduced form (Kd ~ 10–5–10–6 M), although it can be shown that with increasing alkyl chain length, even the ferric form can exhibit dissociation constants in the micromolar range (Simonneaux and Bondon 2000).
Given the established value of 13CH3-methyl probes for NMR structural and dynamical studies of large proteins (Pellecchia et al. 2002; Showalter et al. 2007; Tugarinov et al. 2006), our interest in alkyl isocyanides is limited to methyl isocyanide (13CH3NC). As the smallest member of the alkyl isocyanides, it is the one least likely to interfere with substrate or inhibitor binding (being only slightly larger than the native ligand, dioxygen). Also, the lack of adjacent methylene protons avoids dipolar relaxation effects, which would lead to line broadening. The 13C-label allows for selective observation of the attached methyl protons, which provides a chemical shift perturbation probe of structural (and potentially dynamical) changes in the binding pocket, due to binding of substrate or inhibitor. Moreover, because of the short internal correlation time of the rapidly rotating methyl group, even when bound to the heme, the proton linewidth should remain narrow—like those of a small molecule. Taken together, these features potentially allow structural and dynamic information to be extracted from NMR experiments such as: 2D 1H-13C HSQC, 13C-filtered 1H 1D, 13C-edited (or filtered) NOESY, methyl dynamics (ex. using 13CHD2, 13CH2D (Showalter et al. 2007; Cavanagh et al. 2007), and various other experiments (Cavanagh et al. 2007). Given the large size of CYP enzymes, especially when complexed to micelles, most of these NMR experiments would not normally be feasible. Herein is presented the synthesis, initial characterization and application of the 13C-methyl isocyanide probe, to enable such studies. This probe will permit the first such targeted NMR studies of CYP binding sites, and provide evidence for active site changes near the heme, due to the presence or absence of substrates, inhibitors or other cellular components, especially lipids or electron donating protein partners.
Materials and methods
CYP2B4, myoglobin and biochemical reagents
Myoglobin (horse heart, >90% pure), benzphetamine, DEAE resin, and other reagents were from Sigma-Aldrich, unless specified otherwise. CYP2B4 was expressed in C41 E. coli cells using the pLW01-P450 2B4 mem plasmid, and purified as described previously (Miroux and Walker 1996; Bridges et al. 1998). 13C-methyl iodide and d8-glycerol were from Isotec, and d38-DPC (dodecylphosphocholine-d38) was from Cambridge Isotope Laboratories.
Expression and purification of P450cam
P450cam was expressed in E. coli (BL21DE3*) using a pCWori expression construct (a kind gift from Dr. Paul Ortiz de Montellano) and purified largely as described previously (Sibbesen et al. 1996; Yao et al. 2007), with some changes to the protocol. Briefly, 6 L of cell culture were grown at 37°C in 1 L batches to an OD600 of 0.7–0.9, before adding 1 mM IPTG (Isopropyl-β-D-Thiogalactopyranoside) for induction and 30 μM δ-ALA (δ-aminolevulinic acid) as a heme precursor, and allowing protein expression to continue for 20–22 h. After the cells were harvested they were frozen at –80°C for several hours, before resuspending in lysis buffer (50 mM Tris*HCl, pH 7.5 (room temp.), 25 mM KCl, 1–2 mM camphor, 1 mM EDTA, 1 mM PMSF, 1 lM leupeptin, 1 μM pepstatin, 32 U/ml DNase, 3 U/ml RNase, 1 mg/ml lysozyme) and sonicating. The cell debris was spun down at 50,000g and the supernatant was collected to be loaded onto a 50-ml bed volume DE-52 anion exchange column that had been pre-equilibrated with running buffer (50 mM Tris*HCl, pH 7.5 (4°C), 25 mM KCl, 2 mM camphor). After washing with two column volumes of running buffer, protein was eluted using a linear gradient from 0 to 100% of elution buffer (50 mM Tris*HCl, pH 7.5 (4°C), 300 mM KCl, 2 mM camphor). All fractions with an RZ ratio (A392/A280) > 0.5 were pooled and concentrated.
Ammonium sulfate was added to the pooled fractions to 25% saturation (~143 mg/ml), and they were then loaded onto a phenyl sepharose column (50 ml bed volume), after it had been pre-equilibrated with running buffer (50 mM Tris*Cl, pH 7.5, 50 mM KCl, 25% NH4SO4, 100 μM camphor). After washing, elution was achieved with a linear gradient of 0 to 100% of elution buffer (50 mM Tris*HCl (pH 7.5), 50 mM KCl, 1 mM camphor). All fractions having an Rz ratio ≥ 1.4 were pooled, and purity was confirmed with an SDS gel (Supplementary Fig. S1). Protein (500 μM) was dialyzed into 30 mM phosphate buffer (pH 7.4), 100 mM KCl, 2 mM camphor, and flash frozen in 12 1-ml aliquots and stored at –80°C. Typical yield was ~ 100 mg/6 L cell culture.
Synthesis of 13C-methyl isocyanide (13CH3NC)
13CH3NC was synthesized with modifications to a previously reported procedure (Freedman and Nixon 1972). 1 mmol of 13C-methyl iodide and 2 mmol of silver cyanide were placed in a thick walled glass tube and sealed. The mixture was heated for 1 h at 95°C in an oil bath and the tube was then brought to room temperature slowly. The sealed tube was opened and the brown solid was quenched for 12 h in 5 ml of an 8 M KCN solution. The resultant mixture was distilled under vacuum to obtain crude 13C-methyl isocyanide, which was purified by bulb-to-bulb distillation. Yield was 21%, and the 1H NMR spectrum (Fig. 2) confirmed the structure. The pair of 1:1:1 triplets resulting from 13C-1H scalar coupling (1J coupling, producing the large doublet) and 14N-1H scalar coupling (2J coupling, producing the triplet) was only apparent in d4-methanol and D2O; in other solvents that were used, including d3-acetonitrile, d1-chloroform and d6-benzene, the spectrum only showed the large doublet splitting from the 13C-1H coupling. The proton chemical shift also varied depending upon the solvent used, and generally shifted upfield with increasing nonpolarity of the solvent (for example, the methyl protons had a chemical shift of 1.1 ppm in d6-benzene).
Fig. 2.

NMR spectrum of 13C-methyl isocyanide in d4-methanol. The 13C-coupled 1H resonances from 13CH3NC flank the solvent resonance. The inset shows the further splitting (1:1:1 triplet) of the two 13C-split proton resonances. This triplet splitting is due to 2J coupling between the quadrupolar 14N nucleus (I = 1) and the methyl protons. Spectrum acquired at 300 MHz
NMR spectral analysis of 13CH3NC bound to heme proteins
All NMR spectra were collected at 25°C on a 600 MHz Varian spectrometer equipped with a triple-resonance cryoprobe. 1H-13C HSQC spectra were collected with a spectral width of 12,000 and 28,000 Hz and digitized with 1920 and 256 complex points for the 1H and 13C dimensions, respectively. Spectra were referenced directly to H2O (δ = 4.76 ppm at 25°C) in the 1H dimension and indirectly in the 13C dimension. For the samples with P450cam as the heme protein, the protein concentration was 150 lM, as confirmed spectrophotometrically, using an extinction coefficient of 105 μM–1 cm–1 for the Fe3+-substrate free protein with λmax = 418 nm (O'Keeffe et al. 1978). Buffer conditions were 30 mM potassium phosphate (pH 7.4), 100 mM KCl, with saturating camphor (for samples that contained substrate) at 2 mM. When CYP2B4 was used as the heme protein, the concentration was 75 μM, determined using the extinction coefficient for the CYP2B4-CO complex of Δε450–490 = 91 mM–1 cm–1 (Omura and Sato 1964). Buffer was 100 mM potassium phosphate (pH 7.4) with either 20% (v/v) d8-glycerol or 50 mM d38-DPC, with maximum benzphetamine concentrations (for samples that contained substrate) of 1.6 mM, close to its solubility limit at pH 7.4. All samples, after having argon gas bubbled through the solution for 15 min to purge any oxygen, were reduced with dithionite, followed by addition of an equimolar amount of 13CH3NC to form the ferrous heme protein-13CH3NC complex.
Results and discussion
Synthesis and characterization of 13CH3NC: coupling to 14N and effects of solvent polarity
The 13CH3NC probe was prepared from 13C-methyl iodide and silver cyanide. Surprisingly, the proton NMR spectrum reveals that in some solvents (esp. polar solvents), a 2-bond coupling to the cyanide 14N atom is present. Such coupling to nitrogen is not typically observed due to quadrupolar relaxation from 14N (which is I = 1). But the axial symmetry of the molecule, along with a very short correlation time, appears to be decreasing the extent of quadrupolar relaxation. This is because the quadrupolar relaxation rate is proportional to the correlation time, and also has a squared dependence on the asymmetry parameter η = (EXX – EYY)/EZZ (Nelson 2003), which should be zero (EXX – EYY = 0) for an axially symmetric molecule like CH3NC. Perhaps in nonpolar solvents, where this coupling is no longer observed, there is some sort of intermolecular interaction between molecules that breaks this symmetry (EXX – EYY ≠ 0), thereby restoring the quadrupolar relaxation effects that usually decouple 14N coupling. In this regard, it is interesting that gas phase (argon matrix) infrared spectroscopy studies (Freedman and Nixon 1972) have indicated that methyl isocyanide forms a dimer, whereby the large CN dipoles (see the isonitrile resonance structure in Scheme 1) align in an antiparallel orientation. Such pairing to neutralize dipole moments might also be favored in a nonpolar solvent, and could produce an upfield shift for the methyl protons, as shown in Scheme 2.
Scheme 2.
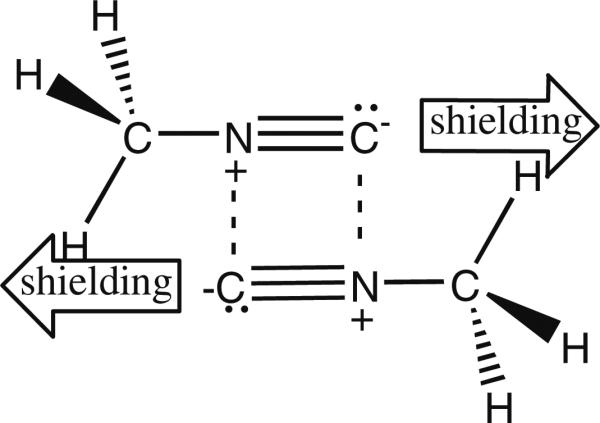
Proposed methyl isocyanide dimer in nonpolar solvent
However, the situation could certainly be different in solution relative to the gas phase. One must also consider the possibility of higher order structures, at least in solution, and even cyclic trimers that might form due to stabilizing dative bonds between molecules in the imine resonance form, shown in Scheme 1. Whatever the source of this interesting phenomenon, it is not relevant for protein studies, since the 14N coupling is no longer observed when 13CH3NC is bound to heme iron.
Application of the 13CH3NC probe to studies of a bacterial CYP: P450cam
Initial studies were done with cytochrome P450cam (P450cam) which, being a soluble and well-studied P450 protein, serves as an excellent model system. In Fig. 3a is shown the 1H-13C HSQC spectrum of 13C-labeled methyl isocyanide (13CH3NC) in buffer, without protein present. The appearance of the spectrum is the same in the presence or absence of camphor, indicating that no significant interactions occur between substrate and probe in solution, in the absence of P450cam. The 1H-13C HSQC spectrum of 13CH3NC in the presence of an equimolar concentration of reduced (ferrous) P450cam (Fig. 3b) shows a large upfield shift due to heme binding. The signal of free 13CH3NC has disappeared and a new signal with a 1Hδ of 1.2 ppm and 13Cδ of 21.4 ppm has appeared. The upfield shift in both dimensions presumably reflects the fact that the methyl group of the probe, when bound to the heme-iron, now sits directly above the aromatic porphyrin ring, resulting in increased shielding of the relevant resonances due to ring current effects (Fig. 1). Figure 3c is a spectrum of the same sample shown in Fig. 3b, but now with excess 13CH3NC, clearly showing that the free and bound forms are in slow exchange on the chemical shift timescale. Finally, the 1H-13C HSQC spectrum of the 13CH3NC/Fe2+-P450cam/camphor ternary complex (Fig. 3d) reveals two new major crosspeaks (and a third less intense resonance), distinct from both free and bound (- camphor). It appears therefore that camphor is either binding in two orientations, or that the probe itself occupies two major orientations (possibly linear and bent, but now in slow exchange on the NMR timescale, due to steric clash with camphor?). In any case, this experiment clearly identified camphor as binding in the active site, thereby validating the use of 13CH3NC for identifying such molecules in NMR-based screening.
Fig. 3.

P450cam 1H-13C HSQC spectra showing (a) unbound 13CH3NC, (b) stoichiometric 13CH3NC bound to Fe2+-P450cam, (c) 13CH3NC bound to Fe2+-P450cam in the presence of excess 13CH3NC, and (d) 13CH3NC bound to Fe2+-P450cam in the presence of a saturating amount of camphor. The additional peaks in panels B, C, and D at 60–75 ppm are due to natural abundance glycerol 1H-13C signal
Application of the 13CH3 NC probe to studies of a mammalian CYP: CYP2B4
The same binding studies were done with native, full-length CYP2B4 in 100 mM potassium phosphate buffer (pH 7.4) and 20% (v/v) d8-glycerol (see Fig. S2). This time, upon addition of an equimolar amount of CYP2B4, the bound form of the 13CH3NC probe had a 1Hδ of 3.13 ppm and a 13Cδ of 76.8 ppm. That is, there is now a dramatic downfield shift for carbon (but not hydrogen). When substrate (benzphetamine) was then added, the methyl crosspeak shifted to have a 1Hδ of 3.13 ppm and a 13Cδ of 49.5 ppm. The chemical shifts of the unbound 13CH3NC probe (not shown) are the same as those reported above, in the absence of d8-glycerol (Fig. 3a). These data indicate that 13CH3NC is an extremely sensitive chemical shift probe that can be used to identify occupancy of active sites for solubilized mammalian CYPs like CYP2B4.
While the above studies are of great value for identifying active site binders, they still suffer from the same limitations that most structural studies of mammalian CYPs do—they are of the enzyme in a relatively artificial state, unbound to lipid. Because CYP2B4 is normally bound to a phospholipid bilayer in vivo, we repeated the above experiments, but now in the presence of d38-DPC micelles. In the control spectrum taken in the absence of both protein and substrate, there now appeared a new peak for the methyl of 13CH3NC, in addition to the one previously observed for free 13CH3NC (Fig. 3a). This new resonance is shifted upfield in the proton dimension with a 1Hδ of 1.17 ppm, but is largely unchanged in the carbon dimension (13Cδ of 30.3 ppm). This new peak is likely due to 13CH3NC that has been taken up into the nonpolar interior of the micelle, and is similar to the chemical shift change we saw for probe in nonpolar solvents like d6-benzene (vide supra). When CYP2B4 protein is then added, one new peak appears, presumably from 13CH3NC that binds the heme iron of micelle-bound CYP2B4. This peak is upfield shifted (Table 1). Finally, when benzphetamine was added, the intensity of this crosspeak decreased and three new peaks appeared with 1Hδs of 2.64, 2.73 and 2.80 ppm and 13Cδs of 37.5, 27.15 and 38.5 ppm, respectively (Table 1). These data suggest that 13CH3NC-bound CYP2B4 can bind benzphetamine, and that multiple conformations exist for this CYP2B4:13CH3NC:benzphetamine ternary complex. Most importantly, these chemical shift changes are very different than what was observed for the free CYP2B4 (with glycerol present), indicating at least qualitatively that the heme is in a very different electronic state when CYP2B4 is bound to micelles versus when it is solubilized with glycerol. Currently, there are no structural methods to probe active site changes in mammalian CYPs, due to binding to lipid. Given that all current crystal structures are of free CYPs, it is therefore vitally important to determine if the active site structure changes in the presence of lipid.
Table 1.
1H-13C HSQC crosspeak chemical shifts for various 13CH3NC complexes
| Sample | 1Hδ | 13Cδ |
|---|---|---|
| 13CH3NC in buffer A* | 3.13 | 0.5 |
| 13CH3NC/P450cam complex in buffer A | 1.21 | 21.4 |
| 13CH3NC/P450cam/camphor complex in buffer A | 3.22 | 34.2 |
| 2.66 | 27.4 | |
| 2.51 | 27.3 | |
| 2.82 | 30.7 | |
| 13CH3NC in buffer B* | 3.13 | 30.5 |
| 13CH3NC/CYP2B4 complex in buffer B | 3.13 | 76.8 |
| 13CH3NC/CYP2B4 complex in buffer B | 3.13 | 76.8 |
| 13CH3NC in buffer C* | 3.13 | 30.5 |
| 13CH3NC/CYP2B4 complex in buffer C | 1.17 | 30.3 |
| 13CH3NC/CYP2B4 complex in buffer C | 1.17 | 30.3 |
| 0.89 | 24.8 | |
| 2.64 | 37.5 | |
| 2.73 | 27.2 | |
| 2.81 | 38.5 |
Buffer A: 100 mM KCl, 30 mM potassium phosphate, pH 7.4; Buffer B: 20% (v/v) glycerol, 100 mM potassium phosphate, pH 7.4; Buffer C: 50 mM DPC, 100 mM potassium phosphate, pH 7.4
Application of 13CH3NC probe using 1D spectra—towards higher throughput
The above studies establish the utility of the 13CH3NC probe for screening to identify molecules that bind in CYP active sites. But, 2D 1H-13C HSQC experiments are too time-consuming (1–4 h) for higher throughput screening efforts, especially if lower protein concentrations are used (≤150 μM). Fortunately, since only one crosspeak is being monitored, 2D experiments are not necessary. Instead, a much shorter 13C-filtered 1H 1D spectrum (Fig. 4) could be acquired, or a 13C 1D using a cryoprobe with cryocooled 13C preamp (Kovacs et al. 2005), depending upon whether the change of interest is occurring in the 1H or the 13C dimension. This, coupled with the fact that very little chemical probe is actually needed (~5 μg for a 0.5 ml sample of 250 μM protein), makes its use in a high-throughput assay even more attractive, as it should be able to provide reliable, fast and inexpensive information about a compound's ability to bind to a given heme protein. Furthermore, advances in microflow probe technology (Olson et al. 2004) would permit assays to be done on volumes as low as 10 μl, making such screening even more feasible in terms of protein consumption (requiring only ~100 μg protein/assay, so ~10 mg to screen a 96-well plate of compounds), at least as a secondary assay. Of course, since protein concentration would be ~150 μM, one would need to have at least 50 νM of the compound to be screened present to see a significant effect on the methyl chemical shift. Finally, subsequent use of the 13CH3NC probe to obtain structural information from NOE data will be limited to molecules that bind in fast exchange (i.e. using transferred NOE experiments), because it is usually not practical to obtain concentrated solutions of mammalian CYP enzymes, given their hydrophobic nature.
Fig. 4.

13C-filtered 1H 1D spectra of (a) unbound 13CH3NC and (b) 13CH3NC bound to Fe2+-P450cam
Interpretation of chemical shift changes
Besides the qualitative information on a compound's ability to bind to CYP active sites, the magnitude of the chemical shift perturbations could possibly be used to calculate a binding orientation for the probe relative to the central heme iron, and the largely planar and aromatic porphyrin ring system (Fig. 1). Furthermore, anisotropy effects from a bound CYP substrate or inhibitor (which are often aromatic) could help to position it relative to probe. Indeed, chemical shift values are increasingly being used to refine protein structures (Xu and Case 2001). Unfortunately, these algorithms are not adequately parameterized at present to deal properly with the highly anisotropic effects from the CH3NC triple bond, the heme pyrrole rings and the diverse array of aromatic ring systems likely to be present in CYP substrates or inhibitors.
At one level, since it is reasonable to assume the dominant resonance structure of 13CH3NC free in solution is the isonitrile (Scheme 1), then any deviation of the 1Hδ from that of its unbound solution form must be due to effects from the heme anisotropy and/or from increasing the population of the bent orientation. In the case of CYP2B4 in 20% glycerol, the 1Hδ remained the same as that of the unbound probe, both in the absence and presence of saturating amounts of substrate, while the 13Cδ was first shifted dramatically downfield ~45 ppm and then back upfield ~25 ppm, still distant from its original position. One possible interpretation of these unusual shifts is that, due to the structural/steric constraints of the binding pocket (or heme electronic changes leading to more backbonding), the probe is forced to adopt a more bent orientation relative to the porphyrin ring. It is surprising, though, that the methyl protons wouldn't shift downfield like the methyl carbon does, if the change is simply due to a more bent orientation. The reasons for these large chemical shift changes are not entirely clear, but will be a function of: (a) the high level of anisotropy coming from the heme pyrrole rings and the cyanide triple bond (Fig. 1), (b) the extent of backbonding from iron (a function of heme electronic state), which would alter the relative population of the linear and bent forms of the probe, (c) the physical perturbation/steric clash between a substrate or inhibitor and the 13CH3NC probe, which could also alter the relative population of linear and bent forms (and its position in this highly anisotropic environment), and (d) any anisotropic effects from the substrate/inhibitor itself, if it has aromatic rings as benzphetamine does. The combination of these effects would make any structure-based calculation of chemical shift changes difficult at present, but clearly the probe's unique position in a highly anisotropic environment makes it exquisitely sensitive to even modest structural or electronic perturbations, and thereby makes it a very sensitive chemical shift probe. As a final point on structural inferences that could be made, it is tempting to speculate that when the probe gives two new crosspeaks for a complex with substrate or inhibitor, this might indicate a steric clash is occurring, which slows the exchange between the linear and bent structures (Scheme 1 and Fig. 1). The data with camphor bound to P450cam and benzphetamine bound to micelle-bound CYP2B4 are consistent with this interpretation, but more experiments are needed to prove that this is in fact the source of the multiple crosspeaks.
Application of the13CH3 NC probe to other heme proteins: myoglobin
Regardless of the actual underlying structural changes occurring within these CYP complexes, and the source of the chemical shift changes, the NMR data do indicate that the 13CH3NC probe will be useful as a screening tool for monitoring active site changes in CYPs. But, might the probe also be useful for exploring other heme proteins, beyond the CYPs? As a first step towards answering this question, spectra of 13CH3NC bound to 500 μM myoglobin (a non-P450 heme protein) have been obtained (Fig. S3), and they show a similar upfield chemical shift in both 1H and 13C dimensions. This suggests the probe might be useful for studying other heme protein active sites as well.
In summary, we have developed 13CH3NC as the first NMR-based probe of cytochrome P450 active sites. This probe binds to both bacterial and mammalian P450s, even membrane-bound P450s, and may have broader utility with other heme proteins. It can be used to detect structural and electronic changes in active sites. In particular, it can be used to screen for P450 substrates or inhibitors using a reasonably high throughput assay, by acquiring a simple 13C-filtered 1D spectrum and monitoring chemical shift changes for the methyl protons. Since this could be done with microflow NMR (ex. Protasis CapNMR) at volumes as low as ~10 μl, the assay would consume only ~100 μg protein, making it reasonably sensitive for an NMR assay.
Supplementary Material
Acknowledgements
We thank professors Rajendra Rathore, Milo Westler and Bill Donaldson for helpful discussions. This research was supported, in part, by Marquette University and by National Institutes of Health/National Science Foundation Instrumentation Grants S10 RR019012 and CHE-0521323, and by NIH (R01-GM3553) and VA Merit Review grants to L.W., and also made use of the National Magnetic Resonance Facility at Madison (NIH: P41RR02301, P41GM66326). Remote connectivity was with Abilene/Internet2, funded by NSF (ANI-0333677).
Abbreviations
- δ-ALA
δ-Aminolevulinic acid
- cam
Camphor
- CYP
Cytochrome P450
- DEAE
Diethylaminoethyl
- DPC
Dodecylphosphocholine
- EDTA
Ethylenediaminetetraacetic acid
- HSQC
Heteronuclear single quantum coherence
- IPTG
Isopropyl-β-D-thiogalactopyranoside
- PMSF
Phenylmethanesulonyl fluoride
- Tris
tris(hydroxymethylethyl)aminomethane
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10858-009-9300-8) contains supplementary material, which is available to authorized users.
Contributor Information
Christopher R. McCullough, Chemical Proteomics Facility at Marquette, Department of Chemistry, Marquette University, P.O. Box 1881, Milwaukee, WI 53201-1881, USA
Phani Kumar Pullela, Chemical Proteomics Facility at Marquette, Department of Chemistry, Marquette University, P.O. Box 1881, Milwaukee, WI 53201-1881, USA.
Sang-Choul Im, Department of Anesthesiology, University of Michigan and VA Medical Center, Ann Arbor, MI 48105, USA.
Lucy Waskell, Department of Anesthesiology, University of Michigan and VA Medical Center, Ann Arbor, MI 48105, USA.
Daniel S. Sem, Chemical Proteomics Facility at Marquette, Department of Chemistry, Marquette University, P.O. Box 1881, Milwaukee, WI 53201-1881, USA
References
- Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem. 1998;273:17036–17049. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, III, Rance M, Skelton NJ. Protein NMR spectroscopy. 2nd edn. Elsevier Academic Press; New York: 2007. [Google Scholar]
- Freedman TB, Nixon ER. Matrix isolation studies of methyl cyanide and methyl isocyanide in solid argon. Spectrochim Acta Biochim. 1972;28A:1375–1391. [Google Scholar]
- Furge LL, Guengerich FP. Cytochrome P450 enzymes in drug metabolism and chemical toxicology. Biochem Mol Biol Educ. 2006;34:66–74. doi: 10.1002/bmb.2006.49403402066. [DOI] [PubMed] [Google Scholar]
- Gelb MH, Heimbrook DC, Malkonen P. Stereochemistry and deuterium isotope effects in camphor hydroxylation by the cytochrome P450cam monoxygenase system. Biochemistry. 1982;21:370–377. doi: 10.1021/bi00531a026. [DOI] [PubMed] [Google Scholar]
- Kanaeva IP, Dedinskii IR, Skotselyas ED, Krainev AG, Guleva IV, Sevryukova IF, Koen YM, Kuznetsova GP, Bachmanova GI, Archakov AI. Comparative study of monomeric reconstituted and membrane microsomal monooxygenase systems of the rabbit liver. I. Properties of NADPH-cytochrome P450 reductase and cytochrome P450 LM2 (2B4) monomers. Arch Biochem Biophys. 1992;298:395–402. doi: 10.1016/0003-9861(92)90427-x. [DOI] [PubMed] [Google Scholar]
- Katagiri M, Ganguli BN, Gunsalus IC. A soluble cytochrome P450 functional in methylene hydroxylation. J Biol Chem. 1968;243:3543–3546. [PubMed] [Google Scholar]
- Kovacs H, Moskau D, Spraul M. Cryogenically cooled probes-a leap in NMR technology. Prog in Nucl Magn Reson Spectrosc. 2005;46:131–155. [Google Scholar]
- Lee DS, Park SY, Yamane K, Obayashi E, Hori H, Shiro Y. Structural characterization of n-butyl-isocyanide complexes of cytochromes P450nor and P450cam. Biochemistry. 2001;40:2669–2677. doi: 10.1021/bi002225s. [DOI] [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Nelson JH. Nuclear magnetic resonance spectroscopy. Pearson Education, Inc; New Jersey: 2003. p. 196. [Google Scholar]
- O'Keeffe DH, Ebel RE, Peterson JA. Purification of bacterial cytochrome P-450. Methods Enzymol. 1978;52:151–157. doi: 10.1016/s0076-6879(78)52017-x. [DOI] [PubMed] [Google Scholar]
- Olson DL, Norcross JA, O'Neil-Johnson M, Molitor PF, Detlefsen DJ, Wilson AG, Peck TL. Microflow NMR: concepts and capabilities. Anal Chem. 2004;76:2966–2974. doi: 10.1021/ac035426l. [DOI] [PubMed] [Google Scholar]
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- Pellecchia M, Meininger D, Dong Q, Chang E, Jack R, Sem DS. NMR-based structural characterization of large protein-ligand interactions. J Biomol NMR. 2002;22:165–173. doi: 10.1023/a:1014256707875. [DOI] [PubMed] [Google Scholar]
- Poulos TL, Johnson EF. Structures of cytochrome P450 enzymes. In: Ortiz de Montellano P, editor. Cytochrome P450: structure, function and biochemistry. 3rd edn. Plenum Press; New York: 2005. [Google Scholar]
- Poulos TL, Finzel BC, Howard AJ. Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
- Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD. An open conformation for mammalian cytochrome P450 2B4 at 1.6 Å resolution. Proc Natl Acad Sci USA. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showalter SA, Johnson E, Rance M, Bruschweiler R. Toward quantitative interpretation of methyl side-chain dynamics from NMR and molecular dynamics simulations. J Am Chem Soc. 2007;129:14146–14147. doi: 10.1021/ja075976r. [DOI] [PubMed] [Google Scholar]
- Sibbesen O, De Voss JJ, Ortiz de Montellano PR. Putidaredoxin reductase-putidaredoxin-cytochrome P450cam triple fusion protein. J Biol Chem. 1996;271:22462–22469. doi: 10.1074/jbc.271.37.22462. [DOI] [PubMed] [Google Scholar]
- Simonneaux G, Bondon A. Isocyanides and phosphines as axial ligands in heme proteins and iron porphyrin models. The Porphyrin Handbook. 2000:299–311. [Google Scholar]
- Tomita T, Ogo S, Egawa T, Shimada H, Okamoto N, Imai Y, Watanabe Y, Ishimura Y, Kitigawa T. Elucidation of the differences between the 430- and 455-nm absorbing forms of P450-isocyanide adducts by resonance Raman spectroscopy. J Biol Chem. 2001;276:36261–36267. doi: 10.1074/jbc.M104932200. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kanelis V, Kay LE. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Sridar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monoxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Ward A, Angove HC, Matak Vinkovic D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:46–464. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structure of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- Xu X-P, Case DA. Automated prediction of 15N, 13Cα, 13Cβ and 13C’ chemical shifts in proteins using a density functional database. J Biomol NMR. 2001;21:321–323. doi: 10.1023/a:1013324104681. [DOI] [PubMed] [Google Scholar]
- Yao H, McCullough CR, Costache AD, Pullela PK, Sem DS. Structural evidence for a functionally relevant second camphor binding site in P450cam: model for substrate entry into a P450 active site. Proteins. 2007;69:125–138. doi: 10.1002/prot.21508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.