

Abstract
Diethanolamine complexed heterocyclic boronic acids (DABO boronates) are air-stable reagents that can be used directly in Suzuki-Miyaura reactions in the presence of water or a protic co-solvent. Interestingly, heterocyclic DABO boronates can be stored for extended periods of time at room temperature with no noticeable degradation, unlike their boronic acid counterparts. Heterocyclic DABO boronates constitute an operationally simple and efficient alternative to other boronic acid derivatives as coupling partners in palladium catalyzed cross-coupling reactions under standard Suzuki-Miyaura conditions.
Keywords: Suzuki-Miyaura reaction, cross-coupling, palladium catalysis, heterocyclic boronate, aryl boronate
Boronic acids are versatile building blocks essential to a variety of important carbon-carbon bond forming reactions. Key examples of their application include the Suzuki-Miyaura cross-coupling reaction,1,2 asymmetric conjugate addition,3 and hydroarylation of alkynes,4 among others.5 While boronic acid couplings have many desirable characteristics such as nontoxic byproducts, high functional group tolerance, and many commercially available building blocks,5 cross-coupling reactions with labile boronic acids pose a challenge.6,7 Herein, we describe a new class of heterocyclic boronic acid analogues that can be directly applied to Suzuki-Miyaura reactions.
Boronic acids are often unstable in air and can form cyclic trimers with loss of water, troubling characteristics that complicate reaction stoichiometry and reproducibility. Conversion of boronic acids into pinacol boronic esters provides more stable reactants, but their isolation and purification often requires distillation or chromatography, and the transmetalation of pinacol boronic esters is slower than that of boronic acids, which can lead to sluggish reactions.5 Vinyl,8 aliphatic,9 and heteroaryl6 potassium trifluoroborate salts are air-stable, crystalline solids that can be used in coupling reactions. Their isolation and purification, however, can be tedious, often requiring extended Soxhlet extraction. N-Methyliminodiacetic acid (MIDA) has been shown to react with boronic acids10 to form complexes that function as a protecting groups for boronic acids under anhydrous conditions.11,12 Their synthesis, however, requires heating in DMF for 2–18 h with the azeotropic removal of water, followed by column chromatography purification. Although boronic acids complexes have been used in coupling reactions, there is room for the development of more convenient forms of labile boronic acids.
Various N-alkyldiethanolamine adducts of arylboronic acids, including methyl13,14 and phenyl,15 or N,N-diethanolaminomethyl polystyrene,16 have been developed to improve stability and ease of use of boronic acids, but most of these derivatives are not crystalline and their use in coupling reactions often requires copper additives.13,15,16 In contrast, simple diethanolamine (DEA) complexes of boronic acids, or 2,8-dioxa-5-aza-1-bora-bicyclo[3:3:0]octanes (DABO boronates) are air and water stable adducts that allow for easy isolation, characterization, and storage,17,18,19 DEA is the least expensive auxiliary for stabilizing boronic acids when compared to MIDA, KHF2, and pinacol.20 DABO boronates are known to hydrolyze to the boronic acid or ester in aqueous solutions and protic solvents21 commonly used in coupling reactions, suggesting that they would be viable candidates for Suzuki-Miyaura cross-coupling reactions (Scheme 1). In the past DEA complexes have been used to facilitate the handling of aryl boronates, but a separate hydrolysis step was considered necessary prior to a transition metal mediated coupling reaction.22 The recent report from the Gras group on the direct coupling of aryl DABO boronates prompted us to report our findings with much more diverse boronate structures.23
Scheme 1.
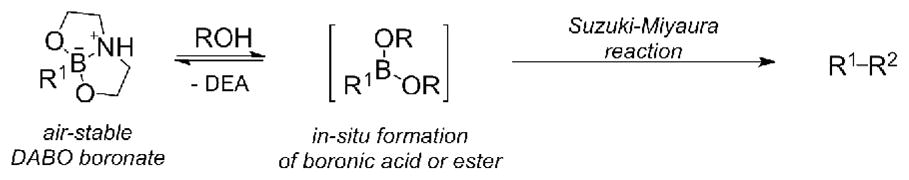
Proposed reactivity of DABO boronates
The synthesis of aryl and heteroaryl DABO boronates from the corresponding boronic acid is simple and efficient (Table 1). Stirring the boronic acid with DEA and dichloromethane in an open flask at room temperature provides a solid that can be isolated in pure form by filtration and/or recrystallization.24 This procedure allows for the isolation of aryl DABO boronates 1–6 in high yield. Complexation of diethanolamine with 2,6-dimethylphenylboronic acid did not yield the DABO boronate; steric hindrance by the adjacent methyl groups apparently prevented formation of a tetrahedral boronate species. Substitution only at the 2-position, however, did not adversely affect formation of 2-methoxyphenyl DABO (4). Heteroaryl DABO boronates 7–12 were isolated in pure form from the corresponding labile boronic acids as air-stable solids.25 Both electron rich and electron poor boronates were isolated in high yields. In addition, vinyl DABO boronate 13 was isolated as a crystalline solid, illustrating the versatility of the DEA auxiliary. Syntheses of vinyl and heteroaryl DABO boronates are remarkably straightforward because these complexes are poorly soluble in organic solvents and usually precipitate from the reaction solution.
Table 1.
Preparation of Diverse DABO Boronates
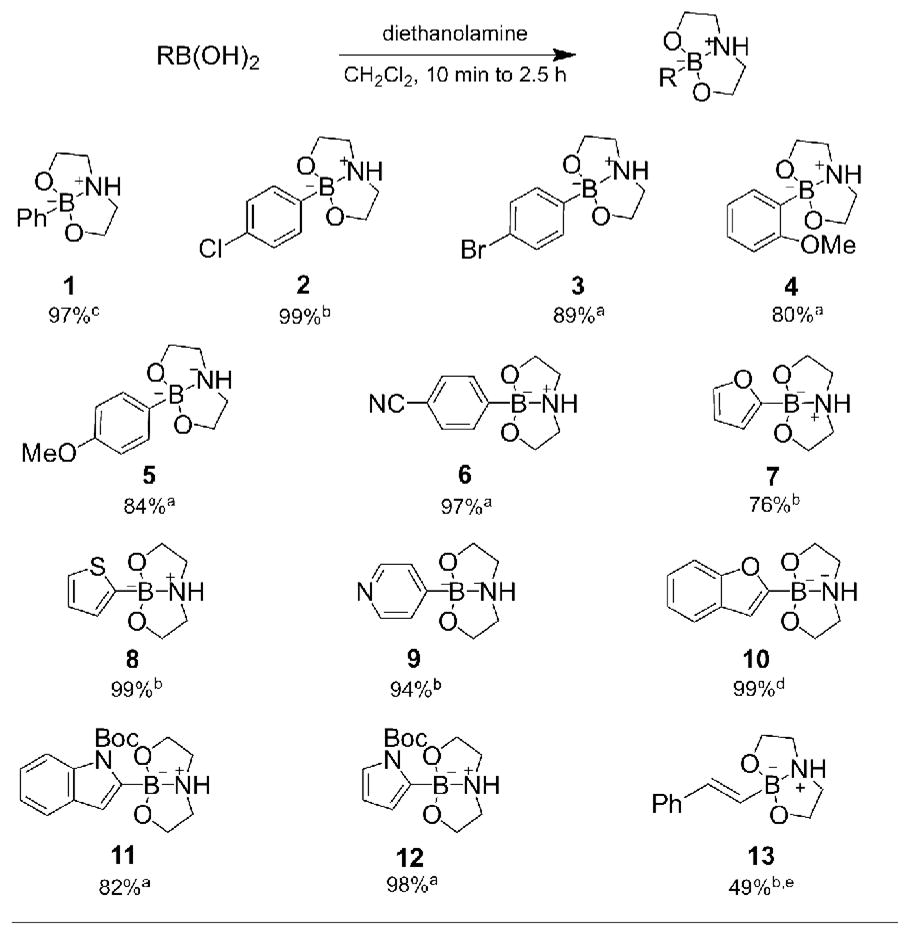
|
Synthesis was conducted on a 2.5–9.5 mmol scale.
Synthesis was conducted on a 11–28 mmol scale.
Synthesis was conducted on a 42 mmol scale.
Synthesis was conducted on a 123 mmol scale.
Ethyl acetate was used instead of CH2Cl2.
The Suzuki coupling reaction with phenyl DABO 1 and 4-bromoanisole was optimized as outlined in Table 2. Initially several ligands were screened; both dppf and JohnPhos worked very well in the coupling, but all the phosphine ligands gave acceptable yields. The Pd(OAc)2/JohnPhos was selected because the palladium source was stable and easily handled, and the combination gave the best yield. Screening the base showed that Cs2CO3 was optimal (entry 5), but all the bases worked well. Other solvents and temperatures were explored, and all of the conditions were reasonably successful. In this system copper was not necessary for excellent reactivity.
Table 2.
Optimization of Suzuki-Miyaura Coupling with DABO Boronate 1

| |||
|---|---|---|---|
| entry | cat./ligand | base | yield (%NMR)a |
| 1 | Pd(dppf)Cl2 | Cs2CO3 | 91 |
| 2 | Pd(PPh3)4 | Cs2CO3 | 67 |
| 3 | Pd2(dba)3 | Cs2CO3 | 37 |
| 4 | Pd(PPh3)Cl2 | Cs2CO3 | 70 |
| 5 | Pd(OAc)2/JohnPhos | Cs2CO3 | 98 |
| 6 | Pd(OAc)2/JohnPhos | CsF | 74 |
| 7 | Pd(OAc)2/JohnPhos | K2CO3 | 92 |
| 8 | Pd(OAc)2/JohnPhos | K2PO3 | 74 |
| 9 | Pd(OAc)2/JohnPhos | tBuOK | 87 |
| 10 | Pd(OAc)2/JohnPhos | NaOH | 73 |
An internal standard, 4-methylbiphenyl, was added to the crude reaction mixture after it was filtered through Celite rinsing with CH2Cl2 and concentrated. The 1H NMR ratio of the internal standard Me and the product OMe were compared to provide an NMR yield.
The Suzuki-Miyaura coupling reaction was first optimized with phenyl DABO 1 and 4-bromoanisole. Pd(OAc)2 was chosen as a catalyst because it was inexpensive and air-stable. JohnPhos provided the highest reactivity, and was thus used in subsequent reactions. All reactions were set up in air and evacuated and refilled with argon before the addition of liquid reagents and solvents. Reaction with various aryl halides led to the formation of the desired coupled product in good yields (Table 3). Individual reactions, however, were unoptimized. These reactions do not require the addition of a copper cocatalyst.23 In these palladium cross-coupling reactions, DEA did not adversely affect the catalytic cycle or the availability of the boronic acid for transmetalation. Reduced yields were observed under anhydrous (or aprotic) conditions, suggesting that the formation of the boronic acid (or ester) is necessary for efficient transmetalation. The reaction of sterically hindered 2,4,6-triisopropylbromobenzene was slow under the original conditions, and a higher temperature and longer reaction time were adopted to promote formation of 15 in moderate yield (Entry 2). DABO boronates are effective in Suzuki-Miyaura coupling reactions with no additional catalysts.
Table 3.
Suzuki-Miyaura Cross-Coupling Reactions with DABO Boronatesa

| |||||
|---|---|---|---|---|---|
| Entry | DABO | DABO equiv | RX | Product | Yield(%) |
| 1 | 1 | 1.5 |

|
 14 |
92 |
| 2 | 1 | 2.5 |
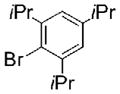
|
 15 |
53 |
| 3 | 10 | 1.5 |
|
16 |
97 |
| 4 | 11 | 2.0 |

|
 17 |
97 |
| 5 | 7 | 1.3 |
 18 |
 19 |
99b |
| 6c | 9 | 1.5 |

|
20 |
92 |
| 7 | 12 | 2.0 |

|
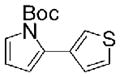 21 |
65 |
| 8 | 8 | 2.0 |

|
22 |
94 |
| 9 | 13 | 1.2 |

|
 23 |
82 |
| 10 | 13 | 1.2 |

|
 24 |
87 |
Reaction condition changes and details are denoted in the experimental section.
XPhos was used instead of JohnPhos.
The solvent was anhydrous n-butanol, and the base was K3PO4. JohnPhos = (2-biphenyl)di-t-butylphosphine; XPhos = 2-dicyclohexyl-phosphino-2′,4′,6′-triisopropylbiphenyl
Heterocyclic DABO boronates were successful coupling partners in Suzuki-Miyaura reactions (Table 2). Coupling reactions with 2-furyl boronic acids can be challenging;6 however, the coupling of 2-furyl DABO (7) with sterically hindered vinyl triflate 18, which is reported to be a poor substrate for Pd-catalyzed coupling reactions, provided vinyl furan 20 in excellent yield using XPhos as the ligand (Entry 6).26 Coupling reactions were conducted with a variety of aryl halides to give heterobiaryl products 16–17 and 19–22 in good yields. Although excess DABO boronates were used to drive these test reactions to completion, upon further optimization it was found that only a modest excess of the DABO boronate was necessary, vide infra. The coupling of 4-pyridyl DABO 9 with 3-bromothiophene using Buchwald’s conditions6 led to the formation of 20 in good yield despite evidence that pyridines are challenging coupling partners.6 This coupling reaction was run under anhydrous conditions in n-butanol; presumably the DABO complex exchanges with the solvent to form a reactive dibutyl boronate ester. Vinyl DABO boronate 13 coupled nicely with both 4-bromobenzonitrile and 3-bromothiophene to provide vinylogous products 23 and 24, respectively. Heteroaryl and vinyl DABO boronates thus proved to be effective boronic acid surrogates for the Suzuki-Miyaura reaction.
The viability of large-scale DABO boronate cross-coupling reactions was demonstrated using only 1.1 equiv of benzofuran DABO boronate 10 (eq 1).27 The catalyst loading was reduced to 0.5 mol % palladium for greater economy, and triphenylphosphine, an inexpensive ligand, was employed. A 44 mmol coupling reaction under these conditions provided 8.9 g of coupled product 16 in 89% yield.
Equation 1.

Large-scale Suzuki-Miyaura cross-coupling reaction
Aliphatic boronates were also found to be effective in cross-coupling reactions. The synthesis of DABO boronate 25 was less efficient that most examples because the product is less crystalline than other DABO boronates, and the preparation was run on a small scale. Aliphatic DABO boronate 25 coupled effectively under classic Suzuki conditions with palladium dppf catalyst system.28 When conducted in anhydrous THF, a significant amount of homocoupled product, biphenyl-4,4′-dicarbonitrile, was observed. The addition of water as a cosolvent led to significantly decreased amount of homocoupled product and increased yield of 26, supporting the hypothesis that water enhances the availability of the boronate for transmetalation. No β-hydride elimination was observed in the crude reaction mixture, consistent with previous findings.28 Ethylbenzene was also absent from the crude reaction mixture, which suggests that protodeboronation is not a significant decomposition pathway.
DABO boronates exhibit excellent stability in air and provide a convenient and inexpensive alternative to boronic acids. We have established that these complexes can be used directly in Suzuki-Miyaura cross-coupling reactions in conjunction with water or protic solvent to provide biaryl products. Heterocyclic DABO boronates can be stored open to the air for months with no noticeable decomposition. Aryl, vinyl, heterocyclic and even aliphatic DABO boronates are practical replacements for boronic acids in Suzuki-Miyaura cross-coupling reactions, and are superior to other masked boronic acids based on ease of formation, convenience in handling, and cost.
Synthesis of 2-Benzofuranyl DABO boronate 10
To a suspension of 2-benzofuranyl boronic acid (20.0 g, 123 mmol, 1 equiv) in methylene chloride (500 mL) in a 1-neck 1 L round bottom flask was added diethanolamine (12.0 mL, 123 mmol, 1 equiv) via syringe. A plastic cap was placed on top of the flask to minimize solvent evaporation. The suspension changed color from white to off-white and was stirred for 20 min under an atmosphere of air at room temperature to provide a suspension of the DABO boronate. The resulting suspension was vacuum filtered to provide the crude DABO boronate with some excess diethanolamine as a powdery solid (33.8 g). Excess diethanolamine was removed by triturating the solid in 60 mL of ethyl acetate for 5 min and vacuum filtering, rinsing with ethyl acetate. The filtration provided DABO boronate 10 as a white solid (28.2 g, 122 mmol, 99% yield). Decomposition point 225–227 °C; 1H NMR (500 MHz, CD3CN) δ 7.52 (d, J = 7.5 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.19 (dt, J = 1.5, 7.5 Hz, 1H), 7.14 (dt, J = 1.5, 8.0 Hz, 1H), 6.74 (s, 1H), 5.56 (br s, 1H), 3.97 (m, 2H), 3.89 (m, 2H), 3.25 (m, 2H), 2.86 (m, 2H); 1H NMR (500 MHz, DMSO) δ 7.51 (d, J = 7.0 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.30 (br s, 1H), 7.16–7.10 (m, 2H), 6.70 (s, 1H), 3.86 (m, 2H), 3.81 (m, 2H), 3.14 (m, 2H), 2.86 (m, 2H); 13C NMR (125 MHz, DMSO) δ 156.2, 128.9, 122.7, 121.6, 120.2, 110.7, 110.1, 62.7, 50.4 (missing boronate carbon); IR (KBr) 3093 (br), 2871, 1950, 1908, 1639, 1552 cm−1.
Suzuki-Miyaura reaction to provide tert-Butyl 2-(thiophen-3-yl)-1H-pyrrole-1-carboxylate (21)
To a reaction vial equipped with a stir bar was added all solid reactants, including Pd(OAc)2 (7.9 mg, 0.035 mmol, 7 mol %), JohnPhos (12 mg, 0.035 mmol, 7 mol %), Cs2CO3 (489 mg, 1.5 mmol, 3.0 equiv), and DABO complex (1.0 mmol, 2.0 equiv). A PFT crimp-on septum cap was then placed on the reaction vial, and the system was evacuated on high vacuum and backfilled with argon twice. Tetrahydrofuran (3.0 mL) and H2O (0.6 mmol) were added via syringe followed by 3-bromothiophene (0.047 mL, 0.5 mmol, 1.0 equiv). The vial was then placed in a pre-heated heating block at 60 °C. After 6 h, the vial was removed from the heating block and allowed to cool to room temperature. The reaction mixture was filtered through a pad of Celite, rinsing with methylene chloride. The resulting orange/brown solution was concentrated by rotary evaporation to give a viscous liquid. Purification by chromatography on silica gel using 80:1 hexanes/ethyl acetate as the eluant provided the product as a mixture with tert-butyl 1H-pyrrole-1-carboxylate, resulting from protodeboronation of the boronic acid in solution. The impurity was removed by subjecting the clear, colorless liquid to high vacuum to provide tert-butyl 2-(thiophen-3-yl)-1H-pyrrole-1-carboxylate 21 as a clear, colorless oil (81 mg, 0.32 mmol, 65% yield). The sample was subjected to GC-MS, and the chromatogram contained one peak. Doubling in the 1H NMR spectrum was attributed to rotamers: 1H NMR (500 MHz, CDCl3) δ 7.34 (m, 1H), 7.28–2.24 (m, 2H), 7.10 (d, J = 4.0 Hz, 1H), 6.22–6.18 (m, 2H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 149.4, 134.4, 129.9, 129.5, 124.1, 123.0, 122.5, 114.8, 110.6, 83.7, 28.0; IR (thin film) 3151, 3109, 2981, 2935, 2873, 1747 cm−1; HRMS (ES/MeOH) m / z calcd for C13H15NO2SNa (M + Na)+ 272.0721, found 272.0722.
Supplementary Material
Scheme 2.

Synthesis and cross-coupling of aliphatic DABO boronate 25
Acknowledgments
Support was provided by the National Institute of General Medicine (NIGMS GM-43854) and the University of California, Irvine.
Footnotes
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synlett.
References
- 1.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 2.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 3.Hayashi T, Yamasaki K. Chem Rev. 2003;103:2829–2844. doi: 10.1021/cr020022z. [DOI] [PubMed] [Google Scholar]
- 4.Lin P-S, Jeganmohan M, Cheng C-H. Chem Eur J. 2008;14:11296–11299. doi: 10.1002/chem.200801858. [DOI] [PubMed] [Google Scholar]
- 5.Hall DG. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 6.Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- 7.Molander GA, Canturk B, Kennedy LE. J Org Chem. 2009;74:973–980. doi: 10.1021/jo802590b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molander GA, Bernardi CR. J Org Chem. 2002;67:8424–8429. doi: 10.1021/jo026236y. [DOI] [PubMed] [Google Scholar]
- 9.Molander GA, Sandrock DL. Curr Opin Drug Discov. 2009;12:811–823. [PMC free article] [PubMed] [Google Scholar]
- 10.Mancilla T, Contreras R. J Organomet Chem. 1986;307:1–6. [Google Scholar]
- 11.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716–6717. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- 12.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alessi M, Larkin AL, Ogilvie KA, Green LA, Lai S, Lopez S, Snieckus V. J Org Chem. 2007;72:1588–1594. doi: 10.1021/jo0620359. [DOI] [PubMed] [Google Scholar]
- 14.Bouillon A, Lancelot J-C, Santos JS, Callot V, Bovy PR, Rault S. Tetrahedron. 2003;59:10043–10049. [Google Scholar]
- 15.Hodgson PB, Salingue FH. Tetrahedron Lett. 2004;45:685–687. [Google Scholar]
- 16.Gros P, Doudouh A, Fort Y. Tetrahedron Lett. 2004;45:6239–6241. [Google Scholar]
- 17.Davies AG, Roberts BP, Ramsay W. Phys Org, J Chem Soc (B) 1967:17–22. [Google Scholar]
- 18.Korcek S, Watts GB, Ingold KU. J Chem Soc, Perkin II. 1972:242–248. [Google Scholar]
- 19.Crystallographic data: Rettig SJ, Trotter J. Can J Chem. 1975;53:1393–1401.
- 20.MIDA is over 100 times more expensive. KHF2 is similarly priced, but 3.0 equiv are required for the synthesis of heterocyclic potassium trifluoroborates. Pinacol is about 10 times more expensive than DEA. Prices were obtained from the Sigma-Aldrich online catalogue.
- 21.2-Methoxyphenylboronic acid and 2-furyl DABO boronate 7 were mixed at room temperature in THF for 3 h to give an equilibrium mixture of both boronic acids and their DABO counterparts. A similar product mixture was observed beginning with 2-methoxyphenyl DABO boronate 4 and 2-furylboronic acid.
- 22.Caron S, Hawkins JM. J Org Chem. 1998;63:2054–2055. [Google Scholar]
- 23.Bonin H, L–YRMarchiori B, Demonchaux P, Gras E. Tetrahedron Lett. 2011;52:1132–2235. [Google Scholar]
- 24.Excess diethanolamine can be removed by triturating the crude solid in ethyl acetate and filtering. DABO complexes could not be characterized by HRMS, and unidentifiable boron-containing masses were observed by electron ionization. Elemental analysis was attempted, but the carbon percentage was consistently below the theoretical value by about 10.
- 25.No decomposition of 2-furyl DABO boronate 7 was observed by NMR analysis after 72 days of storage in open vials at room temperature. The average temperature and humidity were 24 °C and 65%, respectively, during this period. In contrast, the commercial 2-furylboronic acid completely decomposed in less than one week under these conditions. We recommend storing DABO boronates at room temperature in a closed vial.
- 26.Inoue M, Frontier AJ, Danishefsky SJ. Angew Chem, Int Ed. 2000;39:761–764. [PubMed] [Google Scholar]
- 27.Wang Z, Elokdah H, McFarlane G, Pan S, Antane M. Tetrahedron Lett. 2006;47:3365–3369. [Google Scholar]
- 28.Molander GA, Yun C-S. Tetrahedron. 2002;58:1465–1470. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.