

Abstract
A key feature of the CNS is structural plasticity, the ability of neurons to alter their morphology and connectivity in response to sensory experience and other changes in the environment. How this structural plasticity is achieved at the molecular level is not well understood. We provide evidence that changes in sensory experience simultaneously trigger multiple signaling pathways that either promote or restrict growth of the dendritic arbor; structural plasticity is achieved through a balance of these opposing signals. Specifically, we have uncovered a novel, activity-dependent signaling pathway that restricts dendritic arborization. We demonstrate that the GTPase Rem2 is regulated at the transcriptional level by calcium influx through L-VGCCs and inhibits dendritic arborization in cultured rat cortical neurons and in the Xenopus laevis tadpole visual system. Thus, our results demonstrate that changes in neuronal activity initiate competing signaling pathways that positively and negatively regulate the growth of the dendritic arbor. It is the balance of these opposing signals that leads to proper dendritic morphology.
Keywords: activity-regulated, dendrite, Rem2
Introduction
Each type of neuron in the mammalian CNS has a distinctive dendritic arbor that is typically extensively branched, and that instructs the specific function of the neuron in part by ensuring that the proper synaptic connections are formed (Elston, 2000). In addition, the nervous system has the amazing capacity to transform sensory experience from the environment into changes in neuronal activity that, in turn, cause long-lasting alterations in synaptic connections and dendritic arborization (Wong and Ghosh, 2002; Van Aelst and Cline, 2004; Hensch, 2005). One mechanism through which this structural plasticity could be achieved is via activity-dependent changes in gene expression (Loebrich and Nedivi, 2009; West and Greenberg, 2011). For example, a number of molecules whose mRNA expression is activity regulated and which promote dendritic complexity have been described, such as Cpg15 and Bdnf (McAllister et al., 1995; Nedivi et al., 1998; Zhou et al., 2006). However, negative mediators of experience-dependent dendritic outgrowth that are activity regulated at the transcriptional level are less well understood.
Our previous work has shown that the GTPase Rem2 restricts dendritic complexity (a term that describes both the length and degree of branching of the dendritic arbor) in primary cultures of hippocampal neurons (Ghiretti and Paradis, 2011; Ghiretti et al., 2013). Rem2 is a member of the Rad/Rem/Rem2/Gem/Kir (RGK) protein family, a Ras-related subfamily of small GTPases. Mounting evidence suggests that Rem2 and other RGK family members may not function like canonical Ras-like GTPases, whose activity levels are controlled by nucleotide hydrolysis (Correll et al., 2008; Sasson et al., 2011; Reymond et al., 2012). In addition, mRNA expression of Rem2 and other RGK family members is upregulated by extracellular stimuli such as neuronal depolarization, mitogens, and glucose (Maguire et al., 1994; Finlin et al., 2005; Paradis et al., 2007), suggesting that this may be a defining feature of the family. Recently, we demonstrated that Rem2 is a novel substrate for CaMKII, and that phosphorylation of key serine residues by CaMKII is required for Rem2 inhibition of dendritic complexity (Ghiretti et al., 2013). Thus, Rem2 function is regulated by neuronal activity in at least two ways: Rem2 transcription is upregulated in response to neuronal depolarization, and Rem2 is a downstream target of CaMKII signaling.
Therefore, we sought to explore the relationship between neuronal activity, changes in Rem2 mRNA expression, and negative regulation of neuronal growth processes. Using a combination of in vitro and in vivo approaches, we demonstrate that Rem2 transcription is upregulated in neurons in response to calcium influx selectively through L-type voltage-gated calcium channels (L-VGCCs). Using an RNAi-based approach to knock down Rem2 expression in primary cultures of cortical neurons, we define a signaling pathway whereby Rem2 inhibits calcium-dependent dendritic complexity. Finally, we extend these findings to the optic tectum of Xenopus laevis tadpoles, where we demonstrate that increased Rem2 expression in response to neuronal activity inhibits dendritic complexity in an intact circuit. Thus, Rem2 represents a key activity-regulated molecule that functions to limit the dendritic arborization of neurons in response to changes in sensory experience.
Materials and Methods
Plasmids and pharmacological reagents.
GFP and Rem2 plasmids were as described previously (Paradis et al., 2007; Ghiretti and Paradis, 2011). KCl, nifedipine, nimodipine, and DNQX were purchased from Sigma. D/L APV was purchased from Sigma or Tocris Bioscience. Tetrodotoxin (TTX) was purchased from Sigma or Abcam. Ryanodine was purchased from Abcam.
Neuronal culture stimulation and treatment, RNA extraction, and assay of gene expression by quantitative PCR.
For KCl depolarization of neurons in Figure 1, A and B, 7 day in vitro (7 DIV) rat neurons were quieted overnight with 1 μm TTX. Cells were then incubated for 0 h (control), 0.5 h, 1 h, 2 h, 4 h, or 6 h in 55 mm KCl, or pretreated with a selected reagent: EGTA (5 mm, 30 min pretreatment), APV (100 μm, 1 h pretreatment), DNQX (50 μm, 1 h), nifedipine (5 μm, 1 h), and/or ryanodine (50 μm, 1 h), and then treated with KCl + reagent for 2 h.
Figure 1.
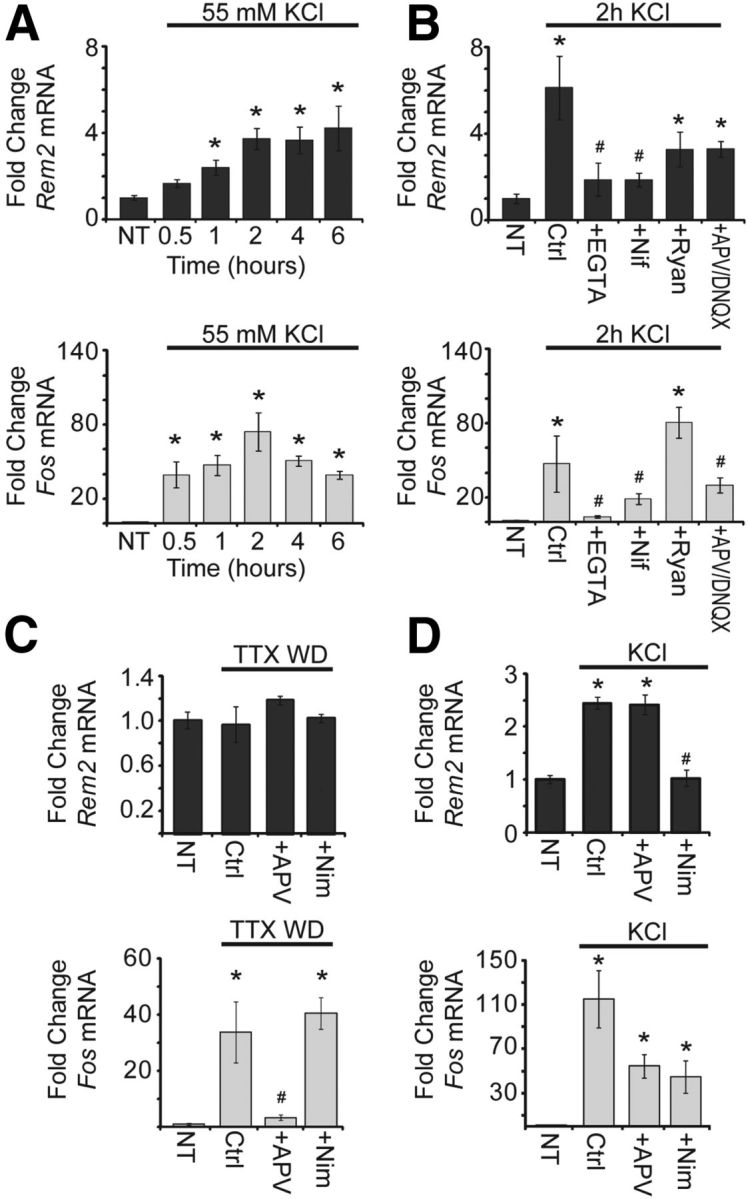
Rem2 mRNA expression is upregulated in response to calcium entry through L-VGCCs. A, Fold change in Rem2 (top, dark gray) and Fos (bottom, light gray) mRNA expression in 8 DIV cultured cortical neurons in response to KCl stimulation (55 mM) for the indicated times. All Rem2 and Fos levels were first normalized to Actb levels, then normalized to the No Treatment (NT) condition, and presented as mean ± SEM. *p ≤ 0.01 from NT by one-way ANOVA with Dunnett's post hoc test. In A and B, n = 4 biological replicates. B, Fold change in Rem2 (top) and Fos (bottom) mRNA expression in 8 DIV cortical neurons after: 0 (NT) or 2 h of KCl (55 mM) treatment alone (Ctrl; Control); or 2 h of KCl treatment plus: EGTA (5 mM), nifedipine (5 μm), ryanodine (50 μm), or APV (100 μm) and DNQX (50 μm). *p ≤ 0.01 from NT, #p ≤ 0.01 from Control by one-way ANOVA with Dunnett's post hoc test. C, Fold change in Rem2 (top) and Fos (bottom) mRNA expression in 7 DIV cortical neurons after: 0 (NT), 3 (Fos), or 6 (Rem2) h of TTX withdrawal (TTX WD) alone (Ctrl), or plus APV (100 μm) or nimodipine (5 μm) treatment. All Rem2 and Fos levels were first normalized to Gapdh levels, then normalized to the No Treatment condition, and presented as mean ± SEM. *p ≤ 0.01 from NT, #p ≤ 0.01 from Control by one-way ANOVA with Dunnett's post hoc test. In C and D, n = 3 biological replicates for Rem2 and two biological replicates for Fos. D, Fold change in Rem2 (top) and Fos (bottom) mRNA expression in 7 DIV cortical neurons after: 0 (NT), 3 (Fos), or 6 (Rem2) h KCl (55 mM) treatment alone (Ctrl) or plus APV (100 μm) or nimodipine (5 μm). *p ≤ 0.01 from NT, #p ≤ 0.01 from Control by one-way ANOVA with Dunnett's post hoc test.
For TTX withdrawal (Fig. 1C), 5 DIV mouse neurons were treated with 1 μm TTX. At 7 DIV, neurons were pretreated for 2 min with 100 μm APV or 5 μm nimodipine, then washed in fresh NB/B27 (also containing APV or nimodipine as required) to remove TTX. For KCl depolarization in Figure 1D, mouse neurons were treated with 1 μm TTX at 5 DIV, then pretreated with 100 μm APV or 5 μm nimodipine at 7 DIV, then depolarized with 55 mm KCl (plus APV or nimodipine as required) for 3 h or 6 h as indicated in the figure legend. At the concentrations used, both nifedipine and nimodipine completely block L-VGCC currents (Shah and Haylett, 2000).
Cortical neurons were harvested from E18 rats or E16 mice of either sex and plated at high density (6,000,000 neurons/well) in a 6-well plate. At 7 DIV (Fig. 1A,B) or 8 DIV (Fig. 1C,D), RNA was extracted after the designated treatment using Absolutely RNA Purify Kit (Agilent) or Trizol Reagent (Invitrogen). DNase-free RNA was prepared and then reverse transcribed using a Random Primer Mix (New England Biologicals). Quantitative PCR (qPCR) was performed using SYBR Green SuperMix (Clontech) on a Rotor Gene thermal cycler (Roche) or using the Applied Biosystems 7300 qPCR System. Primers (sequences provided below; Table 1) were verified by examination of melt curves to ensure reliability. PCR products from genes of interest were normalized to Actb (β-actin) or Gapdh (GAPDH) and presented as fold change over baseline using the delta-delta CT method (Schmittgen and Livak, 2008). Fos (c-Fos) expression was used as a positive control, as Fos is robustly induced by both KCl and TTX withdrawal; note that Fos induction occurs on a much more rapid timescale than that of Rem2 (Morgan and Curran, 1986). “n” represents the number of biological replicates used. Data were compiled from independent biological replicates (Fig. 1A,B, 3 replicates; Fig. 1C,D, 3 replicates Rem2, 2 replicates Fos). A one-way ANOVA followed by a Dunnett's test was used to compare experimental conditions to control or stimulation.
Table 1.
| Rem2 | Forward | TGAGAGACGGATCATGGTGGACAA |
|---|---|---|
| Reverse | AGCCGAAGAAGGGTTTCTGGAACT | |
| Fos | Forward | ATGGGCTCTCCTGTCAACACAC |
| Reverse | ATGGCTGTCACCGTGGGGATAAAG | |
| Actb | Forward | ACGGTCAGGTCATCACTATCG |
| Reverse | AGCCACCAATCCACACAGA | |
| Gapdh | Forward | CATGGCCTTCCGTGTTCCT |
| Reverse | TGATGTCATCATACTTGGCAGGTT | |
| rem2 | Forward | GAGAAGTCAGCATGGAAGAG |
| Reverse | TCACAGTACTGAAAGGTCATG | |
| fos | Forward | CAAGAGCCTGTAGCAGAG |
| Reverse | CGTGTCTACAGACAGAGTG | |
| tubb | Forward | CGTTACCTGACTGTAGCCAC |
| Reverse | AGATCATTCATGTTGCTCTCG |
Rodent cortical culture, transfection, and pharmacological treatment for morphology analysis.
Cortical neurons were cultured at low density on an astrocyte feeder layer, as previously described (Ghiretti and Paradis, 2011). Briefly, dissociated cortical neurons from E18 rats of either sex were plated in 24-well plates at a density of ∼80,000/well onto a layer of confluent astrocytes and grown in Neurobasal media with B27 supplement (Invitrogen).
Neurons were transfected by the calcium phosphate method (Xia et al., 1996), at 2 DIV. The relatively low transfection efficiency (<10%, A. E. Ghiretti and S. Paradis, unpublished observations) of this method allows for a transfected neuron to develop in the context of a network of otherwise relatively unaffected cells. Note that the cotransfection efficiency of up to three plasmids is >93% at both 14 DIV (Ghiretti and Paradis, 2011) and the 5 DIV time point used here (data not shown). All conditions were transfected with a GFP plasmid at 500 ng/well. For control conditions, neurons were also transfected with empty pSuper vector at 33 ng/well or 100 ng/well. For Rem2 RNAi experiments, neurons were also transfected with a pSuper-shRNA plasmid containing an shRNA against Rem2 (Paradis et al., 2007; Ghiretti and Paradis, 2011; Moore et al., 2013) at 33 ng/well. For Rem2 overexpression (OE) experiments, neurons were also transfected with a pCMV-myc-Rem2 cDNA (Ghiretti and Paradis, 2011) at 100 ng/well.
For pharmacological experiments, 4 DIV neurons were quieted overnight in 1 μm TTX, then treated for 6 h before fixing with KCl (55 mm), nifedipine (20 μm), or both KCl and nifedipine at the given concentrations.
Sholl analysis.
At 5 DIV, the neuronal media was replaced with 1× PBS, and then neurons were fixed with 4% paraformaldehyde/4% sucrose for 8 min at room temperature. Coverslips were then washed three times with 1× PBS for 5 min and mounted on glass slides for imaging. Twelve-bit images were acquired on an Olympus Fluoview300 confocal microscope using a 20× (0.85 NA) oil objective. All images were acquired with identical settings for laser power, detector gain, and amplifier offset as a z-stack of 5–10 optical sections with a step size of 1.0 μm. For each experiment, images of 10–20 GFP-expressing neurons from at least two separate coverslips were acquired and analyzed for each condition, and each condition was analyzed in three independent experiments. All imaging and analysis was done in a blinded manner.
The Sholl analysis was performed on maximum intensity projections of each image by generating a series of 11 concentric circles of increasing radii (10 μm intervals) centered at the cell body, using the ImageJ (National Institutes of Health (NIH)) plugin Concentric Circles. The number of dendrite crossings at each circle for each image was manually counted (Sholl, 1953), and the number of dendrite crossings at each circle for each transfection condition was calculated by averaging the number of crossings from every image. An experiment was deemed valid if the two control conditions (empty pSuper vector at 33 and 100 ng/well) did not differ significantly. Subsequently, only the 100 ng/well control was used for comparison to other transfection conditions. Significance was determined using SPSS Software to run a repeated-measures ANOVA followed by Tukey's post hoc test for significance at each circle radius.
qPCR for assay of X. laevis gene expression.
X. laevis tadpoles were obtained via natural mating (single adult pairs were induced to mate by human chorionic gonadotropin injection) and maintained in 0.1× Modified Barth's Saline (MBS; 88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 0.3 mm CaNO3, 0.41 mm CaCl2, 0.82 mm Mg, and 15 mm Tris-HCl, pH 7.5) at 18–19°. From before eye development until Stage 46, tadpoles were housed in dark containers and all feeding and care was performed in a dark room under red lighting to keep visual experience to a minimum. At Stage 46, tadpoles were split into two groups: one group was kept in the dark for 4 h, while the other group was placed under a direct bright light for visual stimulation for 4 h. For the nifedipine/APV treatment, a subset of tadpoles were reared and sorted into dark or 4 h of light groups as described above, and kept in 0.1× MBS containing 100 μm APV or 20 μm nifedipine during the 4 h of the experiment. After 4 h, tadpoles from all groups were immediately anesthetized in a bath of 0.02% MS222 (tricaine methanesulfonate salt; Sigma) and then placed on ice. Subsequently, the brain of each tadpole was microdissected and immediately placed in Trizol Reagent (Invitrogen). Note that whole brains were isolated despite the fact that the region of interest is the optic tectum. However, (1) the tectum makes up a large fraction of the tadpoles' brain volume and (2) any observed effects due to visual stimulation would only be diluted by the inclusion of other brain regions. RNA was isolated and used in qPCR as described above.
X. laevis Rem2 sequence was isolated from cDNA clone EXL1051–9541274 (Open Biosystems), using primers based on the predicted X. tropicalis Rem2 sequence (GenBank RefSeq XM_002941509.1), with which it shares >90% nucleotide homology. X. laevis fos (c-Fos; AJ224511.1) and tubb (β-tubulin-NM_001087257) sequences are publically available. qPCR primers (sequences provided above; Table 1) were designed to amplify ∼150 bp toward the 3′ end of the open reading frame, and specificity was verified by PCR before use in qPCR.
Morpholinos and single cell-electroporation.
The 5′ UTR of X. laevis Rem2 was derived from the cDNA described above using the 5′ RACE System (Invitrogen). Fluorescein-tagged morpholinos (MOs) were ordered from GeneTools, using their design tool to identify the sequence to be used for the Rem2-targeted MO (CACTTTCTCGTGCCTGCTTTCAGGT). The efficacy of the MO was verified by Western blot (Fig. 4D) using our homemade antibody against rat Rem2 (Ghiretti and Paradis, 2011), which detects Xenopus Rem2. Two- to four-cell stage X. laevis embryos were microinjected with either the Rem2 MO or a control MO. GeneTools's standard negative control was used as the control MO (CCTCTTACCTCAGTTACAATTTATA). The embryos were allowed to develop for 48 h, at which point whole embryo lysates were prepared from 20 to 25 embryos per condition. Lysates from uninjected or control MO-injected embryos showed robust Rem2 expression, while expression was decreased in lysate from Rem2 MO-injected embryos (Fig. 4D). Thus, the MO is effective at knocking down Rem2 protein expression, and this effect persists for at least 48 h after introduction of the MO.
Figure 4.

Rem2 mRNA expression is activity regulated in the X. laevis optic tectum. A, Fold change in rem2 mRNA expression in whole brain of Stage 46 X. laevis tadpoles raised in minimal light and then kept in the dark for 4 h (black bars) or exposed to a light stimulus for 4 h (gray bars) in the absence (No Treatment; NT) or presence of D/L APV (100 μm) or nifedipine (Nif; 20 μm); n = 3 biological replicates of 30 tadpoles each. rem2 levels were first normalized to tubb levels, then to the Dark NT condition, and presented as mean ± SEM. Significance is *p < 0.01 from Dark NT and #p < 0.01 from Light NT, by one-way ANOVA followed by Dunnett's post hoc test. B, Fold change in fos mRNA expression in whole brain of Stage 46 X. laevis tadpoles raised in minimal light and then kept in the dark (black bars) or exposed to a light stimulus for 4 h (gray bars) in the absence (NT) or presence of D/L APV (100 μm) or nifedipine (20 μm); n = 3 biological replicates of 30 tadpoles each. fos levels were first normalized to tubb levels, then to the Dark NT condition, and presented as mean ± SEM. Significance is *p < 0.01 from Dark NT, #p < 0.01 from Light NT, by one-way ANOVA followed by Dunnett's post hoc test. C, Agarose gel showing qPCR products isolated midway through the linear phase of the qPCRs described in A and B. D, Western blot of whole-embryo lysates showing Rem2 expression in Stage 40 tadpoles uninjected or microinjected 48 h earlier with Control or Rem2 MO. β-tubulin is shown as a loading control.
Albino X. laevis tadpoles were maintained in 0.1× MBS in an 18–19° incubator until Stage 46. The incubator is dark, but not light tight, and tadpoles were exposed to minimal light during feeding and 0.1× MBS changes. At Stage 46, tadpoles were anesthetized in a bath of 0.02% MS222 for 5 min, and then single-cell electroporated (Haas et al., 2002; Bestman et al., 2006; Hewapathirane and Haas, 2008) using an Axoporator 800A (Molecular Devices). The micropipette contained a combination of fluorescein or Alexa Fluor 488 10,000 MW dextrans (Invitrogen) and 5′-carboxyfluorescein tagged Control or Rem2 MOs (final concentrations in the micropipette: 1.5 mm dextran and 0.5 mm MO) in Figures 5–7, or a combination of a GFP-expressing plasmid and a plasmid expressing a myc-tagged Rem2 cDNA (both plasmids were initially 1.5 μg/μl) in Figure 8. MO/dextran combinations were delivered using 1 s trains of bipolar pulses for 7 ms each at 70 Hz, while 1 s trains of negative pulses for 1 ms each at 200 Hz were used to deliver plasmids. A 1–1.5 μA current consistently labeled single tectal neurons.
Figure 5.

Rem2 inhibits baseline dendritic complexity in the X. laevis tadpole optic tectum. A, Representative raw images (2 per condition) of X. laevis tadpole tectal neurons electroporated with fluorescein dextran and a Control MO (left) or Rem2-targeted MO. Scale bar, 50 μm. B, Representative skeletonized images of the 3D reconstructions of the neurons shown in A. Asterisks denote the location of the soma. Scale bar, 50 μm. C, Quantification of the number of dendritic branch tips of tectal neurons 48 h after electroporation. Significance (from Control MO) is **p < 0.01 by univariate ANOVA; n = 60 Control MO neurons and 45 Rem2 MO neurons, from 20 to 30 tadpoles per condition, in three biological replicates. D, Quantification of the total dendritic length of tectal neurons 48 h after electroporation. Significance (from Control MO) is *p < 0.05 by univariate ANOVA; n = 60 Control MO neurons and 45 Rem2 MO neurons, from 20 to 30 tadpoles per condition, in three biological replicates. E, Number of branch tips plotted as cumulative probability; plot is shifted to the right for Rem2 MO neurons compared with Control MO neurons, indicating increased branch tip number; p < 0.001 by Kolmogorov–Smirnov test. F, Total dendritic length plotted as cumulative probability; plot is shifted to the right for Rem2 MO neurons compared with Control MO neurons, indicating increased dendritic length. p < 0.001 by Kolmogorov–Smirnov test.
Figure 6.
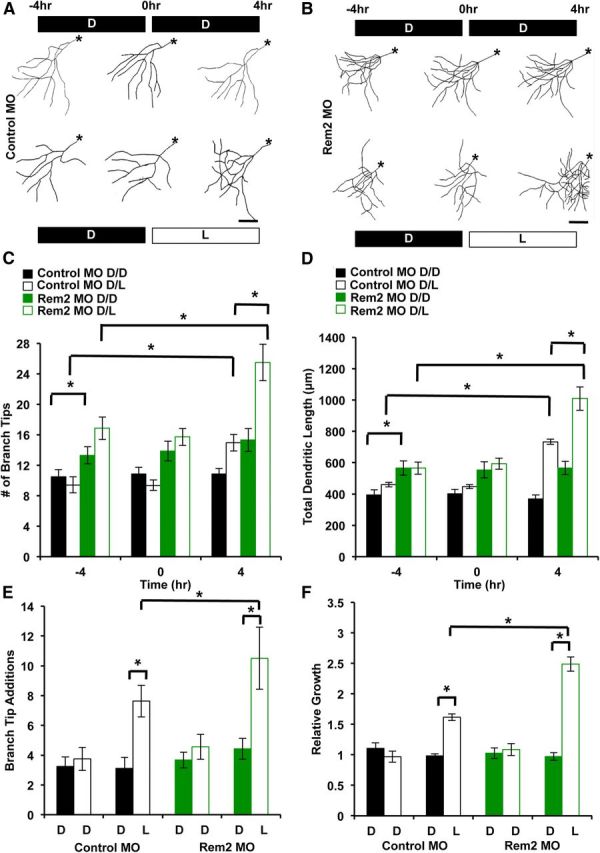
Rem2 is a negative regulator of activity-dependent dendritic complexity in vivo. A, Representative skeletonized images of the 3D reconstructions of X. laevis tadpole tectal neurons coelectroporated with Alexa Fluor 488 dextran and a Control MO. The same neuron was imaged 48 h after electroporation, three times over an 8 h period (at time points denoted as −4, 0, and 4 h). Tadpoles were housed in the dark (D) or under a light stimulus (L) during the periods between imaging as indicated; the top row is D/D, the bottom row is D/L. Asterisks denote the location of the soma. Scale bar, 50 μm. B, Representative skeletonized images of the 3D reconstructions of X. laevis tadpole tectal neurons coelectroporated with Alexa Fluor 488 dextran and a Rem2-targeted MO. The same neuron was imaged 48 h after electroporation, three times over an 8 h period (at time points denoted as −4, 0, and 4 h). Tadpoles were housed in the dark (D) or under a direct light stimulus (L) during the periods between imaging as indicated; the top row is D/D, the bottom row is D/L. Asterisks denote the location of the soma. Scale bar, 50 μm. C, Quantification of the number of dendritic branch tips of tectal neurons 48 h after electroporation, at −4, 0, and 4 h into the experiment. Significance between indicated conditions: *p < 0.05 by two-way ANOVA with Tukey's post hoc test; n ranges from 17 to 26 neurons, from 15 to 25 tadpoles, in three biological replicates, per condition. D, Quantification of the total dendritic length of tectal neurons 48 h after electroporation, at −4, 0, and 4 h into the experiment. Significance between indicated conditions: *p < 0.05 by two-way ANOVA with Tukey's post hoc test; n ranges from 17 to 26 neurons, from 15 to 25 tadpoles, in three biological replicates, per condition. E, Branch tip number (as in C) shown as number of branch tip additions. Branch Tip Additions is the number of new branches that appeared during a given 4 h imaging period. The solid black/green bars are from −4 to 0 h, and the hollow black/green bars are from 0 to 4 h. *p < 0.05 by two-way ANOVA with Tukey's post hoc test. F, Total dendritic length (as in D) shown as relative growth. Relative Growth refers to the total dendritic length added per 4 h imaging period, relative to the initial dendritic length at −4 h. The solid black/green bars are −4 to 0 h, and the hollow black/green bars are 0 to 4 h. *p < 0.05 by two-way ANOVA with Tukey's post hoc test.
Figure 7.

Rem2 mediates activity-dependent changes in dendritic complexity via L-VGCCs but not NMDA receptors. A, Representative skeletonized images of the 3D reconstructions of X. laevis tadpole tectal neurons coelectroporated with Alexa Fluor 488 dextran and a Control MO. The same neuron was imaged 48 h after electroporation, three times over an 8 h period (at time points denoted as −4, 0, and 4 h). Tadpoles were housed in the dark (D) or under a light stimulus (L) during the periods between imaging as indicated. The tadpole solution was untreated (top) or contained 100 μm APV (middle), or 20 μm nifedipine (Nif; bottom). Asterisks denote the location of the soma. Scale bar, 50 μm. B, Representative skeletonized images of the 3D reconstructions of X. laevis tadpole tectal neurons coelectroporated with Alexa Fluor 488 dextran and a Rem2-targeted MO. The same neuron was imaged 48 h after electroporation, three times over an 8 h period (at time points denoted as −4, 0, and 4 h). Tadpoles were housed in the dark (D) or under a light stimulus (L) during the periods between imaging as indicated. The tadpole solution was untreated (top) or contained 100 μm APV (middle), or 20 μm nifedipine (bottom). Asterisks denote the location of the soma. Scale bar, 50 μm. C, Quantification of the number of dendritic branch tips of tectal neurons from tadpoles 48 h after electroporation, shown as number of branch tip additions. Branch Tip Additions is the number of new branches that appeared during a given 4 h imaging period. The solid black/green bars are from −4 to 0 h, and the hollow black/green bars are from 0 to 4 h. Significance between indicated conditions is *p < 0.05 by two-way ANOVA with Tukey's post hoc test; n ranges from 15 to 25 neurons, from 12 to 20 tadpoles, in three biological replicates, per condition. D, Quantification of total dendritic length of tectal neurons from tadpoles 48 h after electroporation, shown as relative growth. Relative Growth refers to the total dendritic length added per 4 h imaging period, relative to the initial dendritic length at −4 h. The solid black/green bars are from −4 to 0 h, and the hollow black/green bars are from 0 to 4 h. Significance between indicated conditions is *p < 0.05 by two-way ANOVA with Tukey's post hoc test; n ranges from 15 to 25 neurons, from 12 to 20 tadpoles, in three biological replicates, per condition.
Figure 8.
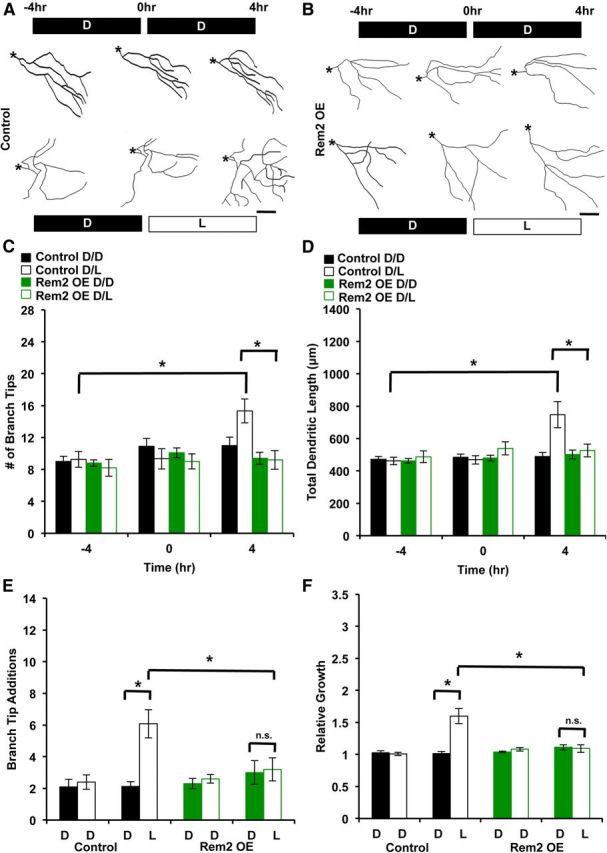
Rem2 OE suppresses activity-dependent dendritic complexity in vivo. A, Representative skeletonized images of the 3D reconstructions of X. laevis tadpole tectal neurons coelectroporated with a GFP-expressing plasmid and an empty vector (Control). The same neuron was imaged 48 h after electroporation, three times over an 8 h period (at time points denoted as −4, 0, and 4 h). Tadpoles were housed in the dark (D) or under a light stimulus (L) during the periods between imaging as indicated: the top row is D/D, the bottom row is D/L. Asterisks denote the location of the soma. Scale bar, 50 μm. B, Representative skeletonized images of the 3D reconstructions of X. laevis tadpole tectal neurons coelectroporated with a GFP-expressing plasmid and a plasmid expressing a Rem2 cDNA (Rem2 OE). The same neuron was imaged 48 h after electroporation, three times over an 8 h period (at time points denoted as −4, 0, and 4 h). Tadpoles were housed in the dark (D) or under a direct light stimulus (L) during the periods between imaging as indicated: the top row is D/D, the bottom row is D/L. Asterisks denote the location of the soma. Scale bar, 50 μm. C, Quantification of the number of dendritic branch tips of tectal neurons 48 h after electroporation, at −4, 0, and 4 h into the experiment. Significance between indicated conditions is *p < 0.05 by two-way ANOVA with Tukey's post hoc test; n ranges from 10 to 15 neurons, from 10 to 15 tadpoles, in three biological replicates, per condition. D, Quantification of the total dendritic length of tectal neurons 48 h after electroporation, at −4, 0, and 4 h into the experiment. Significance between indicated conditions is *p < 0.05 by two-way ANOVA with Tukey's post hoc test; n ranges from 10 to 15 neurons, from 10 to 15 tadpoles, in three biological replicates, per condition. E, Branching (as in C) is shown as number of branch tip additions. Branch Tip Additions is the number of new branches that appeared during a given 4 h imaging period. The solid black/green bars are from −4 to 0 h, and the hollow black/green bars are from 0 to 4 h. *p < 0.05 by two-way ANOVA with Tukey's post hoc test. F, Total dendritic length (as in D) shown as relative growth. Relative Growth refers to the total dendritic length added per 4 h imaging period, relative to the initial dendritic length at −4 h. The solid black/green bars are from −4 to 0 h, and the hollow black/green bars are from 0 to 4 h. *p < 0.05 by two-way ANOVA with Tukey's post hoc test.
Both control and Rem2-targeted MOs contained a 5′-carboxyfluorescein tag to ensure coloading with fluorescent dextrans. The coelectroporation of dextran and fluorescently tagged MO or multiple plasmids has been reported to be 100% (Bestman et al., 2006; Chen et al., 2010), and we verified that the coelectroporation rate in our hands was 98% (35 of 37 neurons) in a subset of experiments in which Alexa Fluor 546 10,000 MW dextran was electroporated with fluorescein-tagged MO (data not shown). After electroporation, tadpoles were returned to fresh 0.1× MBS and recovered within minutes.
Two-photon imaging of baseline effects of Rem2 knockdown in vivo.
Forty-eight hours after electroporation, tadpoles were again anesthetized in 0.02% MS222 and screened for single fluorescent projection neurons under epifluorescence. Projection neurons in the rostral optic tectum were identified by their complex morphology and their long axon that extends out of the tectum. The axon was used for identification purposes only and was not included in subsequent reconstructions. As mature projection neurons have total dendritic length of >400 μm (Wu et al., 1999), projection neurons were excluded if their total dendritic length was less than that. Experiments were performed at Stages 46–48, a time when the vast majority of rostral projection neurons are mature (Wu et al., 1999). Tadpoles with brightly labeled single neurons were placed dorsal side up under an in vivo Prairie Technologies two-photon microscope with Newport/Spectra Physics Mai Tai eHP laser with DeepSee. Neurons were imaged at 780 nm (for Alexa Fluor 488 dextran) or 910 nm (for GFP plasmids) with a 16× (0.8 NA) water-immersion objective and an optical zoom of 4.0 to capture fine details of the entire dendritic arbor. Z-stacks with a 2 μm (Figs. 6–8) or 5 μm (Fig. 5) step size (30–50 sections) were obtained of each neuron. A comparison of images acquired with these step sizes to those with a smaller step size of 0.5 μm did not reveal a significant difference in the magnitude of the changes we observed (data not shown).
Z-stacks were first aligned using the ImageJ (NIH) plugin StackReg, and then semi-automatically reconstructed in three dimensions using the publically available software Neuromantic (http://www.reading.ac.uk/neuromantic/). The software automatically calculates morphological parameters including total dendritic length and number of branch tips. All imaging and analysis were performed by an experimenter blind to the electroporation condition of each tadpole and neuron. The values for four biological replicates were averaged, consisting of 20–30 individual tadpoles and 40–60 labeled neurons total. Statistical significance was determined using a two-way ANOVA followed by Tukey's post hoc test.
Time-lapse imaging of sensory experience-dependent effects on tectal neuron morphology.
Tadpoles were obtained, reared, and single-cell electroporated as described above. Note that the X. laevis tadpole visual system becomes functional at approximately Stage 39/40, and responds only to dramatic on/off stimuli before Stage 45 (Zhang et al., 2000). After screening for Stage 46 tadpoles with brightly labeled single neurons, 48 h after electroporation, successfully labeled tadpoles were placed into two groups for an 8 h experimental paradigm, as developed by Sin et al. (2002). One group was housed entirely in a dark container and kept in minimal light for the entire 8 h of the experiment (D/D). The other group was maintained in a separate dark container for the first 4 h, then moved under direct, bright light stimulation for the second 4 h of the experiment (D/L). It has been previously demonstrated that 4 h of light is sufficient to observe a robust acute, activity-dependent morphological change (Sin et al., 2002).
The same neurons were imaged at 0, 4, and 8 h (corresponding to the −4, 0, and 4 h time points in Figs. 6–8) into the experiment, and individual tadpoles could be repeatedly identified by being placed in separate wells of a 12-well plate within the storage container between imaging sessions. Tadpoles were anesthetized in 0.02% MS222 before each 8 h imaging session, and allowed to recover and behave ad libitum in fresh 0.1× MBS during the periods between imaging sessions. In a subset of experiments (Fig. 7), tadpoles were separated into three additional groups: the 0.1× MBS was untreated for one group, or contained either 20 μm nifedipine or 100 μm APV during the 8 h imaging session.
The imaging parameters, reconstruction, and analyses were performed as described above by an experimenter blind to the electroporation and visual experience conditions (Control MO vs Rem2 MO, Control vs Rem2 OE). The values for three biological replicates were averaged. Statistical significance between conditions of interest was determined using a two-way ANOVA.
Statistics.
Significance was determined using SPSS Software to run a repeated-measures ANOVA (within-subjects: circle radii; between-subjects: transfection condition and experiment date) followed by Tukey's post hoc test for significance at each circle radius for Sholl analysis, or two-way ANOVA between conditions of interest for in vivo experiments. A Kolmogorov–Smirnov test was used to determine significance of cumulative probability plot shifts in Figure 5E–F. For clarity and illustrative purposes, some datasets are plotted in more than one graph within a single figure, with the duplicate conditions indicated in the figure legends. These are Figure 2, C and D (Mock and RNAi conditions), and Figure 3, C and D (Mock and RNAi conditions). In every instance, at least three experiments were performed in parallel and each experiment contained every condition. In addition, the data represented in these figures were compared as a single dataset for statistical analysis, not individually for the conditions represented on each graph.
Figure 2.

Rem2 is a negative regulator of depolarization-induced dendritic outgrowth. A, Representative images of 5 DIV cortical neurons mock transfected with a GFP-expressing plasmid alone or neurons transfected with GFP plus a Rem2 shRNA, untreated (A1, A2) or treated for 6 h with 55 mm KCl alone (A3, A4), 20 μm nifedipine alone (Nif; A5, A6), or in combination (A7, A8). Scale bars, 50 μm. B, Quantification (via Sholl analysis) of dendritic complexity of 5 DIV mock transfected and Rem2 RNAi neurons with or without 55 mm KCl treatment for 6 h. In this and all parts, a bar above or below the curve indicates significance at particular radii compared with Mock, with *p < 0.05 and **p < 0.01 by repeated-measures ANOVA with Tukey's post hoc test. Bars are colored or dashed to match the style/color of the data curve to which they refer; n ranges from 30 to 46 neurons per condition. For clarity, for this part and throughout the rest of Figures 2 and 3, additional significant comparisons are indicated in the legend. RNAi + KCl is also significant (p < 0.05) from both Mock + KCl and RNAi alone at 20–40 μm. C, Quantification (via Sholl analysis) of dendritic complexity of 5 DIV mock transfected and Rem2 RNAi neurons compared with the same conditions treated with 20 μm nifedipine for 6 h. *p < 0.05 from Mock by repeated-measures ANOVA with Tukey's post hoc test; n ranges from 20 to 46 neurons per condition. RNAi + Nif is significant (p < 0.05) from RNAi alone (at 20–40 μm). In this part, Mock and RNAi data from B are replotted for ease of comparison to other conditions. D, Quantification (via Sholl analysis) of dendritic complexity of 5 DIV mock transfected and Rem2 RNAi neurons compared with the same conditions treated with 55 mm KCl and 20 μm nifedipine for 6 h. *p < 0.05 from Mock by repeated-measures ANOVA with Tukey's post hoc test; n ranges from 20 to 46 neurons per condition. RNAi + Nif + KCl is significant (p < 0.05) from RNAi alone (at 20–40 μm), but is not significant from Mock + Nif + KCl. In this part, Mock and RNAi data from B are replotted for ease of comparison to other conditions.
Figure 3.

Rem2 OE suppresses depolarization-induced changes in dendritic morphology. A, Representative images of 5 DIV cortical neurons mock transfected with a GFP-expressing plasmid alone or neurons transfected with GFP plus a plasmid expressing a Rem2 cDNA, untreated (A1, A2) or treated for 6 h with 55 mm KCl alone (A3, A4), 20 μm nifedipine alone (Nif; A5, A6), or in combination (A7, A8). Scale bars, 50 μm. B, Quantification (via Sholl analysis) of dendritic complexity of 5 DIV mock transfected and Rem2 OE neurons with or without 55 mm KCl treatment for 6 h. In this and all parts, a bar above or below the curve indicates significance at particular radii compared with Mock, with *p < 0.05 by repeated-measures ANOVA with Tukey's post hoc test. Bars are colored or dashed to match the style/color of the data curve to which they refer; n ranges from 36 to 52 neurons per condition. For clarity, OE + KCl is not significantly different from Mock or OE, but is significant (p < 0.05) from Mock + KCl (at 20–80 μm). C, Quantification (via Sholl analysis) of dendritic complexity of 5 DIV mock transfected and Rem2 OE neurons compared with the same conditions treated with 20 μm nifedipine for 6 h. *p < 0.05 from Mock by repeated-measures ANOVA with Tukey's post hoc test; n ranges from 36 to 52 neurons per condition. Mock + Nif and OE + Nif are also significant (p < 0.05) from OE (at 20–80 μm). In this part, Mock and RNAi data from B are replotted for ease of comparison to other conditions. D, Quantification (via Sholl analysis) of dendritic complexity of 5 DIV mock transfected and Rem2 OE neurons compared with the same conditions treated with 55 mm KCl and 20 μm nifedipine for 6 h. No conditions were significantly different from Mock; n ranges from 36 to 52 neurons per condition. In this part, Mock and RNAi data from B are replotted for ease of comparison to other conditions.
Results
Rem2 is an activity-regulated gene responsive to calcium influx through L-VGCCs
To determine the precise timing of depolarization-dependent regulation of Rem2 mRNA expression, we performed quantitative real-time PCR to measure the levels of Rem2 mRNA in 8 DIV primary cortical cultures that had been depolarized with high extracellular potassium for the indicated times (Fig. 1A, top). Rem2 mRNA expression was upregulated after as little as 1 h of treatment, and remained high after 2–6 h of treatment (Fig. 1A, top); the mRNA expression of the known activity-regulated gene Fos (Morgan and Curran, 1986) was also increased by 55 mm KCl stimulation over this time course (Fig. 1A, bottom).
We next sought to determine the mechanism(s) through which neuronal depolarization upregulates Rem2 mRNA expression. Importantly, the upregulation of Rem2 mRNA expression following 55 mm KCl treatment was suppressed by treatment with the calcium chelator EGTA (5 mm; Fig. 1B, top), as similarly seen with Fos mRNA expression (Fig. 1B, bottom), suggesting a calcium-dependent mechanism. As there are multiple routes of calcium entry into the neuron, we sought to determine which mode of calcium entry influences Rem2 transcription. We found that the KCl-induced upregulation of Rem2 mRNA expression was blocked by the L-VGCC antagonist nifedipine (10 μm), but not by ryanodine (10 μm) (Fig. 1B, top), which blocks the release of calcium from intracellular stores by antagonizing ryanodine receptors on the endoplasmic reticulum (Berridge, 1998). In addition, the depolarization-induced upregulation of Rem2 mRNA expression was not blocked by the simultaneous addition of APV, an NMDA receptor antagonist, and DNQX, an AMPA receptor antagonist (Fig. 1B, top).
To determine whether Rem2 mRNA expression is sensitive to calcium influx through NMDA receptors, we treated 5 DIV primary cortical cultures with 1 μm TTX, and then washed the neurons with fresh media at 7 DIV to remove the TTX. This “TTX withdrawal” paradigm has been previously demonstrated to cause an increase in action potential firing and synaptic glutamate release that is sufficient to activate NMDA receptor-induced gene transcription (Rao et al., 2006). We found that 6 h of TTX withdrawal did not significantly alter Rem2 mRNA expression (Fig. 1C, top), suggesting that Rem2 transcription is not regulated by calcium entry through NMDA-type glutamate receptors. As expected, treatment of these cultures with D/L APV (100 μm) or the L-VGCC blocker nimodipine (5 μm) had no effect on Rem2 mRNA expression in the TTX withdrawal paradigm (Fig. 1C, top). However, following KCl-mediated depolarization, nimodipine treatment effectively suppressed the KCl-dependent upregulation of Rem2 mRNA, while APV still had no effect (Fig. 1D, top).
As a positive control, we measured expression of Fos, which is known to be upregulated by calcium entry through both NMDA receptors and L-VGCCs (Morgan and Curran, 1986; Zhang et al., 2007; Saha et al., 2011), and found that Fos expression was significantly enhanced following TTX withdrawal (Fig. 1C, bottom). Further, treatment of these cultures with APV during the period of TTX withdrawal fully suppressed the upregulation of Fos, while treatment with nimodipine had no effect (Fig. 1C, bottom). These data validate that TTX withdrawal activates gene transcription via calcium entry through NMDA receptors, as this paradigm induces Fos expression in an NMDA receptor-dependent but not L-VGCC-dependent manner. In contrast, both NMDA receptor blockade and nimodipine treatment effectively suppressed the KCl-dependent upregulation of Fos mRNA (Fig. 1D, bottom), demonstrating the efficacy of the antagonist treatments. Overall, we conclude from these data that Rem2 transcription is selectively activated by calcium entry through L-VGCCs, and is not induced by calcium influx through NMDA receptors.
Neuronal depolarization activates both positive and negative regulators of dendritic complexity
A rise in intracellular calcium associated with increased neuronal activity enhances the complexity of the dendritic arbor of neurons both in vitro and in vivo (Redmond et al., 2002; Sin et al., 2002; Vaillant et al., 2002; Wong and Ghosh, 2002; Wayman et al., 2006; Flavell and Greenberg, 2008). Given that Rem2 is both upregulated by activity and, functionally, a negative regulator of dendritic complexity (Ghiretti and Paradis, 2011; Ghiretti et al., 2013), we reasoned that a calcium-mediated increase in Rem2 expression might serve to restrict an activity-dependent increase in dendritic arborization. To directly test this hypothesis, we depolarized neurons alone or in combination with RNAi-mediated knockdown of endogenous Rem2 or overexpression (OE) of a Rem2 cDNA in primary cortical cultures and assayed the net effect on dendritic morphology. The shRNA against Rem2 and the Rem2 cDNA have been extensively validated (Ghiretti and Paradis, 2011; Ghiretti et al., 2013; Moore et al., 2013). In addition, recapitulation of the Rem2 RNAi dendritic complexity phenotype by morpholino mediated Rem2 knock-out in X. laevis (Fig. 5) further substantiates our RNAi-based findings in cultured neurons.
We transfected and cultured cortical neurons at 2 DIV with a GFP-expressing plasmid and an empty vector plasmid (Mock), a plasmid expressing an shRNA against Rem2 (Rem2 RNAi; Fig. 2), or a plasmid expressing a Rem2 cDNA (OE; Fig. 3). At 5 DIV, we depolarized these neurons by treating with 55 mm KCl for 6 h (Figs. 2A3,A4, 3A3,A4), and/or treated the neurons with nifedipine (Figs. 2A5–A8, 3A5–A8), which has been shown to suppress the effects of depolarization on dendritic complexity (Redmond et al., 2002; Wayman et al., 2004). Subsequently, we fixed and imaged neurons and assessed dendritic complexity via Sholl analysis, which involves superimposing a series of concentric circles of increasing diameter, centered at the soma, on a low-magnification image of a neuron and quantifying the number of dendrites that intersect these circles (Sholl, 1953).
As previous studies of KCl-induced changes in dendritic morphology were performed at 5 DIV (Redmond et al., 2002; Wayman et al., 2004), we first established that the Rem2 RNAi-induced increase in complexity we observed and rescued previously at 14 DIV (Ghiretti and Paradis, 2011) was also apparent at 5 DIV (Fig. 2A2, quantified in B; Ghiretti et al., 2013). Next, we observed that mock transfected neurons treated for 6 h with 55 mm KCl showed a robust increase in complexity compared with untreated neurons (Fig. 2, compare A1 to A3, quantified in B), a phenotype that is consistent with previous findings (Redmond et al., 2002; Fink et al., 2003; Wayman et al., 2004). This result indicates that the net effect of neuronal depolarization is an increase in dendritic complexity. Interestingly, we observed that KCl treatment in combination with RNAi-mediated knockdown of Rem2 resulted in neurons that were more complex than either condition alone (Fig. 2, compare A2, A3 to A4, quantified in B). This suggests that under normal conditions, the increase in complexity following KCl treatment is limited by a simultaneous, depolarization-dependent increase in Rem2 expression that restricts complexity. Thus, when Rem2 levels are sufficiently reduced by RNAi, the KCl-dependent increase in dendritic complexity is even more robust due to loss of a negative regulator, Rem2.
Given that calcium influx through L-VGCCs regulates Rem2 expression (Fig. 1), we next treated neurons with nifedipine (20 μm) and assessed dendritic complexity. We found that nifedipine treatment led to a decrease in dendritic complexity (Fig. 2, compare A1 to A5, quantified in C), in agreement with previous studies (Redmond et al., 2002; Fink et al., 2003). Importantly, treatment of Rem2 RNAi neurons with nifedipine produced the same phenotype as in mock transfected neurons: a decrease in dendritic complexity (Fig. 2, compare A2 to A6, quantified in C). Additionally, nifedipine treatment suppressed the KCl-induced increase in complexity (Fig. 2, compare A3 to A7, quantified in D), demonstrating that calcium entry through L-VGCCs mediates the majority of the neuronal depolarization-dependent effect on dendritic morphology in our culture system. Nifedipine treatment also blocked the additive increase in complexity observed when Rem2 RNAi neurons were treated with KCl (Fig. 2, compare A4 to A8, quantified in D). Together, these results reveal that the increase in complexity observed with Rem2 knockdown is due in large part to the increased activation of a parallel, calcium-dependent pathway(s) that promotes complexity.
Interestingly, we have not observed an effect of Rem2 OE on dendritic complexity to date (Ghiretti and Paradis, 2011; Fig. 3A2). We hypothesized that this might be due to the insufficiency of overexpression of Rem2 in the absence of the activation of additional activity-regulated signaling pathways. For example, a simultaneous increase in activity (such as that provided by KCl-induced depolarization) might be required to initiate signaling pathways upstream of Rem2 that are required for its function, such as the activation of CaMKII (Ghiretti et al., 2013). In support of this hypothesis, neurons that were simultaneously treated with KCl and overexpressing Rem2 did not display as robust an increase in dendritic complexity as that observed with KCl treatment alone (Fig. 3, compare A3 to A4, quantified in B). These results indicate that Rem2 OE is sufficient to negatively regulate dendritic complexity, but only in the context of increased neuronal activity.
Further, treatment of Rem2 OE neurons with nifedipine led to a decrease in complexity similar to that observed with nifedipine treatment alone (Fig. 3, compare A5 to A6, quantified in C). We also observed that cotreatment of either mock or Rem2 OE transfected neurons with KCl and nifedipine resulted in a phenotype that was not significantly different from untreated neurons (Fig. 3, compare A7, A8 to A1, quantified in D), Together, these results suggest calcium entry through L-VGCCs regulates both positive and negative mediators of dendritic complexity. Further, Rem2 represents a novel signaling mechanism through which neurons respond to depolarization by restricting the growth of their dendritic arbor.
Rem2 transcription is upregulated in response to neuronal activity in an intact circuit
A number of studies have demonstrated robust changes in visual system synaptic plasticity, function, and morphology, dendritic complexity, and cortical structure (i.e., columns) in response to changes in visual experience (Wong and Ghosh, 2002; Fox and Wong, 2005). The X. laevis retinotectal circuit is a well established and powerful tool for studying activity-regulated changes in dendritic morphology in vivo for the following reasons: the relative ease of gene knockdown by use of morpholino oligos, which block translation of targeted mRNAs; the accessibility of labeled tectal neurons to in vivo imaging; and robust changes in neuronal connections in response to changes in neuronal activity. In particular, the dendritic complexity of optic tectum neurons in X. laevis tadpoles exposed to a visual stimulus has been demonstrated to be enhanced in an activity-dependent manner (Sin et al., 2002). Thus, we chose to use the X. laevis visual system to ask whether Rem2 mediates activity-dependent changes in dendritic morphology in vivo.
To begin, we asked whether Rem2 is expressed in the brain of X. laevis tadpoles, and further, if rem2 transcription is also upregulated by neuronal activity as we observed in cultures of rat cortical neurons (Fig. 1). Tadpoles were reared in minimal light from before functional eye development until Stage 46, at which point they were split into two groups. One group was kept in minimal light, while the other was exposed to a direct bright light stimulus for 4 h before microdissection of the tadpole brain, a large portion of which is optic tectum. We observed that rem2 mRNA expression was significantly upregulated in the light-exposed group (NT; Fig. 4A,C), and the mRNA expression of the known activity-regulated gene fos was affected similarly under this paradigm (NT; Fig. 4B,C). Based on these results, we conclude that Rem2 is expressed in the X. laevis tadpole brain and further, its mRNA expression is rapidly upregulated in response to visual experience, indicating that rem2 mRNA expression is regulated by physiologically relevant stimuli in vivo.
To uncover the mode of calcium entry through which visual experience increases rem2 mRNA expression in vivo, tadpoles (both dark and light exposed) were further divided into three additional groups during the experiment: they were left untreated, or treated with APV (100 μm) or nifedipine (20 μm). We found that the upregulation of rem2 mRNA expression following light stimulation was robustly suppressed by nifedipine treatment, and partially reduced in magnitude by APV treatment (Fig. 4A,C). In contrast, the upregulation of fos mRNA was efficiently and robustly suppressed by both APV and nifedipine (Fig. 4B,C). Consistent with our results from cultured cortical neurons (Fig. 1C,D), these data indicate that visual experience upregulates rem2 mRNA expression primarily via increased calcium influx through L-VGCCs.
Rem2 restricts dendritic complexity in vivo in response to sensory experience
Next, we asked whether Rem2 mediates the baseline dendritic complexity of tectal neurons in vivo, using a MO to knock down Rem2 expression in the optic tectum of tadpoles that had been reared under minimal light conditions. We microinjected one- and two-cell stage embryos with the Rem2 MO to assess the amount of Rem2 knockdown obtained with the MO and further, whether Rem2 knockdown affected tadpole development. The efficacy of the MO was confirmed by Western blotting of lysates of embryos 48 h after injection (Fig. 4D). In addition, we did not observe any gross morphological defects in the nervous system or tadpole development (as assayed by normal swimming behavior and light microscopy of intact tadpoles through Stage 40; data not shown).
We used single-cell coelectroporation (Haas et al., 2002; Bestman et al., 2006; Hewapathirane and Haas, 2008) of Alexa Fluor 488 dextran dye (to visualize the cells), and either a fluorescein-tagged control MO, or a fluorescein-tagged MO targeting Rem2 (to decrease Rem2 protein expression in fluorescently labeled cells) to label individual neurons within the optic tectum of Stage 46 X. laevis tadpoles. We found that the coelectroporation efficiency of fluorescent dextran and MO was 98% (35 of 37 tadpoles; data not shown; see Materials and Methods) using spectrally separable fluorophores to fill the neuron and tag the MO. After 48 h, two-photon images of single cells were acquired, reconstructed in three dimensions, and the dendritic morphology (both total dendritic length and number of branch tips) was assessed. Tectal neurons that had been electroporated with the Rem2 MO showed a robust increase in both total dendritic length and number of branch tips, leading to an overall increase in dendritic complexity compared with neurons electroporated with the Control MO (Fig. 5). This experiment demonstrates that Rem2 normally functions to restrict the dendritic arbor in vivo. However, as these animals were reared under minimal lighting conditions, this experiment fails to distinguish between a function for Rem2 that may be constitutively expressed versus a function for Rem2 that is expressed specifically in response to increased neuronal activity.
To distinguish between these two possibilities, we tested the hypothesis that Rem2 acts as an activity-dependent, negative regulator of dendritic morphology in vivo using the following experimental paradigm. Stage 46 tadpoles that had been reared in minimal light were electroporated as described above with either Control MO or Rem2 MO, and after an additional 48 h of growth in minimal light, were split into two groups for time-lapse, in vivo two-photon imaging of single-labeled neurons (Sin et al., 2002). Images of the same cell were acquired at the beginning (Fig. 6A,B, −4 h), the halfway point (Fig. 6A,B, 0 h), and the end (Fig. 6A,B, 4 h) of the experiment. A subset of both the Control MO and Rem2 MO-electroporated tadpoles was kept in minimal light during the entire 8 h of the experiment (Fig. 6, D/D groups), or kept in minimal light for 4 h and then exposed to a bright, direct light for visual stimulation for 4 h (Fig. 6, D/L groups). The tadpoles underwent normal swimming behavior between imaging sessions, independent of their visual stimulation status. In this paradigm, a comparison between the −4 and 0 h time points establishes the basal rate of change of the dendritic arbor in the absence of visual stimulus, and then directly tests the effect of sensory experience (in the form of light exposure) on dendritic complexity by a comparison of the 0–4 h time points.
As previously shown in Figure 5, we again found that the MO-mediated decrease in Rem2 expression led to an increase in baseline dendritic complexity (Fig. 6, compare −4 h Control MO and −4 h Rem2 MO). Also, there was a negligible change in this initial complexity of either Control or Rem2 MO-electroporated neurons from tadpoles kept in minimal light for the entire 8 h experiment (Fig. 6, compare −4 and 4 h time points for either Control MO D/D or Rem2 MO D/D). However, consistent with previous results (Sin et al., 2002), we found that 4 h of visual stimulation during the experiment caused a robust increase in the complexity of the dendritic arbor (Fig. 6, at 4 h time point compare Control MO D/D and Control MO D/L), suggesting that the net effect of experience is to increase dendritic complexity.
Interestingly, Rem2 knockdown in combination with visual stimulation led to an even greater increase in dendritic complexity (Fig. 6, at 4 h time point compare Control MO D/L and Rem2 MO D/L). Specifically, a comparison between conditions at the 4 h time point revealed an approximate 30% increase in dendrite length when comparing Control MO D/D versus Control MO D/L (Fig. 6). In contrast, a comparison between the Rem2 MO D/D versus Rem2 MO D/L conditions at 4 h revealed an approximate 60% increase in dendrite length.
We sought to determine whether the increased baseline dendritic complexity observed with Rem2 MO confounded our ability to interpret light-induced effects on dendritic complexity in neurons in which Rem2 expression was decreased. To accomplish this goal, we reanalyzed our raw data to directly quantify the change in total dendritic length and number of branch tips during each 4 h period of the experiment. We regraphed these morphology parameters as Relative Growth and Branch Tip Additions (Fig. 6E,F). This alternate representation of the data confirmed the significant differences we observed with quantification of the raw data (Fig. 6C,D). This result is consistent with a model in which neuronal activity triggers signaling through opposing yet distinct pathways: a Rem2-dependent pathway that functions to restrict dendritic complexity and other, unknown pathway(s) that promote dendritic complexity (Fig. 9).
Figure 9.

Model of signaling pathways that mediate activity-dependent changes in dendritic complexity. Depolarization activates Rem2 signaling in at least two ways: (1) calcium entry through L-VGCCs triggers the transcription of Rem2, and (2) calcium activates CaMKII signaling. This Rem2/CaMKII signaling pathway negatively regulates dendritic complexity (Ghiretti et al., 2013 and this manuscript). Calcium entry through L-VGCCs also activates signal transduction pathway(s) that positively regulate dendritic complexity (“X”) in parallel to Rem2. Thus, the net effect on dendritic morphology is due to the combination of these signaling pathways. Note that a role for constitutively expressed Rem2 in mediating dendritic complexity is not excluded by our results.
Calcium entry through L-VGCCs is required to activate signaling pathways in parallel to Rem2 that promote dendritic complexity
Activity-dependent changes in neuronal structure and function are dependent on calcium entry from two primary sources: L-VGCCs, which open in response to sustained and global cellular depolarization, and NMDA receptors that allow calcium influx to occur at glutamatergic synapses (Bading et al., 1993; Lyons and West, 2011). Activity-regulated genes that mediate changes in neuronal morphology and function may be upregulated by calcium from one or both of these sources (Lerea et al., 1992; Bading et al., 1993). Therefore, we next sought to determine the mode(s) of calcium entry that influenced changes in dendritic morphology observed upon Rem2 knockdown in the presence of visual stimulation in X. laevis tadpole optic tectum. To address this question, we repeated the D/L experiments using tadpoles that had been electroporated 48 h before the D/L experiment with Control or Rem2 MOs as described above, but added either 20 μm nifedipine or 100 μm D/L APV to the 0.1× MBS solution containing the tadpoles during the 8 h imaging session (Fig. 7). We did not include a D/D condition in this experiment, as we were most interested in the effects of nifedipine or APV on the Rem2 and experience-dependent increases in dendritic complexity.
We began by determining the contribution of calcium entry through NMDA receptors to experience-dependent and Rem2-dependent dendritic complexity in tectal neurons. We observed that APV treatment completely suppressed the experience-dependent increase in dendritic complexity observed with Control MO-electroporated neurons (Fig. 7A, compare Untreated to APV at 4 h, quantified in C and D), in agreement with previous work (Sin et al., 2002). These data demonstrate that signaling molecules that promote dendritic arbor growth in response to an acute, 4 h light stimulus can be triggered by calcium entry through NMDA receptors.
Our previous experiments demonstrate that upon Rem2 knockdown for 48 h under minimal light conditions, we observe an increase in baseline dendritic complexity that is further increased by an acute, 4 h light stimulation (Figs. 5, 6). Interestingly, we found that APV treatment in the presence of Rem2 MO failed to suppress this increase in baseline dendritic complexity that occurs as a result of Rem2 knockdown (Fig. 7B, compare Untreated 4 h to APV −44 h, quantified in C and D). However, similar to neurons in the Control MO condition, we found that APV treatment of Rem2 MO-electroporated neurons suppressed the increased dendritic complexity observed upon light stimulation (Fig. 7B, compare Untreated to APV at 4 h, quantified in C and D). The fact that APV treatment can suppress the increased complexity observed with 4 h of light stimulation but not as a result of Rem2 knockdown suggests that different signal transduction pathways mediate the increase in complexity observed upon Rem2 knockdown versus light stimulation.
We next examined the contribution of calcium entry through L-VGCCs to experience-dependent and Rem2-dependent dendritic complexity. First, we asked whether, as in our cultured cortical neurons (Fig. 2), nifedipine treatment was able to block the increase in dendritic complexity that occurs in response to neuronal activity. We observed that nifedipine completely suppressed the experience-dependent increase in dendritic complexity observed with Control MO-electroporated neurons (Fig. 7A, compare Untreated to Nif at 4 h, quantified in C and D). These results demonstrate that experience-dependent changes in dendritic morphology can be regulated by L-VGCC-mediated calcium entry.
Interestingly, we found that nifedipine treatment also decreased the dendritic complexity of Rem2 MO-electroporated neurons following 4 h of visual experience (Fig. 7B, compare Untreated to Nif at 4 h, quantified in C and D), such that the morphology of the Rem2 MO neurons resembled that of Control MO neurons (Fig. 7, compare B, Nif 4 h, A, Untreated at −4 h). Thus, nifedipine treatment blocks the activation of pathways that are calcium dependent, promote increased complexity, and normally oppose Rem2 signaling. Quantification of these data reveals a negative value for branch tip additions compared with Rem2 MO neurons that were in the dark for 4 h (Fig. 7C) and further, a significant decrease in relative growth (Fig. 7D). Therefore, not only are signaling pathways that promote light-dependent dendritic complexity blocked in the presence of nifedipine, but the presence of pathways that promote dendrite retractions is revealed in this condition.
A straightforward interpretation of these data is that the reduction of Rem2 for 48 h under minimal light (i.e., Rem2 MO) allows activity-dependent pathway(s) that promote dendritic complexity to signal unopposed by Rem2, leading to an increase in complexity. Because nifedipine but not APV blocks this dendritic growth (Fig. 7B), we conclude that these parallel, growth-promoting signal transduction pathway(s) are triggered primarily by calcium entry through L-VGCCs, similar to our observations using Rem2 RNAi in cultured neurons (Fig. 2). These results are also consistent with the specificity of activity-dependent Rem2 transcription for calcium entry through L-VGCCS (Figs. 2, 4). In contrast, acute visual experience (i.e., 4 h of light stimulation) triggers both L-VGCC- and NMDA-dependent growth-promoting signal transduction pathways (Fig. 7A,B). It is entirely possible that the L-VGCC-dependent signals that respond to a short visual experience are the same as those that are revealed by a 48 h Rem2 knockdown. However, our data suggest that the NMDA-dependent signaling pathways that promote dendritic complexity are not normally opposed by Rem2 signaling (Fig. 7C,D, compare Rem2 MO + APV with Rem2 MO + Nif).
Rem2 OE suppresses experience-dependent dendritic outgrowth
We next investigated the effect of Rem2 OE in vivo, using our visual stimulation paradigm. We electroporated tadpoles with either a GFP-expressing plasmid and an empty vector (Fig. 8, Control) or both a GFP-expressing plasmid and a plasmid expressing a myc-tagged Rem2 cDNA (Fig. 8, Rem2 OE); 48 h later, the tadpoles were exposed to the same D/L paradigm described above. Interestingly, and similar to our results with cultured neurons (Fig. 3), Rem2 OE had no effect on baseline dendritic morphology (Fig. 8, compare −4 h Control D/D and −4 h Rem2 OE D/D). However, Rem2 OE completely suppressed the experience-induced increase in both total dendritic length and number of branch tips (Fig. 8, compare 4 h Control D/L and 4 h Rem2 OE D/L), again similar to our results in cultured neurons (Fig. 3). This finding suggests that there exists an activity-dependent process, such as CaMKII-mediated phosphorylation of Rem2 (Ghiretti et al., 2013), which is required in conjunction with Rem2 OE for the suppression of dendritic complexity. Further, these data confirm the function of Rem2 as an activity-dependent negative regulator of dendritic morphology in vivo. Together, these results demonstrate that a balance between positive and negative regulation of activity-dependent dendritic complexity occurs to shape the final dendritic arbor of a neuron.
Discussion
In this study, we define Rem2 as a critical regulator of activity-dependent dendritic complexity, as dialing Rem2 expression up or down, in the context of increased sensory experience, decreases or increases dendritic complexity, respectively. This function of Rem2 represents a conserved mechanism of activity-dependent negative regulation of dendritic morphology across vertebrate species, from X. laevis to rodent models. Further, we demonstrate, using these two different experimental paradigms, that neuronal activity simultaneously triggers signaling pathways that both positively and negatively regulate dendritic complexity. Why would an individual neuron concurrently promote and inhibit dendritic outgrowth in response to changes in neuronal activity? Presumably, mechanisms of gene expression in the nervous system have evolved such that both positive and negative regulators of dendritic arborization are expressed to ensure that growth-promoting processes do not proceed unchecked. This type of regulation is reminiscent of diverse biological processes ranging from apoptosis (Wang et al., 1994; Yan and Shi, 2005) to the function of T-cells in the immune system (Singer and Koretzky, 2002), where a balance between positive and negative signal transduction networks achieves the final outcome.
In general, the vast majority of studies examining structural plasticity of neurons have addressed the growth-promoting effects of increased neuronal activity on dendritic complexity both in vitro and in vivo (Redmond et al., 2002; Sin et al., 2002; Vaillant et al., 2002; Fink et al., 2003; Wayman et al., 2006). In contrast, only a few studies have explored the growth-restricting effects of neuronal depolarization. For example, expression of a constitutively active form of the Rho GTPase blocks the light-induced increase in dendritic complexity observed in the X. laevis tadpole tectum (Sin et al., 2002). Further, expression of constitutively active CaMKII in X. laevis tectal neurons restricts dendritic growth (Wu and Cline, 1998), while a loss-of-function study of the activity-regulated MEF2 transcription factor also revealed a role for MEF2 in suppression of dendrite filopodial dynamics (Chen et al., 2012). Last, knock-out of the NR2B subunit of the NMDA receptor in the rodent CNS increases dendritic arborization in certain neuronal subtypes (Lee et al., 2005; Espinosa et al., 2009). These results imply the existence of signaling pathways, such as the one defined by Rem2 in this study, which are responsive to sensory experience yet function to inhibit dendritic complexity.
Overall, our findings suggest the following model (Fig. 9). Neuronal depolarization induces calcium entry through L-VGCCs, which in turn activates signal transduction pathways, such as the one defined by CaMKII (Ghiretti et al., 2013), and increases transcription of activity-regulated genes such as Rem2. Rem2 functions to inhibit dendritic outgrowth, while other signaling molecules function in a parallel pathway to Rem2 to promote dendritic arborization in response to increased neuronal activity and calcium influx through L-VGCCs. Our data demonstrate that while the net effect of neuronal activity is an increase in dendritic complexity, this increase is in fact the result of the simultaneous activation of both positive and negative regulators of the dendritic arbor. Thus, in situations in which Rem2 expression has been decreased and activity increased (Figs. 2, 6), we observe a robust increase in dendritic complexity because we have eliminated an activity-dependent, negative regulator of outgrowth (i.e., Rem2). Conversely, overexpression of Rem2, in the context of increased neuronal activity, is able to suppress activity-dependent increases in dendritic complexity (Figs. 3, 8).
Activity-regulated genes are essential mediators of a variety of neuronal processes, including dendritic outgrowth, synapse formation, and synaptic plasticity. Because of the direct nature of their activation in response to sensory experience, these genes are well poised to rapidly translate changes in neuronal activity into cell autonomous structural changes in individual neurons (Flavell and Greenberg, 2008; Leslie and Nedivi, 2011; West and Greenberg, 2011). To date, much research has focused on activity-regulated genes that act as positive regulators of dendritic morphology (McAllister et al., 1995; Nedivi et al., 1998; Zhou et al., 2006; Hong et al., 2008). In addition, with respect to Bdnf and cpg15, two well studied examples of activity-regulated gene products that regulate dendritic morphology, loss-of-function studies have demonstrated decreased dendritic arborization (Xu et al., 2000; Fujino et al., 2011); however, these studies have not been performed in the context of altered neuronal activity. In fact, a loss-of-function analysis specifically of the activity-regulated Bdnf gene product revealed no effect on dendritic morphology (Hong et al., 2008). Thus, Rem2 is among a small group of activity-regulated genes to be characterized in the context of modulation of sensory experience and further, as negative regulators of dendritic complexity.
Our results indicate that Rem2 mRNA expression is selectively activated by calcium influx through L-VGCCs compared with NMDA-type glutamate receptors both in vitro and in vivo (Figs. 1, 4, 7). While a sustained depolarization of neurons with KCl (Fig. 1) is not representative of the physiological state of cortical neurons in an intact circuit, the fact that Rem2 transcription is rapidly upregulated in response to visual stimulation in vivo (see Fig. 4) demonstrates that physiologically relevant changes in neuronal activity are also sufficient to trigger Rem2 mRNA expression. Numerous studies have demonstrated that the route of calcium entry into the cell influences downstream signal transduction events (Bading et al., 1993; Dolmetsch, 2003), either through assembly of signaling complexes at specific types of channels, or through channel-dependent differences in calcium dynamics (West et al., 2001). Interestingly, overexpression studies have implicated Rem2 and other RGK GTPases in negative regulation of calcium influx through VGCCs, including L-type channels (Chen et al., 2005; Finlin et al., 2005; Moore et al., 2013). However, data supporting a requirement for Rem2 function in promoting calcium influx has yet to be obtained (Wang et al., 2011; Moore et al., 2013). Nonetheless, this suggests the possibility that the Ca2+-dependent upregulation of Rem2 functions in a negative feedback loop to shut off Ca2+ entry into the cell at a certain critical point. Such a mechanism was previously suggested for Rem2 regulation of Ca2+-mediated insulin secretion from pancreatic β cells (Finlin et al., 2005).
In addition to inducing the transcriptional activation of Rem2, we demonstrate that calcium influx through L-VGCCs activates parallel signaling pathway(s) to Rem2 that function to positively regulate dendritic arborization. Thus, Rem2 functions in a negative feedback circuit to keep runaway dendritic growth in check. Our experiments using pharmacological reagents to block calcium entry through NMDA receptors versus L-VGCCs, in conjunction with an analysis of Rem2 mRNA expression (Figs. 1, 4) and dendritic complexity (Fig. 7), begin to differentiate between signaling pathways that promote dendritic outgrowth in response to calcium entry via either L-VGCCs or NMDA receptors. In so doing, these experiments provide novel mechanistic insight into the signaling pathways that mediate dendritic complexity.
Dendritic structural plasticity is crucial to maintaining neuronal circuit function in the face of changes in the environment, such as modulation of sensory experience, hormones, and temperature (McEwen, 2010). Abnormal dendritic morphology and plasticity are components of a number of neurological disorders that have a relatively late onset of symptoms (e.g., Rett and Angelman syndromes), suggesting that experience-dependent processes are aberrant in the nervous system of these individuals (Zoghbi, 2003). In addition, it has been demonstrated that addiction to stimulants such as cocaine causes changes in neuronal structure (Robinson and Kolb, 2004) and further, these structural changes may explain the persistent features of drug addiction, including drug cravings and relapse (Chao and Nestler, 2004; Robinson and Kolb, 2004). Our findings demonstrate that changes in sensory experience concurrently activate signaling pathways that both promote and inhibit dendritic complexity, suggesting that a shift in the balance of these signals must be considered as a possible underlying cause of these neurological disorders.
Footnotes
This work was supported by the Alfred P. Sloan Foundation (S.P.); The Richard and Susan Smith Family Foundation (S.P.); National Institutes of Health (NIH) Grant R01NS065856 (S.P.), NIH Grant F31DA032181 (A.E.G.), NIH Grant T32NS07292 (A.R.M.), and NIH Grant R01DA033610 (A.E.W.); the Searle Scholar Foundation (N.C.L.); NIH Grant R00HD057298 (N.C.L.) and NIH Grant P30NS069339 (S.D.V.); the Massachusetts Life Sciences Center (S.D.V.H.); the Charles H. Hood Foundation (S.D.V.H.); and NIH Grant P30NS45713 (for Core Facilities for Neurobiology at Brandeis University). We thank Dr. Donald Katz for advice regarding statistical analysis and Dr. Michael Marr for sharing of reagents. We also thank Dr. Gina Turrigiano for critical reading of this manuscript.
The authors declare no competing financial interests.
References
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/S0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bestman JE, Ewald RC, Chiu SL, Cline HT. In vivo single-cell electroporation for transfer of DNA and macromolecules. Nat Protoc. 2006;1:1267–1272. doi: 10.1038/nprot.2006.186. [DOI] [PubMed] [Google Scholar]
- Chao J, Nestler EJ. Molecular neurobiology of drug addiction. Annu Rev Med. 2004;55:113–132. doi: 10.1146/annurev.med.55.091902.103730. [DOI] [PubMed] [Google Scholar]
- Chen H, Puhl HL, 3rd, Niu SL, Mitchell DC, Ikeda SR. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J Neurosci. 2005;25:9762–9772. doi: 10.1523/JNEUROSCI.3111-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Tari PK, She K, Haas K. Neurexin-neuroligin cell adhesion complexes contribute to synaptotropic dendritogenesis via growth stabilization mechanisms in vivo. Neuron. 2010;67:967–983. doi: 10.1016/j.neuron.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Chen SX, Cherry A, Tari PK, Podgorski K, Kwong YK, Haas K. The transcription factor MEF2 directs developmental visually driven functional and structural metaplasticity. Cell. 2012;151:41–55. doi: 10.1016/j.cell.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Correll RN, Pang C, Niedowicz DM, Finlin BS, Andres DA. The RGK family of GTP-binding proteins: regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell Signal. 2008;20:292–300. doi: 10.1016/j.cellsig.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R. Excitation-transcription coupling: signaling by ion channels to the nucleus. Sci STKE. 2003;2003:PE4. doi: 10.1126/stke.2003.166.pe4. [DOI] [PubMed] [Google Scholar]
- Elston GN. Pyramidal cells of the frontal lobe: all the more spinous to think with. J Neurosci. 2000;20:RC95. doi: 10.1523/JNEUROSCI.20-18-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JS, Wheeler DG, Tsien RW, Luo L. Uncoupling dendrite growth and patterning: single-cell knockout analysis of NMDA receptor 2B. Neuron. 2009;62:205–217. doi: 10.1016/j.neuron.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/S0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Finlin BS, Mosley AL, Crump SM, Correll RN, Ozcan S, Satin J, Andres DA. Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J Biol Chem. 2005;280:41864–41871. doi: 10.1074/jbc.M414261200. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K, Wong RO. A comparison of experience-dependent plasticity in the visual and somatosensory systems. Neuron. 2005;48:465–477. doi: 10.1016/j.neuron.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Fujino T, Leslie JH, Eavri R, Chen JL, Lin WC, Flanders GH, Borok E, Horvath TL, Nedivi E. CPG15 regulates synapse stability in the developing and adult brain. Genes Dev. 2011;25:2674–2685. doi: 10.1101/gad.176172.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiretti AE, Paradis S. The GTPase Rem2 regulates synapse development and dendritic morphology. Dev Neurobiol. 2011;71:374–389. doi: 10.1002/dneu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiretti AE, Kenny K, Marr MT, 2nd, Paradis S. CaMKII-dependent phosphorylation of the GTPase Rem2 is required to restrict dendritic complexity. J Neurosci. 2013;33:6504–6515. doi: 10.1523/JNEUROSCI.3861-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas K, Jensen K, Sin WC, Foa L, Cline HT. Targeted electroporation in Xenopus tadpoles in vivo–from single cells to the entire brain. Differentiation. 2002;70:148–154. doi: 10.1046/j.1432-0436.2002.700404.x. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hewapathirane DS, Haas K. Single cell electroporation in vivo within the intact developing brain. J Vis Exp. 2008;11:pii:705. doi: 10.3791/705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of BDNF transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LJ, Lo FS, Erzurumlu RS. NMDA receptor-dependent regulation of axonal and dendritic branching. J Neurosci. 2005;25:2304–2311. doi: 10.1523/JNEUROSCI.4902-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerea LS, Butler LS, McNamara JO. NMDA and non-NMDA receptor-mediated increase of c-fos mRNA in dentate gyrus neurons involves calcium influx via different routes. J Neurosci. 1992;12:2973–2981. doi: 10.1523/JNEUROSCI.12-08-02973.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JH, Nedivi E. Activity-regulated genes as mediators of neural circuit plasticity. Prog Neurobiol. 2011;94:223–237. doi: 10.1016/j.pneurobio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebrich S, Nedivi E. The function of activity-regulated genes in the nervous system. Physiol Rev. 2009;89:1079–1103. doi: 10.1152/physrev.00013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MR, West AE. Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol. 2011;94:259–295. doi: 10.1016/j.pneurobio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J, Kelly K. Gem: an induced, immediate early protein belonging to the Ras family. Science. 1994;265:241–244. doi: 10.1126/science.7912851. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-X. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress, sex, and neural adaptation to a changing environment: mechanisms of neuronal remodeling. Ann N Y Acad Sci. 2010;1204(Suppl):E38–E59. doi: 10.1111/j.1749-6632.2010.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Ghiretti AE, Paradis S. A loss-of-function analysis reveals that endogenous rem2 promotes functional glutamatergic synapse formation and restricts dendritic complexity. PLoS One. 2013;8:e74751. doi: 10.1371/journal.pone.0074751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Role of ion flux in the control of c-fos expression. Nature. 1986;322:552–555. doi: 10.1038/322552a0. [DOI] [PubMed] [Google Scholar]
- Nedivi E, Wu GY, Cline HT. Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science. 1998;281:1863–1866. doi: 10.1126/science.281.5384.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Harrar DB, Lin Y, Koon AC, Hauser JL, Griffith EC, Zhu L, Brass LF, Chen C, Greenberg ME. An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron. 2007;53:217–232. doi: 10.1016/j.neuron.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VR, Pintchovski SA, Chin J, Peebles CL, Mitra S, Finkbeiner S. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat Neurosci. 2006;9:887–895. doi: 10.1038/nn1708. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/S0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Reymond P, Coquard A, Chenon M, Zeghouf M, El Marjou A, Thompson A, Ménétrey J. Structure of the GDP-bound G domain of the RGK protein Rem2. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:626–631. doi: 10.1107/S1744309112013541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Saha RN, Wissink EM, Bailey ER, Zhao M, Fargo DC, Hwang JY, Daigle KR, Fenn JD, Adelman K, Dudek SM. Rapid activity-induced transcription of Arc and other IEGs relies on poised RNA polymerase II. Nat Neurosci. 2011;14:848–856. doi: 10.1038/nn.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasson Y, Navon-Perry L, Huppert D, Hirsch JA. RGK family G-domain:GTP analog complex structures and nucleotide-binding properties. J Mol Biol. 2011;413:372–389. doi: 10.1016/j.jmb.2011.08.017. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Shah M, Haylett DG. Ca(2+) channels involved in the generation of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2554–2561. doi: 10.1152/jn.2000.83.5.2554. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sin WC, Haas K, Ruthazer ES, Cline HT. Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature. 2002;419:475–480. doi: 10.1038/nature00987. [DOI] [PubMed] [Google Scholar]
- Singer AL, Koretzky GA. Control of T cell function by positive and negative regulators. Science. 2002;296:1639–1640. doi: 10.1126/science.1071551. [DOI] [PubMed] [Google Scholar]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/S0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, Cline HT. Rho GTPases and activity-dependent dendrite development. Curr Opin Neurobiol. 2004;14:297–304. doi: 10.1016/j.conb.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Wang HG, Wang C, Pitt GS. Rem2-targeted shRNAs reduce frequency of miniature excitatory postsynaptic currents without altering voltage-gated Ca currents. PLoS One. 2011;6:e25741. doi: 10.1371/journal.pone.0025741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/S0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- West AE, Greenberg ME. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol. 2011;3:a005744. doi: 10.1101/cshperspect.a005744. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002;3:803–812. doi: 10.1038/nrn941. [DOI] [PubMed] [Google Scholar]
- Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- Wu GY, Zou DJ, Rajan I, Cline H. Dendritic dynamics in vivo change during neuronal maturation. J Neurosci. 1999;19:4472–4483. doi: 10.1523/JNEUROSCI.19-11-04472.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/S0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu Rev Cell Dev Biol. 2005;21:35–56. doi: 10.1146/annurev.cellbio.21.012704.131040. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Tao HW, Poo M. Visual input induces long-term potentiation of developing retinotectal synapses. Nat Neurosci. 2000;3:708–715. doi: 10.1038/76665. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schütz G, Delucinge-Vivier C, Descombes P, Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent BDNF transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]