

Abstract
Learned associations between environmental cues and morphine use play an important role in the maintenance and/or relapse of opioid addiction. Although previous studies suggest that context-dependent morphine treatment alters glutamatergic transmission and synaptic plasticity in the hippocampus, their role in morphine conditioned place preference (CPP) and reinstatement remains unknown. We investigated changes in synaptic plasticity and NMDAR expression in the hippocampus after the expression, extinction, and reinstatement of morphine CPP. Here we report that morphine CPP is associated with increased basal synaptic transmission, impaired hippocampal long-term potentiation (LTP), and increased synaptic expression of the NR1 and NR2b NMDAR subunits. Changes in synaptic plasticity, synaptic NR1 and NR2b expression, and morphine CPP were absent when morphine was not paired with a specific context. Furthermore, hippocampal LTP was impaired and synaptic NR2b expression was increased after extinction of morphine CPP, indicating that these alterations in plasticity may be involved in the mechanisms underlying the learning of drug–environment associations. After extinction of morphine CPP, a priming dose of morphine was sufficient to reinstate morphine CPP and was associated with LTP that was indistinguishable from saline control groups. In contrast, morphine CPP extinguished mice that received a saline priming dose did not show CPP and had disrupted hippocampal LTP. Finally, we found that reinstatement of morphine CPP was prevented by the selective blockade of the NR2b subunit in the hippocampus. Together, these data suggest that alterations in synaptic plasticity and glutamatergic transmission play an important role in the reinstatement of morphine CPP.
Keywords: CPP, hippocampus, LTP, morphine, NMDA, plasticity
Introduction
Morphine, a widely used opioid analgesic drug, exerts diverse behavioral and molecular effects. Addiction to morphine and other opioids is a major public health issue, with 11 million Americans having engaged in opioid abuse (Compton and Volkow, 2006). Although significant advances in the treatment of opioid addiction have been made through the development of pharmacotherapies (for review, see Bart, 2012), relapse to opioid use after periods of abstinence continues to impede successful treatment, highlighting the need for continued efforts to dissect the mechanisms of opioid-dependent changes in brain plasticity.
Associations between drug use and the drug administration environment can lead to cravings that promote continued use and can facilitate relapse (Daglish et al., 2001; See, 2002). Mice can develop a conditioned place preference (CPP) in which the rewarding properties of morphine are paired with a drug administration environment (Kim et al., 1996; Ribeiro Do Couto et al., 2005; Benturquia et al., 2007; Solecki et al., 2009). This behavior can be extinguished by pairing the drug-associated context with saline and then reinstated by a priming dose of morphine (Ribeiro Do Couto et al., 2005). Animal models of morphine addiction have revealed that drug-associated contexts can lead to changes in glutamatergic signaling (Xia et al., 2011; Fakira et al., 2013). The administration of NMDA receptor (NMDAR) antagonists can block morphine CPP (Ribeiro Do Couto et al., 2005; Ma et al., 2007, 2011a; Zarrindast et al., 2007; Li et al., 2011), suggesting that NMDARs play a critical role in associations between morphine and contextual stimuli.
Evidence suggests that repeated drug treatment can lead to adaptations in hippocampal synaptic plasticity that may play a crucial role in drug–context associations. Several forms of synaptic plasticity in the hippocampal Schaffer collateral–CA1 pathway, such as long-term potentiation (LTP), are dependent on NMDAR-mediated Ca2+ influx (Muller et al., 1988; Shapiro and Eichenbaum, 1999; Miyamoto, 2006). Furthermore, previous morphine CPP studies have demonstrated increased NR2b NMDAR subunit expression in the hippocampus (Ma et al., 2006, 2007) and have shown that hippocampal infusions of an NMDAR antagonist can block morphine CPP (Zarrindast et al., 2007). Together, these data suggest that NMDAR-mediated signaling in the hippocampus may be crucial for morphine CPP and may lead to changes in synaptic plasticity (i.e., LTP). Morphine-induced alterations in hippocampal LTP may have consequences for drug dependence and craving because contextual-driven memories have been associated with drug craving in abstinent opioid abusers (Daglish et al., 2001; Ersche et al., 2006; Prosser et al., 2006). However, few studies have investigated the role of the hippocampus in morphine CPP and morphine-induced reinstatement, which may provide a new target to prevent relapse. In addition, no studies have been conducted on the role of NMDAR on reinstatement of morphine CPP. Thus, in the present study, we used a multidisciplinary approach to directly investigate the changes in hippocampal synaptic NMDAR expression, basal synaptic transmission, and LTP after expression, extinction, and reinstatement of morphine CPP. Together, our results identify the importance of hippocampal synaptic plasticity and NMDAR expression in associations between morphine and environmental context that ultimately lead to reinstatement of morphine CPP.
Materials and Methods
Subjects
Subjects were 7- to 10-week-old male C57BL/6J mice (25–30 g; Harlan) that were maintained on a 12 h light/dark cycle and were habituated to the colony room for 1 week before the start of experiments. All experimental protocols were approved by the Institutional Animal Care and Use Committee at Columbia University and Washington University School of Medicine, according to the U.S. Department of Agriculture Animal Welfare Act (Public Law 89-544) and the National Institutes of Health Guide for the Care and Use of the Laboratory Animals (Department of Health, Education, and Welfare Publication 85-23, revised 1985).
Apparatus
CPP to morphine was assessed using four identical conditioning boxes (63 × 26 × 30.5 cm) that contained two chambers for drug conditioning (26 × 26 × 30.5 cm) and a central compartment (8.5 × 26 × 30.5 cm) that was not paired with saline or morphine treatment. The drug conditioning chamber walls had 2.5-cm-wide horizontal or vertical black and white stripes that were used to distinguish the two chambers from each other, whereas the walls in the central chamber were made of white nonreflective plastic. The floors of the two conditioning chambers were lined with cob bedding (Enrich-O' Cobs; Andersons Lab Bedding). The total amount of time spent in all three chambers was determined with an array of photobeams (Med Associates) that were positioned at the base of each chamber. Morphine CPP experiments conducted at the Washington University School of Medicine used identical CPP chambers as those described above, but chamber preference was determined using a video tracking system (Ethovision; Noldus).
Drug administration
Morphine sulfate (National Institute on Drug Abuse Drug Program or Sigma-Aldrich) was dissolved in saline at the concentration of 1 mg/ml. For all morphine CPP experiments, mice were injected subcutaneously with daily escalating doses of morphine (5, 8, 10, and 15 mg/kg) or equivalent volume of saline and then immediately placed in the conditioning chamber. Animals were killed immediately after either morphine CPP (24 h after the final morphine injection), extinction of morphine CPP (7 d after morphine treatment), or reinstatement of morphine CPP (8 d after initial morphine treatment and 15 min after morphine reinstatement injection). After each of these tests, hippocampi were immediately dissected and either used for electrophysiological experiments or stored flash frozen at −80°C until use.
Cannulae implantation surgical procedure
After acclimatizing to the holding facility for 7 to 9 d, the animals were anesthetized in an induction chamber (4% isoflurane) and mounted on a stereotaxic frame under sterile conditions (model 1900; David Kopf Instruments) in which they were maintained at 1–2% isoflurane for the duration of the surgery. A craniotomy was performed, and two cannulae (C315G/SPC, 1.5 mm; Plastics One) were lowered bilaterally into the hippocampus (−1.7 mm anteroposterior; ±1.5 mm mediolateral; −1.8 mm dorsoventral from bregma; Shirayama et al., 2004; Land et al., 2009; Knoll et al., 2011; Smith et al., 2012). The cannulae were secured using two bone screws (catalog #743102; CMA/Microdialysis) and affixed with dental cement (Lang Dental). Mice were allowed to recover for at least 7 d before behavioral testing.
To confirm cannulae placement, immunohistochemistry was performed as described previously (Bruchas et al., 2007a,b; Land et al., 2008; Lemos et al., 2011; Al-Hasani et al., 2013b). Mice were anesthetized with pentobarbital and perfused intracardially with ice-cold 4% paraformaldehyde in phosphate buffer (PB). Brains were dissected, postfixed 24 h at 4°C, cryoprotected with a solution of 30% sucrose in 0.1 m PB at 4°C for at least 24 h, cut into 30 μm sections, and processed for immunostaining. Sections were washed twice in PBS and blocked in PBS containing 0.1% Triton X-100 (TX-100; G-Biosciences) for 10 min, followed by an 1 h incubation with fluorescent Nissl stain (1:400; Neurotrace; Invitrogen). Sections were then washed twice in PBS and mounted on glass slides with hard set Vectashield (Vector Laboratories). All sections were imaged with an epifluorescent microscope (Olympus BX61) in the Washington University Pain Center.
Experimental design
Handling and habituation.
As described previously (Portugal and Gould, 2009), before all experiments, mice were habituated for 3 d (6 h/d) to the room in which behavioral experiments would occur, and each mouse was handled twice daily. During handling sessions, mice were gently restrained as though in preparation for a subcutaneous injection and were released after several seconds. This process was repeated for ∼1 min, and then mice were returned to the home cage. Mice that exhibited signs of increased anxiety (i.e., shaking, vocalizations) received additional handling sessions until signs of anxiety were reduced.
Morphine CPP.
At the start of morphine CPP training, initial preferences for the three chambers were determined in a 15 min preconditioning session, in which mice were placed in the central chamber and allowed to explore all three chambers (for a schematic of the experiment, see Fig. 1A). Mice exhibited no initial preference (bias) for the horizontal or vertical striped chamber (t(64) = 1.51; p > 0.05), and therefore the morphine-paired chamber was randomly assigned for each mouse in the morphine CPP group, in a counterbalanced unbiased manner. After preconditioning (pretest), mice received eight conditioning sessions (four morning and four evening sessions; Land et al., 2009; Bruchas et al., 2011; Einstein et al., 2013). During conditioning, mice in the morphine test group received a subcutaneous injection of saline in the morning and were immediately confined to one chamber (saline-paired conditioning chamber) for 30 min. Four hours later, in the afternoon, mice in the morphine CPP group received a subcutaneous injection of morphine and were immediately confined to the other chamber (morphine-paired conditioning chamber) for 30 min. In contrast, mice in the saline CPP group received saline during both conditioning sessions. To rule out the effects of 24 h withdrawal from morphine treatment on NMDAR expression and synaptic plasticity, a morphine-unpaired group was included in which morphine was administered alternately in both conditioning chambers during the afternoon conditioning sessions. For the morphine-unpaired control group, the location of morphine administration was alternated between days (i.e., horizontal chamber in the afternoon on day 1, vertical chamber in the afternoon on day 2), and thus any changes in NMDAR expression and synaptic plasticity observed in morphine-unpaired mice could not be attributable to an association between morphine treatment and a specific drug conditioning chamber. On the test day (24 h after final morphine injection), mice were placed in the central chamber and were allowed to explore all three chambers for 15 min. Place preference scores were calculated as described previously by subtracting the time spent in the drug-paired side during preconditioning from the time spent in the drug-paired side during postconditioning (Tzschentke, 2007; Al-Hasani et al., 2013a,b).
Figure 1.

Morphine CPP is associated with increased basal synaptic transmission and impaired LTP. A, A schematic of the experimental design is shown for the training and testing of morphine CPP. B, Treatment with escalating doses of morphine produces CPP. Mice treated with escalating doses of morphine-paired with a novel context (morphine-paired group) exhibited a significant increase in preference for the morphine-paired chamber relative to saline-treated mice (saline group) and mice that received morphine in both drug conditioning chambers (morphine-unpaired group; n = 9–14 per group; *p < 0.05). Preference score was determined by subtracting the time spent in the morphine-paired side during preconditioning from the time spent in the morphine-paired side during testing (expressed as seconds). C, Mice trained and tested for morphine CPP exhibited a significantly enhanced fEPSP slope compared with saline-treated mice (n = 4 per group; one-way ANOVA; *p < 0.05, **p < 0.01). Insets show fEPSP–FV relationships. D, The expression of morphine CPP was associated with disrupted LTP when compared with saline-treated mice and morphine-unpaired mice (n = 3–4 per group; one-way ANOVA; *p < 0.05). Insets show comparisons of fEPSP slopes during the final 10 min of recording.
Extinction and reinstatement of morphine CPP.
Extinction of morphine CPP commenced 24 h after morphine CPP (for a schematic of this experiment, see Fig. 4A). Mice in all groups received 10 extinction training sessions (five morning and five evening sessions), in which a subcutaneous injection of saline was administered and mice were confined to the saline-paired compartment for 30 min. During afternoon extinction training, mice received a second subcutaneous injection of saline and were confined to the morphine-paired chamber for 30 min. Testing of extinction occurred 24 h after the final conditioning session (7 d after morphine treatment); mice were placed in the central chamber and allowed to explore all three chambers for 15 min. Preference scores were calculated in the same manner as described above, and extinction of CPP was defined as a preference score that was <15% of the initial preference score. As with the experiments described above, a morphine-unpaired group was included in extinction experiments to rule out whether changes in NMDAR expression and synaptic plasticity that could be attributable to withdrawal from morphine treatment. Mice in the morphine-unpaired group received morphine in both chambers on alternating days during the training of morphine CPP, and extinction experiments were conducted in the same manner as described above.
Figure 4.

Extinction of morphine CPP is associated with impaired LTP. A, A schematic of the experimental design is shown for the training and testing of morphine CPP and extinction of morphine CPP. B, Treatment with escalating doses of morphine produces CPP and can be extinguished by pairing saline with both drug conditioning chambers. Mice exhibited CPP when morphine was paired with a novel context (morphine-paired group; n = 10–16 per group; one-way ANOVA; *p < 0.05), whereas no preference was observed when mice received morphine in both conditioning chambers (morphine-unpaired group). After extinction training in which mice received saline in both drug conditioning chambers, mice no longer showed a preference for the morphine-paired chamber. C, After extinction of morphine CPP, no changes were observed in fEPSP slope. Insets show fEPSP/FV relationships. D, The extinction of morphine CPP was associated with impaired LTP when compared with saline-treated mice and morphine-unpaired mice (n = 3–5 per group; one-way ANOVA; *p < 0.05, **p < 0.01). Insets show comparisons of fEPSP slopes during the final 10 min of recording.
The reinstatement of morphine CPP occurred 24 h after the CPP extinction test (for a schematic of this experiment, see Fig. 5A). Mice that showed extinction of morphine CPP or saline controls were treated subcutaneously with 5 mg/kg morphine, placed in the central chamber, and allowed to explore all three chambers for 15 min. As with previously described experiments, a third group of mice was included in which morphine treatment was not paired with the context during CPP training (morphine-unpaired group). During reinstatement testing, mice in the morphine-unpaired group were administered 5 mg/kg morphine, placed in the central chamber, and explored all three chambers for 15 min. A fourth group of mice was also included that were trained and tested for both morphine CPP and morphine CPP extinction as described above. However, during the reinstatement test, mice in this group received subcutaneous saline instead of morphine and were allowed to explore the three chambers for 15 min. Morphine preference was calculated in the same manner as in the morphine CPP experiments.
Figure 5.
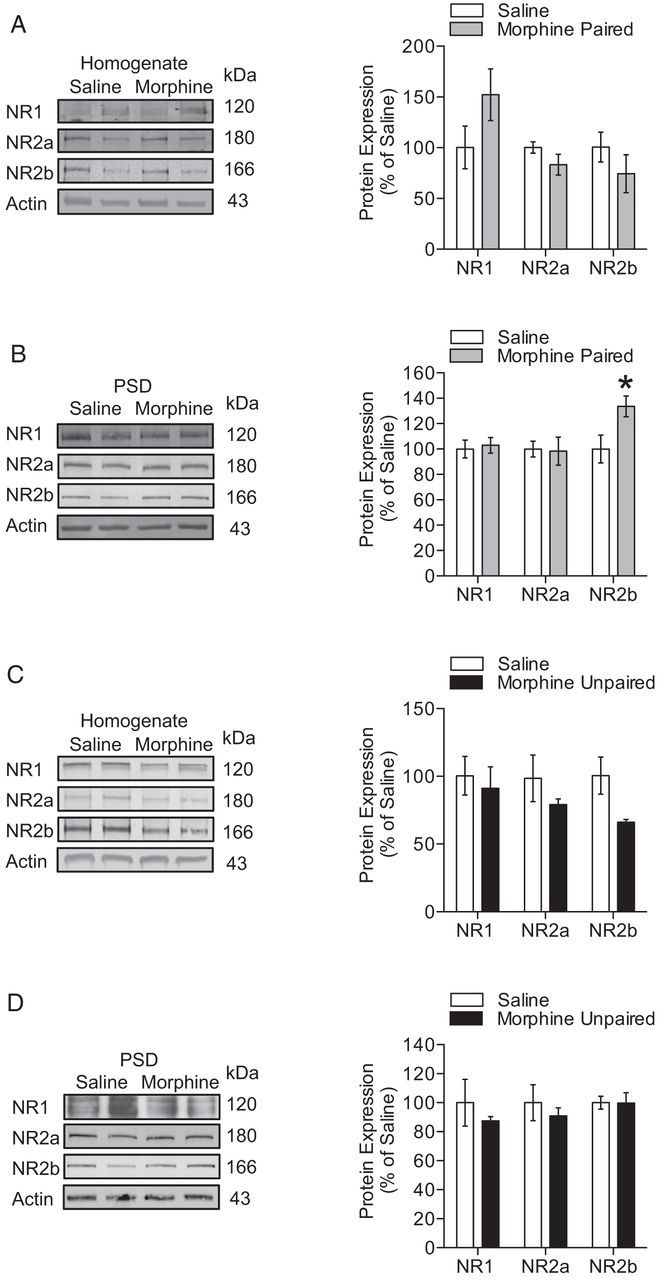
Alterations in synaptic NMDAR subunit expression after extinction of morphine CPP. A, B, After extinction of morphine CPP, NR2b NMDAR subunit expression was increased at the PSD (n = 4 per group; unpaired t test; *p < 0.05). C, D, In contrast, mice showed no changes in NMDAR subunit expression after extinction from a design of unpaired morphine treatment.
Mice implanted with cannulae in the hippocampus, which showed extinction of morphine CPP (see criteria above), were given dummy injections using tailored injectors (C3151/SPC; Plastics One) to acclimate the mice to injectors and infusion tubing. On the reinstatement day, the mice were infused with either aCSF or ifenprodil (2 μg/1 μl) into their hippocampus (Zhang et al., 2013) using an infusion pump (Pump 11 Elite; Harvard Apparatus). Mice were returned to their home cage for 30 min after infusion. Mice were then injected with morphine (5 mg/kg), then placed in the central chamber, and allowed to explore all three chambers for 15 min to determine the time spent on the drug-paired side.
Subcellular fractionation of postsynaptic densities.
Subcellular fractionation was used to isolate the postsynaptic density (PSD) for immunoblotting experiments and was performed as described previously (Morón et al., 2007; Billa et al., 2009, 2010; Xia et al., 2011). Total hippocampi from individual mouse brains were carefully dissected and homogenized in a 0.32 m sucrose solution containing 0.1 mm CaCl2, protease, and phosphatase inhibitors (Sigma-Aldrich). A 50 μl sample of homogenate was obtained, was diluted with 50 μl of the 0.32 m sucrose solution, and was used as the total homogenate fraction for immunoblotting experiments. The homogenate was adjusted to a final concentration of 1.25 m sucrose by adding 2 m sucrose dissolved in dH2O. The homogenate was then overlaid with a 1 m sucrose solution dissolved in 0.1 mm CaCl2 and was centrifuged at 100,000 × g for 3 h at 4°C. Eight milliliters of synaptosomal fraction were collected at the interface between 1.25 m/1 m sucrose solutions and was then solubilized in 20 mm Tris-HCl, pH 6.0, buffer containing 1% TX-100 for 20 min at 4°C. The synaptic junctions were precipitated from the synaptosomal fraction by centrifugation at 40,000 × g for 30 min at 4°C. Synaptic junctions were resuspended in 20 mm Tris-Cl, pH 8, 1% TX-100, and 0.1 mm CaCl2 and incubated at 4°C for 20 min to separate the presynaptic fraction. Finally, the PSD fraction was precipitated by centrifugation at 40,000 × g for 30 min at 4°C, dissolved in 1% SDS, and kept at −80°C until use.
Immunoblotting.
Equal amounts of either total homogenate or PSD fractions (10 μg) were loaded and separated in 10% SDS-PAGE for 120 min. Protein was then transferred to nitrocellulose membranes via electrophoresis at 4°C for 90 min. Membranes were blocked in 5% nonfat evaporated milk for 30 min and were incubated with primary antibodies selective to NR1 (1:500; incubated overnight at 4°C; PA3–102; Pierce), NR2a (1:1000; incubated 1 h at room temperature; AB1555; Millipore), or NR2b (1:1000; incubated 1 h at room temperature; catalog #75-097; NeuroMab) or β-actin (1:5000; incubated 1 h at room temperature; MAB1501; Millipore). Next, the membranes were incubated with either IRDye 680 anti-mouse or IRDye 800 anti-rabbit (1:5000) followed by scanning with Odyssey (LI-COR Biosciences) or with anti-rabbit HRP (1:5000) followed by Amersham ECL Plus detection (GE Healthcare). The pixel intensity of NR1, NR2a, NR2b, and β-actin was measured using AlphaEase software, and the intensity of NR1, NR2a, and NR2b was normalized to β-actin to ensure equal protein loading between groups. β-Actin was used as a loading control because its expression did not change across any drug treatment for all experiments and because it is enriched in PSD fractions (Matus et al., 1982). Morphine CPP and unpaired morphine groups were normalized to saline-treated controls to calculate the percentage of change in intensity relative to saline-treated controls.
Electrophysiology.
Mice were decapitated immediately after the postconditioning testing phase of morphine CPP, extinction of morphine CPP, and reinstatement of morphine CPP. Brains were quickly removed in a semi-frozen sectioning solution that contained 175 mm sucrose, 7 mm MgCl2, 2.5 mm KCl, 1.2 mm NaH2PO4, 0.5 mm CaCl2, 26 mm NaHCO3, and 25 mm glucose. Brains were mounted and sliced at 450 μm thickness while submerged in an ice-cold sectioning solution. Sagittally cut slices were transferred to beakers that contained aCSF and incubated at 30 ± 1°C for at least 30 min. Slices were then transferred to the recording chamber that was maintained at 30 ± 1°C, and 95% O2/5% CO2 was bubbled continuously during dissection, incubation, and recording. A concentric bipolar stimulating electrode was placed in the Schaffer collateral pathway to evoke synaptic responses, and a glass Ag/AgCl electrode filled with aCSF was placed in the stratum radiatum of CA1 to record extracellular field EPSPs (fEPSPs). Slices were allowed to stabilize for 15 min, and then a stimulus–response curve was constructed by recording fEPSPs after stimulating the slice with increasing voltages (3–9 V). For LTP recordings, a voltage that elicited 50% of the maximum fEPSP was used (between 5 and 7 V in all recordings). Baseline synaptic responses were recorded for 20 min, and then LTP was induced by two high-frequency stimulation trains (100 Hz, 1 s duration) separated by a 20 s intertrain interval. Data acquisition and analysis were performed using Clampex and Clampfit 8.2 (Molecular Devices).
Data and statistical analyses.
Results are expressed as the mean ± SEM. Data from behavioral and electrophysiological experiments with three groups were analyzed using one-way ANOVAs, followed by Tukey's or Games–Howell post hoc comparisons, whereas data from immunoblotting experiments were analyzed with two-tailed unpaired Student's t tests. Linear regression was used to compare the slopes of fiber volley (FV)/fEPSP ratios. Significance was set at p < 0.05. Statistical analyses of data were generated by using Prism software (version 5.0; GraphPad Software).
Results
Basal synaptic transmission is increased whereas hippocampal LTP is selectively impaired after expression of morphine CPP
To directly investigate the effects of morphine CPP on basal synaptic transmission and hippocampal LTP, mice were conditioned in an unbiased morphine CPP paradigm that paired escalating doses of morphine (5, 8, 10, and 15 mg/kg, s.c.) with a neutral context, and electrophysiological changes in basal synaptic transmission and LTP were recorded immediately after CPP testing. Mice trained in the CPP procedure showed a significant increase in time spent in the drug conditioning chamber that was paired with morphine relative to all other groups (Fig. 1B; n = 9–14 per group; F(2,30) = 12.98; p < 0.05, Tukey's post hoc test).
After expression of morphine CPP, hippocampi were collected and prepared for use in electrophysiological field recordings. Mice that exhibited morphine CPP showed significant increases in basal synaptic transmission (Fig. 1C; n = 4 per group; 6 V: F(2,8) = 4.30, p < 0.05, 7 V: F(2,8) = 5.49, p < 0.05; 8 V: F(2,8) = 8.17, p < 0.01). Tukey's post hoc comparisons revealed the application of 6–8 V stimulation produced significant increases in basal synaptic transmission in morphine conditioned mice when compared with saline conditioned mice (p < 0.05); no other significant differences were found between the three groups. As a control measure, all fEPSP slopes were also plotted against presynaptic FV amplitude to determine whether differences in basal synaptic transmission were related to differences in the positioning of the stimulating electrode. Morphine CPP significantly increased fEPSP slopes at similar presynaptic FV amplitudes when compared with all other groups (Fig. 1C, inset; n = 4 per group; linear regression comparisons; morphine-paired vs saline: F(1,38) = 45.65, p < 0.05; morphine-paired vs morphine-unpaired, F(1,38) = 8.21, p < 0.01). These data suggest that morphine CPP selectively enhances basal synaptic transmission, leading to significant alterations in hippocampal synaptic plasticity.
We demonstrated previously that treatment with escalating doses of morphine can alter hippocampal synaptic plasticity associated with context-dependent behavioral sensitization (Xia et al., 2011), but it remains unknown whether other forms of context-dependent morphine treatment, such as morphine CPP, also lead to changes in hippocampal synaptic plasticity. The CPP model is suitable for examining context-dependent drug effects and reinstatement of the conditioned responses after extinction. Therefore, we examined changes in hippocampal LTP after the expression of morphine CPP. Comparisons of fEPSP slopes between groups were made during the final 10 min of recording (50–60 min after the start of recording). Mice that were trained and tested for morphine place preference showed a profound and significant decrease in hippocampal LTP (Fig. 1D; n = 3–4 per group; F(2,27) = 114.9, p < 0.05, Tukey's post hoc test) when compared with both groups.
It is possible that changes in basal synaptic transmission and/or synaptic plasticity may be directly attributable to 24 h of withdrawal from morphine administration instead of an effect that was observed only when morphine was paired with a distinct context. Therefore, an unpaired control morphine CPP group was included that received morphine in both drug conditioning chambers. Mice trained in the morphine-paired design had a significant increase in place preference relative to morphine-unpaired mice (Fig. 1B; p < 0.05, Tukey's post hoc test), and morphine-unpaired mice showed no changes in preference when compared with saline controls. Furthermore, no changes in basal synaptic transmission or LTP were observed in mice that received unpaired morphine treatment (Fig. 1D), indicating that changes in hippocampal synaptic plasticity observed in the morphine CPP group are not attributable to mechanisms related to a 24 h withdrawal from morphine exposure. Furthermore, these data suggest that these effects on synaptic transmission and LTP are likely not attributable to the morphine exposure itself because mice treated in an unpaired design did not show place preference, increased basal synaptic transmission, or impaired LTP. Together, these data suggest that alterations in basal synaptic transmission and synaptic plasticity only occur in animals that have acquired and are expressing a drug–context association.
Finally, we conducted studies to examine whether the impaired LTP is observed after withdrawal of morphine conditioning. To this end, a separate group of mice were conditioned and tested for morphine CPP as described above. After the CPP test, mice were kept in their home cages for 7 d (Fig. 2A). We found that LTP in morphine-treated mice was indistinguishable from saline-injected mice 7 d after morphine conditioning (Fig. 2B,C; n = 5–6 per group), indicating that alterations in synaptic plasticity in the hippocampus are no longer observed when animals are not reexposed to the previously drug-paired chambers 7 d after the initial CPP test.
Figure 2.

Impaired LTP after morphine CPP is a consequence of the reexposure to the drug-paired chamber. A, A schematic of the experimental design is shown. B, C, Mice trained and tested for morphine CPP that are kept at their home cages for 7 d show LTP similar to saline-treated mice. C, Shows comparisons of fEPSP slopes during the final 10 min of recording.
Increased synaptic NR1 and NR2b expression is associated with expression of morphine place preference
Changes to basal synaptic transmission and hippocampal LTP that occur after the expression of morphine CPP may be attributable to changes in the expression of NMDARs, because NMDARs are essential for hippocampal synaptic plasticity (Muller et al., 1988; Shapiro and Eichenbaum, 1999; Miyamoto, 2006). Therefore, we next determined whether changes in NMDARs in the total homogenate and PSD were present biochemically after the expression of morphine CPP. Mice that were trained and tested in morphine CPP had increased expression of NR1 and NR2a in the total homogenate when compared with saline controls (Fig. 3A; n = 4–8 per group; NR1, t(13) = −2.84, p < 0.05; NR2a, t(13) = −2.57, p < 0.05), whereas we found that expression levels of all three subunits were increased in the PSD (Fig. 3B; n = 4–8 per group; NR1, t(14) = −3.37, p < 0.01; NR2a, t(6) = −3.10, p < 0.05; NR2b, t(6) = −2.88, p < 0.05). In contrast, mice that were conditioned and tested with an unpaired design only showed increased NR2a expression in the total homogenate (Fig. 3C; n = 4–8 per group; NR2a, t(6) = −2.97, p < 0.05) and PSD (Fig. 3D; n = 4–8 per group; NR2a, t(14) = −3.67, p < 0.01) relative to saline-treated mice. Given that NR2a expression in the total homogenate was increased in both morphine CPP and unpaired morphine groups, it is likely that this increase we measured is attributable to either treatment with morphine or the 24 h withdrawal from morphine exposure. However, increased expression of NR1 and NR2b in the PSD was only observed in morphine-conditioned mice, strongly suggesting that the alterations of NR1 and NR2b occur only when morphine is directly paired with a novel context. Together, these data suggest that the expression of morphine CPP is associated with a selective increase in synaptic NR1 and NR2b expression, whereas increases in total synaptic NR2a expression occur as a direct result of morphine exposure.
Figure 3.
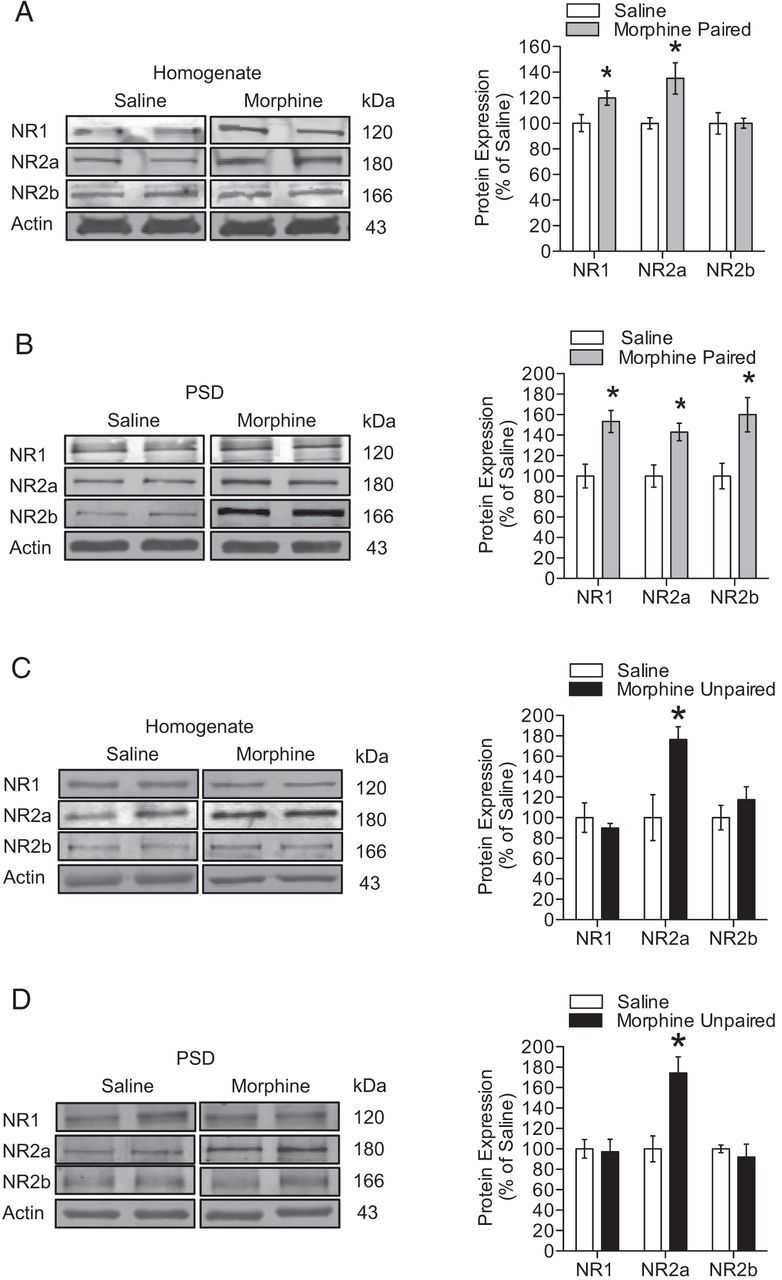
Alterations in synaptic NMDAR subunit expression after expression of morphine CPP. A, After the expression of morphine CPP, NR1 and NR2a NMDAR subunits were increased in the total homogenate (n = 4–8 per group; unpaired t test; *p < 0.05). B, NR1, NR2a, and NR2b subunit expression was increased at the PSD (n = 4–8 per group; unpaired t test; *p < 0.05). C, Only NR2a subunit expression was increased in the total homogenate (n = 4–8 per group; unpaired t test; *p < 0.05). D, PSD (n = 4–8 per group; unpaired t test; *p < 0.01) when mice received morphine in both drug conditioning chambers (morphine-unpaired group).
Hippocampal LTP is impaired after the extinction of morphine CPP
The results from the previous experiments suggest that morphine CPP is associated with increased basal synaptic transmission, disrupted LTP, and increased synaptic NR1 and NR2b expression (Table 1). However, it is also possible that these alterations in synaptic plasticity and NMDAR expression may also be observed after extinction of morphine CPP, because extinction training engages the learning of a new drug–environment association. Therefore, we conditioned mice in an unbiased morphine CPP procedure as described above, and morphine place preference was extinguished with repeated extinction sessions (five times, 30 min each) in which saline was administered in the morphine-paired chamber in place of morphine (Fig. 4A). Mice conditioned in the morphine CPP procedure exhibited increased preference for the morphine-paired compared with all other groups (Fig. 4B; n = 10–16 per group; F(2,35) = 16.98, p < 0.05). In contrast, no significant preference for the former drug-paired context was observed when mice were tested for preference after repeated extinction training with saline injections.
Table 1.
Changes in synaptic plasticity during morphine expression, extinction, and reinstatement
| Expression |
Extinction |
Reinstatement |
||||||
|---|---|---|---|---|---|---|---|---|
| Morphine paired | Morphine unpaired | Morphine paired | Morphine unpaired | Saline/morphine | Morphine paired/saline | Morphine paired/morphine | Morphine unpaired/morphine | |
| fEPSP | ↑ | = | = | = | = | = | = | = |
| LTP | ↓ | = | ↓ | = | = | ↓ | = | = |
| Homogenate | ||||||||
| NR1 | ↑ | = | = | = | ||||
| NR2a | ↑ | ↑ | = | = | ||||
| NR2b | = | = | = | = | ||||
| PSD | ||||||||
| NR1 | ↑ | = | = | = | ||||
| NR2a | ↑ | ↑ | = | = | ||||
| NR2b | ↑ | = | ↑ | = | ||||
Summary of changes in fEPSP, LTP, and NMDAR subunit expression during the three phases of morphine CPP, extinction, and reinstatement.
Electrophysiological recordings were conducted immediately after the behavioral test extinction CPP testing. Recordings of basal synaptic transmission revealed no differences between groups when fEPSPs were directly examined and when fEPSPs were normalized to presynaptic FV amplitude (Fig. 4C and inset in C). Although no changes in basal synaptic transmission were observed, recordings of hippocampal LTP revealed that mice showing extinguished morphine CPP had significantly impaired LTP compared with saline-treated and unpaired morphine groups (Fig. 4D; n = 3–5 per group; F(2,27) = 157.3, p < 0.05 and p < 0.01, respectively). In contrast, mice that received unpaired morphine treatment and extinction training exhibited robust LTP that was not significantly different from saline-treated mice, suggesting that the disrupted LTP observed after extinction of morphine CPP is not attributable to the 1 week of withdrawal from unpaired morphine treatment. These data demonstrate that hippocampal LTP is impaired after extinction of morphine CPP. Given that extinction reflects an active learning process, similar to that during morphine CPP, these data suggest that disrupted LTP may be related to the acquisition of a drug–environment association.
Increased synaptic NR2b expression is associated with extinction of morphine CPP
Increases in synaptic NR2b expression may be important for the alterations of synaptic plasticity that were observed after morphine CPP expression and extinction. Using synaptosomal preparations, we found that overall expression of NMDAR subunits NR1, NR2b, and NR2a in total homogenates were not significantly altered after morphine CPP extinction training or in the morphine-unpaired group that underwent extinction training (Fig. 5A,C; n = 4). Furthermore, expression of NR2b in the PSD was significantly increased after extinction of morphine CPP relative to saline-injected mice (Fig. 5B; NR2b, t(6) = 2.879; F(3,3) = 1.801, p < 0.05; n = 4). In contrast, no changes in NMDAR expression were observed in mice that were extinguished from unpaired morphine treatment compared with saline-treated mice (Fig. 5D). Together, these results indicate that extinction from morphine CPP is associated with increased synaptic expression of the NR2b NMDAR subunit. Interestingly, this increase was absent in mice that received similar extinction training but underwent morphine-unpaired treatment. In addition, these data further support the notion that the effects we observed are not attributable to 1 week withdrawal from morphine treatment.
Drug-induced reinstatement of morphine CPP is associated with increased hippocampal LTP
Although we found that extinction of morphine CPP is associated with disrupted LTP, it is unknown whether the reinstatement of morphine CPP with a priming dose of morphine could also lead to alterations in LTP. Therefore, mice were conditioned and tested in an unbiased morphine CPP procedure and then morphine CPP was extinguished as described above (for a description of animal groups, see Fig. 6A). Twenty-four hours after testing for the extinction of morphine CPP, mice received a subcutaneous priming dose of 5 mg/kg morphine and were allowed to explore all three chambers for 15 min (morphine-paired/morphine rein) while preference for the drug-paired chamber was recorded. Additionally, control groups were included that received only saline during CPP and extinction training but were administered 5 mg/kg morphine during reinstatement (saline group) and a group that received unpaired morphine treatment during CPP, saline during the extinction phase, and 5 mg/kg morphine during reinstatement (morphine-unpaired group). A final group was included that received morphine CPP, saline during extinction, but received saline during reinstatement (morphine-paired/saline rein). Mice in the morphine-paired/morphine and morphine-paired/saline groups both showed a significant preference for the morphine-paired chamber during the test for CPP, whereas the other control groups did not form a significant place preference for a particular context (Fig. 6B; n = 8–12 per group; F(3,38) = 8.73, p < 0.05). After extinction, no significant preference score was observed in any of the four groups. Morphine CPP extinguished mice that received a priming dose of morphine showed significant reinstatement to morphine CPP relative to all other groups (Fig. 6B; n = 8–12 per group; F(3,38) = 12.21, p < 0.05). In contrast, mice in the morphine CPP/saline group showed no change in their preference scores after a priming dose of saline, indicating a selective effect of morphine-prime injection on reinstatement of CPP.
Figure 6.

Reinstatement of morphine CPP is associated with robust enhancement of LTP. A, A schematic of the experimental design is shown for the training and testing of morphine CPP, extinction of morphine CPP, and reinstatement of morphine CPP. B, Drug-induced reinstatement of morphine CPP. Mice exhibited a CPP when morphine was paired with a novel context (morphine-paired; n = 8–12 per group; one-way ANOVA; *p < 0.05), whereas no preference change was observed after extinction of morphine CPP. After extinction of morphine CPP, mice received a priming dose of either saline or morphine and were allowed to explore all three chambers. Mice that received a priming dose of morphine showed a significant preference for the morphine-paired chamber relative to all other groups (n = 8–12 per group; one-way ANOVA; *p < 0.05). C, No changes were observed in fEPSP slope after reinstatement of morphine CPP. Insets show fEPSP–FV relationships. D, The reinstatement of morphine CPP was associated with LTP was similar to saline-treated mice and morphine-unpaired mice. In contrast, mice that received a saline priming dose did not exhibit reinstatement of morphine CPP and had disrupted LTP when compared with all other groups (n = 3–4 per group; one-way ANOVA; *p < 0.05, **p < 0.01). Insets show comparisons of fEPSP slopes during the final 10 min of recording.
After reinstatement of morphine CPP, hippocampi were dissected and sliced for use in electrophysiological experiments. No changes in basal synaptic transmission were observed between groups when fEPSPs were directly examined (Fig. 6C) or when fEPSPs were normalized to presynaptic FV amplitude. Recordings of hippocampal LTP after reinstatement or morphine CPP revealed that mice in the morphine-paired/saline group, which did not show reinstatement of morphine CPP, had significantly impaired LTP when compared with all other groups (Fig. 6D; n = 3–4 per group; F(3,36) = 37.3, p < 0.05). In contrast, mice in the morphine CPP/morphine group showed reinstatement of morphine CPP and had LTP that was indistinguishable from LTP observed in saline-treated mice and morphine-unpaired mice. Together, these data suggest that morphine-primed reinstatement may lead to a restoration of hippocampal LTP, which in turn may be a critical synaptic mechanism for the reinstatement of expressed morphine CPP.
We observed that NR2b expression was increased after the expression and the extinction of morphine CPP, suggesting a possible role for NR2b in reinstatement of morphine CPP. To investigate this, mice were bilaterally cannulated in the hippocampus. These mice were conditioned and tested in an unbiased morphine CPP procedure (Fig. 7A) and then morphine CPP was extinguished as described above. Twenty-four hours after testing for the extinction of morphine CPP, mice were infused with either aCSF or ifenprodil (2 μg/1 μl), placed in the home cage for 30 min, and then received a subcutaneous priming dose of 5 mg/kg morphine. Mice were then allowed to explore all three chambers for 15 min while preference for the drug-paired chamber was recorded. Mice infused with ifenprodil did not show reinstatement to morphine CPP (Fig. 7B; n = 5 per group; F(4,3) = 2.763, p < 0.05), whereas the aCSF-injected control groups showed a typical robust morphine-primed reinstatement of CPP. In addition, locomotor activity was measured during the reinstatement phase, and no significant difference in activity was observed between mice infused with aCSF or ifenprodil, demonstrating that the ifenprodil inhibition of reinstatement is not attributable to decreased activity after infusion of drug (Fig. 7C). Together, these data suggest that blockade of NR2b with local administration of ifenprodil in the hippocampus prevents morphine-induced reinstatement.
Figure 7.

Local antagonism of the NR2b subunit in the hippocampus blocks reinstatement of morphine CPP. A, A schematic of the experimental design is shown for the training and testing of morphine CPP, extinction of morphine CPP, ifenprodil infusion, and reinstatement of morphine CPP. B, Drug-induced reinstatement of morphine CPP. After extinction of morphine CPP, mice were infused with either aCSF or ifenprodil 30 min before a priming dose of morphine and were allowed to explore all three chambers. Ifenprodil significantly blocked morphine-primed reinstatement (n = 5 per group; Student's t test; *p < 0.05). C, Locomotor activity measured in 5 min bins during reinstatement phase. Data represent mean ± SEM distance traveled (centimeters), n = 5. D, Schematic of three sections spanning the target region of cannulae placement; filled black circles represent cannulae placement in each mouse (n = 5 per group).
Discussion
Dissecting the association between hippocampal-dependent mechanisms, for drugs such as morphine, with environmental stimuli are an important and understudied component of addiction. Drug-associated environmental stimuli can lead to cravings, which in turn contribute to maintenance of drug use or lead to relapse (Daglish et al., 2001; See, 2002; Rukstalis et al., 2005; Gould, 2006). Previous studies have indicated that alterations of glutamatergic transmission in the hippocampus are crucial for behavioral measures of context-dependent morphine treatment, such as morphine CPP, behavioral sensitization, and cue or context-induced reinstatement of self-administration (Bossert et al., 2006; Zarrindast et al., 2007; LaLumiere and Kalivas, 2008; Zhou and Kalivas, 2008; Knackstedt and Kalivas, 2009; Xia et al., 2011), but it has remained unclear whether morphine CPP is associated with changes to NMDAR-dependent forms of synaptic plasticity, such as LTP. In the present study, we demonstrate for the first time that morphine CPP, extinction of morphine CPP, and reinstatement of morphine CPP are all associated with robust changes in hippocampal synaptic plasticity. Expression of morphine CPP was associated with increased basal synaptic transmission, impaired hippocampal LTP, and increased synaptic expression of the NR1 and NR2b NMDAR subunits. These changes in NMDAR expression and synaptic plasticity were not observed when morphine treatment was not associated with a specific context. After extinction of morphine CPP, hippocampal LTP was impaired and synaptic NR2a and NR2b expression was increased, suggesting that some changes in hippocampal synaptic plasticity and synaptic NMDAR expression observed during expression of morphine CPP may be similar to those observed after extinction training (Table 1). Interestingly, hippocampal LTP was indistinguishable from saline controls when a priming dose of morphine was used to reinstate morphine CPP. In contrast, a priming injection of saline did not reinstate morphine CPP and was associated with disrupted LTP. Finally, the blockade of the NR2b subunit with ifenprodil in the hippocampus prevented morphine-induced reinstatement of CPP.
The expression of CPP is a widely used pavlovian behavioral paradigm that models the association between the rewarding effects of drugs and contextual stimuli that occur in opioid addicts (Bardo and Bevins, 2000). Results from both the present study and previous findings suggest that hippocampal glutamatergic transmission may be crucial for the development and maintenance of context-dependent associative behaviors. In the present study, NR1 and NR2a NMDAR subunit expression was increased in the homogenate after the expression of morphine CPP, whereas expression of NR1, NR2a, and NR2b was increased in the PSD. In contrast, only NR2a expression was increased when morphine treatment was not paired with a particular context. Thus, the increased NR2a expression observed in the morphine CPP group and the unpaired morphine group suggests that this change in NMDAR subunit expression may be attributable to treatment with morphine or 24 h withdrawal from morphine. The results reported here are similar to previous studies that report an increase in NR2b subunit expression in the hippocampus or nucleus accumbens after morphine CPP or that have used a selective NR2b antagonist (ifenprodil) to block morphine CPP (Ma et al., 2006, 2011b; Xu et al., 2012). However, no studies to date have directly determined how hippocampal NMDAR expression is affected in the PSD after a morphine CPP paradigm. Furthermore, previous studies using other behavioral models have demonstrated that context-dependent morphine treatment requires glutamatergic transmission in the hippocampus (Ma et al., 2006, 2011b; Kao et al., 2011; Xu et al., 2012). Studies of other drugs of abuse, such as cocaine, heroin, and nicotine, indicate that glutamatergic transmission may also be important for drug–context associations (Gould and Lewis, 2005; Bossert et al., 2006; Crombag et al., 2008), but this hypothesis requires additional examination.
In the present study, we observed that morphine CPP was associated with changes in NMDAR expression, which may also lead to alterations of hippocampal synaptic plasticity. After the expression of morphine CPP, basal synaptic transmission was enhanced and LTP was significantly impaired in the hippocampus. These effects were not observed when morphine was not paired with a specific context. Therefore, it is unlikely that changes in hippocampal synaptic plasticity are attributable to withdrawal from unpaired morphine treatment. Furthermore, we also observed that this reduction in LTP was not observed when animals were kept in their home cages 7 d after the initial morphine CPP test, suggesting that the alterations in synaptic plasticity may be a consequence of the reexposure to the drug-paired context. The increased basal synaptic transmission observed after morphine CPP expression may in fact be related to increased NR1, NR2a, and NR2b subunit expression observed in the PSD. Consistent with this interpretation, previous studies have reported that increased NMDAR subunit expression at the synapse could lead to increased calcium permeability and could potentiate hippocampal synaptic responses (MacDermott et al., 1986; Jahr and Stevens, 1987; Malenka et al., 1988). Furthermore, it is possible that increased synaptic NMDAR expression after morphine CPP could lead to a saturation of NMDARs at the synapse, which in turn could produce an occlusion of LTP in the hippocampus (Nicoll and Malenka, 1999). Conversely, a growing body of evidence suggests that the NR2b subunit plays a critical role in plasticity (Barria and Malinow, 2005; Foster et al., 2010; Kiraly et al., 2011). Indeed, it has been described that the NR2b subunit forms a complex with CaMKII and that reduction of this complex should produce a disruption of synaptic plasticity (Sanhueza et al., 2011). Based on our findings, one possible explanation is that the increase in the expression of NR2b at the PSD observed after the expression and the extinction of morphine CPP could interfere with the formation of the NR2b–CaMKII complex and thus disrupt the induction and maintenance of LTP. Together with our current results, this suggests that context-dependent morphine treatment produces enhanced basal synaptic transmission and impaired LTP, and these effects may be attributable to enhanced glutamatergic transmission at hippocampal synapses.
The extinction of drug-associated contextual memories may help reduce the likelihood of relapse (Peters et al., 2008b), but changes in neural function that are resistant to extinction are important because these changes could lead to failure of extinction and may facilitate relapse of drug-seeking behavior (Ovari and Leri, 2008; Peters et al., 2008a). We found that, after extinction of morphine CPP, hippocampal LTP was impaired and NR2b subunit expression was increased in the PSD. In contrast, no changes in LTP or NMDAR subunit expression were observed when mice received unpaired morphine treatment. Although the results observed during extinction were similar to those after expression of morphine CPP, basal synaptic transmission was not altered after extinction but it was increased after expression of morphine CPP. The lack of effect on basal transmission after extinction training may be attributable to the fact that, unlike NR2b, synaptic levels of NR2a subunits returned to normal. It has been shown previously by our laboratory and others that the NR2a subunit is a major contributor to NMDAR currents in CA1 hippocampus (Bartlett et al., 2007; Foster et al., 2010; Fakira et al., 2013). Thus, the fact that no changes in NR2a expression were observed after extinction training may account for the lack of effect on basal synaptic transmission.
Because extinction training reflects an active learning process that may not be different from that which is engaged when animals are acquiring the initial morphine–environment association, it is not surprising that the impaired LTP is observed after both the expression and extinction of CPP. However, it is likely that the impaired LTP observed after extinction may require different neural mechanisms from those required for the expression of CPP. This is because only NR2b subunit expression was increased after extinction and basal synaptic transmission remained unchanged.
Reinstatement of morphine CPP can be used as a high throughput behavioral model of relapse to drug use. In the present study, a priming dose of morphine was sufficient to reinstate morphine CPP, but the infusion of the NMDAR blocker ifenprodil directly into the hippocampus blocked morphine reinstatement. These behavioral findings correlate with the increase in NR2b, which may be associated with changes/restoration of LTP via CaMKII (Sanhueza et al., 2011). Interestingly, recordings of hippocampal LTP in mice that showed morphine-primed reinstatement were similar to saline controls. In contrast, mice showed impaired hippocampal LTP after a priming dose of saline that did not lead to reinstatement of morphine CPP. This restoration of LTP after morphine-induced reinstatement may be attributable to the fact that hippocampal plasticity becomes “opioid dependent” after drug conditioning. Indeed, previous studies have shown that long-term chronic morphine treatment leads to impaired hippocampal LTP (Pu et al., 2002; Bao et al., 2007). Interestingly, this impaired LTP was restored by reexposure of the animals to opioids, suggesting that this hippocampal plasticity has been adapted to the presence of opioids. Given the role of the cAMP pathway in the synaptic effects of opioids, Pu et al., (2002) show that this opioid-dependent hippocampal plasticity is regulated by the activation of the cAMP pathway such that overactivation of the cAMP pathway after chronic opioid exposure seems to be responsible for the reduction in LTP. Overactivation of this pathway can then be suppressed by the reexposure to the opioid, which results in restoration of LTP.
Studies of heroin self-administration have revealed that mGluR agonists and AMPA antagonists in the nucleus accumbens can block reinstatement of heroin self-administration (Bossert et al., 2006; LaLumiere and Kalivas, 2008; Zhou and Kalivas, 2008). Together, these results suggest that hippocampal glutamatergic signaling may be important for both reinstatement of morphine CPP and changes in hippocampal LTP that occur after morphine CPP. Investigations of the mechanisms underlying these changes to behavior and synaptic plasticity are important extensions of the current work. A better understanding of the molecular mechanisms involved in LTP impairment observed during morphine CPP is necessary to identify molecular targets that enhance the extinction of morphine CPP or prevent reinstatement of CPP. These studies provide useful information for the potential development of new pharmacotherapeutics for treating opioid addiction.
Footnotes
This work was supported by National Institute of Health Grants DA025036, DA027460 (J.A.M.), R00DA025182 (M.R.B.), and MH101956 (J.G.M.) and McDonnell Center for Systems Neuroscience (M.R.B.).
The authors have no conflicts of interest to declare.
References
- Al-Hasani R, McCall JG, Bruchas MR. Exposure to chronic mild stress prevents kappa opioid-mediated reinstatement of cocaine and nicotine place preference. Front Pharmacol. 2013a;4:96. doi: 10.3389/fphar.2013.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, McCall JG, Foshage AM, Bruchas MR. Locus coeruleus kappa opioid receptors modulate reinstatement of cocaine place preference through a noradrenergic mechanism. Neuropsychopharmacology. 2013b;38:2484–2497. doi: 10.1038/npp.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart G. Maintenance medication for opiate addiction: the formation of recovery. J Addict Dis. 2012;31:207–225. doi: 10.1080/10550887.2012.694598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao G, Kang L, Li H, Li Y, Pu L, Xia P, Ma L, Pei G. Morphine and heroin differentially modulate in vivo hippocampal LTP in opiate-dependent rat. Neuropsychopharmacology. 2007;32:1738–1749. doi: 10.1038/sj.npp.1301308. [DOI] [PubMed] [Google Scholar]
- Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52:60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Benturquia N, Le Guen S, Canestrelli C, Lagente V, Apiou G, Roques BP, Noble F. Specific blockade of morphine- and cocaine-induced reinforcing effects in conditioned place preference by nitrous oxide in mice. Neuroscience. 2007;149:477–486. doi: 10.1016/j.neuroscience.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Billa SK, Sinha N, Rudrabhatla SR, Morón JA. Extinction of morphine-dependent conditioned behavior is associated with increased phosphorylation of the GluR1 subunit of AMPA receptors at hippocampal synapses. Eur J Neurosci. 2009;29:55–64. doi: 10.1111/j.1460-9568.2008.06560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billa SK, Liu J, Bjorklund NL, Sinha N, Fu Y, Shinnick-Gallagher P, Morón JA. Increased insertion of glutamate receptor 2-lacking alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors at hippocampal synapses upon repeated morphine administration. Mol Pharmacol. 2010;77:874–883. doi: 10.1124/mol.109.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert JM, Gray SM, Lu L, Shaham Y. Activation of group II metabotropic glutamate receptors in the nucleus accumbens shell attenuates context-induced relapse to heroin seeking. Neuropsychopharmacology. 2006;31:2197–2209. doi: 10.1038/sj.npp.1300977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007a;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007b;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C. Selective p38α MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton WM, Volkow ND. Major increases in opioid analgesic abuse in the United States: concerns and strategies. Drug Alcohol Depend. 2006;81:103–107. doi: 10.1016/j.drugalcdep.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Crombag HS, Bossert JM, Koya E, Shaham Y. Review. Context-induced relapse to drug seeking: a review. Philos Trans R Soc Lond B Biol Sci. 2008;363:3233–3243. doi: 10.1098/rstb.2008.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daglish MR, Weinstein A, Malizia AL, Wilson S, Melichar JK, Britten S, Brewer C, Lingford-Hughes A, Myles JS, Grasby P, Nutt DJ. Changes in regional cerebral blood flow elicited by craving memories in abstinent opiate-dependent subjects. Am J Psychiatry. 2001;158:1680–1686. doi: 10.1176/appi.ajp.158.10.1680. [DOI] [PubMed] [Google Scholar]
- Einstein EB, Asaka Y, Yeckel MF, Higley MJ, Picciotto MR. Galanin-induced decreases in nucleus accumbens/striatum excitatory postsynaptic potentials and morphine conditioned place preference require both galanin receptor 1 and galanin receptor 2. Eur J Neurosci. 2013;37:1541–1549. doi: 10.1111/ejn.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersche KD, Clark L, London M, Robbins TW, Sahakian BJ. Profile of executive and memory function associated with amphetamine and opiate dependence. Neuropsychopharmacology. 2006;31:1036–1047. doi: 10.1038/sj.npp.1300889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakira AK, Portugal GS, Carusillo B, Melyan Z, Morón JA. Increased small conductance calcium-activated potassium type 2 channel-mediated negative feedback on N-methyl-d-aspartate receptors impairs synaptic plasticity following context-dependent sensitization to morphine. Biol Psychiatry. 2013 doi: 10.1016/j.biopsych.2013.04.026. doi: 10.1016/j.biopsych.2013.04.026. Advance online publication. Retrieved November 25, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, McLaughlin N, Edbauer D, Phillips M, Bolton A, Constantine-Paton M, Sheng M. Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J Neurosci. 2010;30:2676–2685. doi: 10.1523/JNEUROSCI.4022-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TJ. Nicotine and hippocampus-dependent learning: implications for addiction. Mol Neurobiol. 2006;34:93–107. doi: 10.1385/MN:34:2:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TJ, Lewis MC. Coantagonism of glutamate receptors and nicotinic acetylcholinergic receptors disrupts fear conditioning and latent inhibition of fear conditioning. Learn Mem. 2005;12:389–398. doi: 10.1101/lm.89105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr CE, Stevens CF. Glutamate activates multiple single channel conductances in hippocampal neurons. Nature. 1987;325:522–525. doi: 10.1038/325522a0. [DOI] [PubMed] [Google Scholar]
- Kao JH, Huang EY, Tao PL. NR2B subunit of NMDA receptor at nucleus accumbens is involved in morphine rewarding effect by siRNA study. Drug Alcohol Depend. 2011;118:366–374. doi: 10.1016/j.drugalcdep.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Kim HS, Jang CG, Park WK. Inhibition by MK-801 of morphine-induced conditioned place preference and postsynaptic dopamine receptor supersensitivity in mice. Pharmacol Biochem Behav. 1996;55:11–17. doi: 10.1016/0091-3057(96)00078-0. [DOI] [PubMed] [Google Scholar]
- Kiraly DD, Lemtiri-Chlieh F, Levine ES, Mains RE, Eipper BA. Kalirin binds the NR2B subunit of the NMDA receptor, altering its synaptic localization and function. J Neurosci. 2011;31:12554–12565. doi: 10.1523/JNEUROSCI.3143-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Kalivas PW. Glutamate and reinstatement. Curr Opin Pharmacol. 2009;9:59–64. doi: 10.1016/j.coph.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, Carroll FI, Nestler EJ, Konradi C, Carlezon WA., Jr Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry. 2011;70:425–433. doi: 10.1016/j.biopsych.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28:3170–3177. doi: 10.1523/JNEUROSCI.5129-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Roth CA, Chavkin C. Signaling events initiated by kappa opioid receptor activation: quantification and immunocolocalization using phospho-selective KOR, p38 MAPK, and K(IR) 3.1 antibodies. Methods Mol Biol. 2011;717:197–219. doi: 10.1007/978-1-61779-024-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang XS, Dai RP, Zhang JY, Zhou XF, Hao W, Li CQ. The activation of NMDA receptor-ERK pathway in the central amygdala is required for the expression of morphine-conditioned place preference in the rat. Neurotox Res. 2011;20:362–371. doi: 10.1007/s12640-011-9250-2. [DOI] [PubMed] [Google Scholar]
- Ma XC, Jiang D, Jiang WH, Wang F, Jia M, Wu J, Hashimoto K, Dang YH, Gao CG. Social isolation-induced aggression potentiates anxiety and depressive-like behavior in male mice subjected to unpredictable chronic mild stress. PLoS One. 2011a;6:e20955. doi: 10.1371/journal.pone.0020955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YY, Guo CY, Yu P, Lee DY, Han JS, Cui CL. The role of NR2B containing NMDA receptor in place preference conditioned with morphine and natural reinforcers in rats. Exp Neurol. 2006;200:343–355. doi: 10.1016/j.expneurol.2006.02.117. [DOI] [PubMed] [Google Scholar]
- Ma YY, Chu NN, Guo CY, Han JS, Cui CL. NR2B-containing NMDA receptor is required for morphine-but not stress-induced reinstatement. Exp Neurol. 2007;203:309–319. doi: 10.1016/j.expneurol.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Ma YY, Yu P, Guo CY, Cui CL. Effects of ifenprodil on morphine-induced conditioned place preference and spatial learning and memory in rats. Neurochem Res. 2011b;36:383–391. doi: 10.1007/s11064-010-0342-9. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Zucker RS, Nicoll RA. Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.CPJ06007X. [DOI] [PubMed] [Google Scholar]
- Morón JA, Abul-Husn NS, Rozenfeld R, Dolios G, Wang R, Devi LA. Morphine administration alters the profile of hippocampal postsynaptic density-associated proteins: a proteomics study focusing on endocytic proteins. Mol Cell Proteomics. 2007;6:29–42. doi: 10.1074/mcp.M600184-MCP200. [DOI] [PubMed] [Google Scholar]
- Muller D, Joly M, Lynch G. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science. 1988;242:1694–1697. doi: 10.1126/science.2904701. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Ovari J, Leri F. Inactivation of the ventromedial prefrontal cortex mimics re-emergence of heroin seeking caused by heroin reconditioning. Neurosci Lett. 2008;444:52–55. doi: 10.1016/j.neulet.2008.08.015. [DOI] [PubMed] [Google Scholar]
- Peters J, LaLumiere RT, Kalivas PW. Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J Neurosci. 2008a;28:6046–6053. doi: 10.1523/JNEUROSCI.1045-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Vallone J, Laurendi K, Kalivas PW. Opposing roles for the ventral prefrontal cortex and the basolateral amygdala on the spontaneous recovery of cocaine-seeking in rats. Psychopharmacol Berl. 2008b;197:319–326. doi: 10.1007/s00213-007-1034-2. [DOI] [PubMed] [Google Scholar]
- Portugal GS, Gould TJ. Nicotine withdrawal disrupts new contextual learning. Pharmacol Biochem Behav. 2009;92:117–123. doi: 10.1016/j.pbb.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser J, Cohen LJ, Steinfeld M, Eisenberg D, London ED, Galynker II. Neuropsychological functioning in opiate-dependent subjects receiving and following methadone maintenance treatment. Drug Alcohol Depend. 2006;84:240–247. doi: 10.1016/j.drugalcdep.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Bao GB, Xu NJ, Ma L, Pei G. Hippocampal long-term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates. J Neurosci. 2002;22:1914–1921. doi: 10.1523/JNEUROSCI.22-05-01914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro Do Couto B, Aguilar MA, Manzanedo C, Rodríguez-Arias M, Miñarro J. NMDA glutamate but not dopamine antagonists blocks drug-induced reinstatement of morphine place preference. Brain Res Bull. 2005;64:493–503. doi: 10.1016/j.brainresbull.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Rukstalis M, Jepson C, Patterson F, Lerman C. Increases in hyperactive-impulsive symptoms predict relapse among smokers in nicotine replacement therapy. J Subst Abuse Treat. 2005;28:297–304. doi: 10.1016/j.jsat.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See RE. Neural substrates of conditioned-cued relapse to drug-seeking behavior. Pharmacol Biochem Behav. 2002;71:517–529. doi: 10.1016/S0091-3057(01)00682-7. [DOI] [PubMed] [Google Scholar]
- Shapiro ML, Eichenbaum H. Hippocampus as a memory map: synaptic plasticity and memory encoding by hippocampal neurons. Hippocampus. 1999;9:365–384. doi: 10.1002/(SICI)1098-1063(1999)9:4<365::AID-HIPO4>3.0.CO%3B2-T. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- Smith JS, Schindler AG, Martinelli E, Gustin RM, Bruchas MR, Chavkin C. Stress-induced activation of the dynorphin/κ-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J Neurosci. 2012;32:1488–1495. doi: 10.1523/JNEUROSCI.2980-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki W, Turek A, Kubik J, Przewlocki R. Motivational effects of opiates in conditioned place preference and aversion paradigm—a study in three inbred strains of mice. Psychopharmacol (Berl) 2009;207:245–255. doi: 10.1007/s00213-009-1672-7. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol. 2007;12:227–462. doi: 10.1111/j.1369-1600.2007.00070.x. [DOI] [PubMed] [Google Scholar]
- Xia Y, Portugal GS, Fakira AK, Melyan Z, Neve R, Lee HT, Russo SJ, Liu J, Morón JA. Hippocampal GluA1-containing AMPA receptors mediate context-dependent sensitization to morphine. J Neurosci. 2011;31:16279–16291. doi: 10.1523/JNEUROSCI.3835-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lv XF, Cui CL, Ge FF, Li YJ, Zhang HL. Essential role of NR2B-containing NMDA receptor-ERK pathway in nucleus accumbens shell in morphine-associated contextual memory. Brain Res Bull. 2012;89:22–30. doi: 10.1016/j.brainresbull.2012.06.012. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Lashgari R, Rezayof A, Motamedi F, Nazari-Serenjeh F. NMDA receptors of dorsal hippocampus are involved in the acquisition, but not in the expression of morphine-induced place preference. Eur J Pharmacol. 2007;568:192–198. doi: 10.1016/j.ejphar.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Zhang XH, Liu SS, Yi F, Zhuo M, Li BM. Delay-dependent impairment of spatial working memory with inhibition of NR2B-containing NMDA receptors in hippocampal CA1 region of rats. Mol Brain. 2013;6:13. doi: 10.1186/1756-6606-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Kalivas PW. N-acetylcysteine reduces extinction responding and induces enduring reductions in cue- and heroin-induced drug-seeking. Biol Psychiatry. 2008;63:338–340. doi: 10.1016/j.biopsych.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]