

Abstract
Background
The production of enzymes by an industrial strain requires a complex adaption of the bacterial metabolism to the conditions within the fermenter. Regulatory events within the process result in a dynamic change of the transcriptional activity of the genome. This complex network of genes is orchestrated by proteins as well as regulatory RNA elements. Here we present an RNA-Seq based study considering selected phases of an industry-oriented fermentation of Bacillus licheniformis.
Results
A detailed analysis of 20 strand-specific RNA-Seq datasets revealed a multitude of transcriptionally active genomic regions. 3314 RNA features encoded by such active loci have been identified and sorted into ten functional classes. The identified sequences include the expected RNA features like housekeeping sRNAs, metabolic riboswitches and RNA switches well known from studies on Bacillus subtilis as well as a multitude of completely new candidates for regulatory RNAs. An unexpectedly high number of 855 RNA features are encoded antisense to annotated protein and RNA genes, in addition to 461 independently transcribed small RNAs. These antisense transcripts contain molecules with a remarkable size range variation from 38 to 6348 base pairs in length. The genome of the type strain B. licheniformis DSM13 was completely reannotated using data obtained from RNA-Seq analyses and from public databases.
Conclusion
The hereby generated data-sets represent a solid amount of knowledge on the dynamic transcriptional activities during the investigated fermentation stages. The identified regulatory elements enable research on the understanding and the optimization of crucial metabolic activities during a productive fermentation of Bacillus licheniformis strains.
Keywords: dRNA-Seq, RNA-based regulation, UTR, ncRNA, sRNA, Antisense RNA, Subtilisin, Transcription start site, Operon prediction, Reannotation
Background
Bacillus licheniformis is a spore-forming soil bacterium closely related to the Gram-positive model organism Bacillus subtilis. The species’ saprophytic life style, based on the secretion of biopolymer-degrading enzymes, predestinates strains of B. licheniformis as ideal candidates for the large-scale industrial production of exoenzymes, such as amylases and peptide antibiotics [1]. Especially its high capacity of secreting overexpressed alkaline serine proteases has made B. licheniformis one of the most important bacterial workhorses in industrial enzyme production [2]. Due to their high stability and relatively low substrate specificity, alkaline serine proteases like subtilisins are crucial additives to household detergents and the greatest share on the worldwide enzyme market [2,3]. Attempts to optimize the productivity have addressed the fermentation process [4,5], protein-engineering [3,6,7], and cellular influences on protein quality and quantity [2,8]. Since the 4.2 Mb circular genome of the type strain B. licheniformis DSM13 was published in 2004 [1,9], several genome-based studies targeting strain improvement have been performed successfully [10,11]. However, genome-based studies are limited to information directly accessible from the DNA sequence and cannot benefit from knowledge of the active transcriptome. Considering that the regulatory network represented by protein- and RNA-based regulators determines the performance of an industrial-oriented fermentation process [12] RNA-Seq data might contribute to further optimization approaches.
RNA-based regulatory elements are involved in the regulation of metabolism, growth processes, the adaptation to stress and varying culture conditions [13] and can be divided into two main categories. The first category comprises non-coding RNAs (ncRNAs). Trans-encoded ncRNAs, often referred to as small RNAs (sRNAs), are encoded independently from protein genes and are able to modulate the mRNA of genes located at distant chromosomal loci or to interact with target proteins [14]. Upon formation of secondary structures, trans-encoded ncRNAs interact with their target RNAs by imperfect base pairing, which is triggered by the binding of a seed region of at least six contiguous nucleotides [15,16]. This mechanism allows the sRNA to address multiple targets, thus orchestrating different members of one regulon [14,17]. It has been shown that sRNAs affect mRNA degradation and translation and modulate protein activity [14,16]. A second class of regulatory ncRNAs is encoded in cis, which means that these ncRNAs are transcribed from the antisense strand of protein-coding genes [18]. Hence, they are complementary in full-length and can therefore form RNA duplexes with the mRNA of the targeted genes [19]. Most described examples of these cis-encoded antisense RNAs (asRNA) range from 100 to 300 nt in size, but some asRNAs are also shown to be substantially longer [18,20]. Antisense RNAs have been proven to either positively or negatively affect transcription, translation and mRNA stability [16]. In addition, a cis-encoded asRNA might work as a trans-encoded sRNA for another target [19]. Antisense transcription has been detected in multiple organisms [21] and, with the growing number of explored species, it is assumed that antisense transcripts can be found for ~10 to 20% of the bacterial genes [22]. A second class of RNA-based regulators encompasses cis-regulatory elements, mainly present at the 5′ untranslated region (5′UTR) of mRNA transcripts, e.g. riboswitches, T-boxes or thermosensors [23]. Whereas both 5′ as well as 3’untranslated regions can bear signals for the initiation and termination of translation [24,25], respectively, 5′UTRs additionally have the ability to fine-tune translation by cis-regulatory elements. They can be prone to RNA-binding proteins or antisense RNAs, carry attenuation systems [14,23] and play a role in mRNA stability [26]. In contrast, 3’UTRs are not as well understood and have escaped the attention of most transcriptomic studies [27]. It is known that long UTRs can be localized antisense to adjacent genes on the opposite strand; in fact some of these overlapping UTRs have been demonstrated to act as negative regulators for genes encoded on the opposite strand [20].
The development of next-generation sequencing techniques including RNA sequencing (RNA-Seq) enabled the genome-wide identification of RNA-based regulatory elements in an unprecedented depth. The high dynamic range of RNA-Seq allows the identification of transcripts which are expressed at vastly different levels. Also, this method does not exhibit background noise and is therefore appropriate for the identification of lowly abundant transcripts [28]. RNA-Seq analyses targeting ncRNA in particular, have been published for e.g. Mycobacterium tuberculosis[29], Streptomyces coelicolor[30] and Sinorhizobium meliloti[31].
The major goal of the project in which this study is embedded is the improvement of production strains and thus ultimately the enhancement of enzyme production. This study is targeted on the identification of active regulatory RNA elements within the different phases of a productive fermentation process. Therefore samples from crucial stages of an industrial-oriented B. licheniformis subtilisin fermentation process have been examined by strand-specific RNA-Seq and differential RNA-Seq (dRNA-Seq) [32]. A comprehensive analysis of the data revealed a multitude of RNA features which correlate to the physiology and the growth phases during the process. The combination of genomic data and RNA features provides an excellent basis to understand the regulatory events within an industrial fermentation process.
Results and discussion
B. licheniformis MW3Δspo, a germination deficient mutant of B. licheniformis DSM13, transformed with an expression plasmid encoding an alkaline serine protease, was grown in fed-batch mode in 6 L cultures. The fermentations were carried out in complex amino acid broth under conditions resembling the parameters used in industrial fermentation processes (Figure 1). To enhance the reliability of the analysis, the experiments were carried out in triplicate (L, R and M). Samples were taken at five selected time points of the fermentation process, which were chosen to follow the initial cell growth (sampling points I, II and III) and to determine the decisive changes within the early (IV) and the late stage (V) of the protease-producing states (Figure 1). Total RNA from each sample was prepared for strand-specific whole transcriptome sequencing [33]. RNA from samples L-I to L-V was additionally prepared for differential RNA-Seq for determination of transcription start sites (TSS), as described by Sharma et al. [32].
Figure 1.

Protease production and process parameters. Process parameters are shown for fermentation L (the parameters for the replicate fermentations R and M are corresponding, data not shown). Temperature T [°C], oxygen partial pressure pO2 [%], glucose concentration cGlucose [g/L], supplied glucose feedGlucose [g/L] and normalized protease activity [%] are displayed on left y-axis, whereas acetate concentration cAcetate [g/L] and carbon dioxide content CO2 [%] are scaled on the right y-axis. Process time t [h] is given on the x-axis. The sampling points I to V are indicated by orange lines.
Whole transcriptome sequencing
Strand-specific deep sequencing of the whole transcriptome of 15 B. licheniformis samples yielded more than 500 million reads with a specific length of 50 nucleotides. The number of reads for each library ranged from 2.4 × 107 to 4.3 × 107.
After the application of a strict quality processing (see Methods), 77.3 to 93.9% of these reads have been found to map to the chromosome and the expression plasmid used in this study (for details see Additional file 1: Figure S1 and Additional file 2: Table S1). Due to repeat regions, 1.45% of the B. licheniformis genome is not precisely mappable when considering the applied read length of 50 nucleotides. Thus, all reads mapping completely to such repeat regions have been excluded from further analysis. This pertains mainly to those 68.5 to 88.8% of reads which map to tRNA and rRNA genes. The majority of these rRNA matching reads can be assigned to 5S rRNA genes, which is in accordance with the fact that the applied depletion targets especially 16S and 23S rRNAs. Also, all reads mapping to the plasmid were removed from the dataset, as this analysis is focused on the transcriptional activity of the chromosome. Finally, 4.4 to 12.0% of the initial reads were taken for further analyses. These reads enabled the identification of transcriptional units and the determination of their boundaries to assign the transcriptional activity of coding as well as non-coding regions of the chromosome (see Methods).
To facilitate the comparison of different transcription levels between samples, we introduce the nucleotide activity per kilobase of exon model per million mapped reads (NPKM) value as single nucleotide-resolution measure of transcriptional activity (see Methods). NPKMs for each RNA feature and for every gene were calculated and are available at Additional file 2: Table S2 and Table S3.
Transcription start site determination and operon prediction
Differential RNA-Seq (dRNA-Seq) has been designed by Sharma et al. [32] to allow selective enrichment of native 5′ ends of transcripts for the determination of transcription start sites (TSS). The method is based on the observation that 5′ triphosphorylated RNA fragments are originating from native 5′ ends. In contrast, 5′ monophosphorylated RNAs are products of RNA decay or processing and do not contain information of transcription initiation. The dRNA-Seq approach includes a treatment with 5′ phosphate-dependent exonuclease (TEX), which results in the depletion of all monophosphorylated transcripts. It has been shown that TSS identification based on dRNA-Seq data is superior to an estimation of transcript boundaries based on whole transcriptome RNA-Seq reads [32].
The differential sequencing of samples L-I to L-V resulted in 22,047,373 reads (Additional file 2: Table S4). A total of 2522 putative TSS was predicted (see Methods), 1500 of which were detected in at least two samples (Additional file 2: Table S5). A comparison of the latter with the transcript boundaries obtained by whole transcriptome sequencing (Additional file 2: Table S6) shows that 412 identified TSS confirm the RNA-Seq data, whereas the other findings introduce TSS not detectable by conventional RNA-Seq. To allow the assignment of the identified TSS to their putative origin, an allocation to four different classes was accomplished (Figure 2A) [34]. Naturally, the affiliation of TSS according to this schema is ambiguous as some TSS sort to multiple classes, e. g. some TSS are located in a promoter region and within the upstream gene as well. The distribution of the identified TSS to each class is shown in Figure 2B. 1092 TSS were detected in promoter regions, 72 genes are bearing more than one putative TSS in this region. The dRNA-Seq data enabled conclusions for TSS determination in cases in which read-through transcription of the upstream gene caused by leaky termination prohibits the identification of downstream TSS by conventional RNA-Seq data. Of the identified 456 intragenic TSS, 267 are not located in the 500 bp promoter region of the downstream gene, reflecting a high number of putative internal promoters. Orphan TSS may indicate potential start sites of yet unknown genes or non-coding RNAs, this is supported by the finding that 76 of the 141 detected orphan TSS could be allocated to identified ncRNAs.
Figure 2.

Classification and distribution of TSS. (A) Classification scheme of transcription start sites adapted from Dötsch et al. [34]. White arrows indicate genes. P: Protein-coding gene-dependent TSS located within a 500 bp range upstream of annotated start codons. I: Intragenic TSS situated within an annotated gene on the same strand. A: TSS localized antisense to an annotated gene. O: Orphan TSS not located in a promoter region or a gene on the same strand. (B) Distribution of transcription start sites identified in this study. Numbers in brackets give the amount of instances for each class. Numbers in the legend give the total amount of every class.
Operon prediction based on RNA-Seq and dRNA-Seq data resulted in 2510 putative operons structuring the genome of B. licheniformis (Additional file 2: Table S7). While most operons are monocistronic (66.8%) or bicistronic (18.3%), seven operons seem to encompass more than ten genes (Additional file 1: Figure S2). This small number of long operons is not in accordance with the operon prediction made by Kristoffersen et al. [35] for B. cereus. The difference is due to the varying operon concept employed here. Especially the consideration of internal TSS in combination with distinct shifts of expression resulted in an increase of shortened operons in this study.
Reannotation
The first genome annotation of B. licheniformis DSM13 has been published in 2004 [1]. It has been shown previously that mapping of RNA-Seq data to genomes allows the correction of open reading frames and supports the identification of not-annotated protein genes [36]. Therefore, we performed a complete reannotation of the genome in order to integrate the RNA-Seq data provided by this study as well as the progress in gene prediction and annotation of the recent years. Distinct transcription start sites determined by dRNA-Seq and RNA-Seq-based whole transcriptome data have been used to identify putative mis-annotated genes (Figure 3A). These findings were validated by length comparisons to genes deposited in public databases and confirmation of ribosomal binding sites and -10 and -35 promoter regions. This approach enabled the correction of reading frames of 23 protein genes, 25 pseudo genes, 21 rRNA genes and two tRNA genes (Additional file 2: Table S8). Moreover, 60 previously not-annotated protein genes were identified based on transcriptional activity and protein conservation (Figure 3B, Additional file 2: Table S9). 52 genes (Additional file 2: Table S10) were removed from the annotation as these previously predicted ORFs could not be verified by detailed genome analysis and comparisons to public databases. In total, the reannotation approach resulted in a dataset containing 4297 ORFs. Comparisons to the annotation of B. licheniformis DSM13 by Rey et al. [9] showed that 16 of the newly annotated genes have not been described for this organism before. 18 of the removed genes were annotated in both genomes. More than 2000 gene annotations have been improved. These improvements mainly comprise former hypothetical proteins now assigned to a function and proteins with altered gene symbols. In addition to gene-associated improvements, seven genomic regions were identified as prophage regions based on GC content deviations, significant similarities to known prophage genes and the presence of insertion repeats. The transcriptional activity of the prophage regions was rather low, which is consistent with the observation that many prophages are induced during SOS response, which should not occur within a fermentation process [37].
Figure 3.
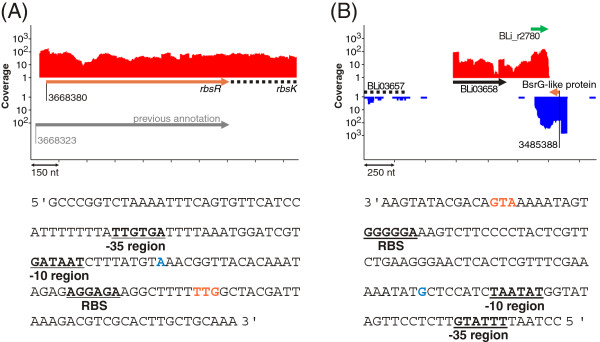
Correction and insertion of annotated genes. (A) Correction of start codons. (Upper panel) Transcriptional activity of pooled RNA-Seq data. The grey arrow displays the coordinates of the ribose operon repressor RbsR (BLi03840) according to Veith et al. [1]. Based on the transcriptional data, the start codon has been reassigned 57 bp downstream of the former position (orange arrow). (Lower panel) The new start codon is marked in orange and the transcription start site in blue. The location of patterns of a ribosomal binding site and -10 and -35 regions of the rbsR- regulating σA upstream of the gene provide additional confirmation to the new annotation. (B) Insertion of new genes. (Upper panel) Transcriptional activities (sample L-I) of BLi03658 (black arrow) and indep RNA BLi_r2780 (green arrow). The previously not detected [1,9] protein gene BLi05038 (orange arrow) was annotated as BsrG-like peptide (see also chapter Comparative transcriptomics). (Lower panel) The start codon of the new gene is marked in orange, and the transcription start site in green. The location of patterns of a ribosomal binding site and a σA -10 and -35 promoter region provide additional confirmation of the new annotation.
5′ and 3′untranslated regions
In this study, 1433 5′untranslated regions (Figure 4) with a mean length of 117 nt (Figure 5A, Additional file 2: Table S6) could be identified. Thirty of these 5′UTRs are shorter than 11 nt, implicating that leaderless transcription, commonly found in many bacteria [38], is not an abundant mechanism in B. licheniformis. Correspondingly, low occurrence of leaderless transcription has also been suggested for other members of the phylum Firmicutes[39]. The most strongly transcribed 5’UTRs ≥150 nt and the 5’UTRs discussed in the following passage are listed in Table 1.
Figure 4.

Classification and distribution of RNA features. (A) Classification scheme of ten RNA feature classes. White arrows indicate CDS and black arrows represent RNA transcripts. All antisense transcripts are framed green. RNA features which are part of an mRNA are denoted 5’UTRs or 3’UTRs. Antisense transcripts that are mRNA-bound were classified as A5’UTR, A3’UTR and Art. Non-coding antisense transcripts were classified as A5, A3 and AI and comprise antisense transcripts opposite to 5’ and 3’UTRs or to protein-coding regions of the mRNA. Amisc designates antisense ncRNAs that target more than one gene or are only partially antisense. Independently transcribed ncRNAs without any antisense localization are designated indep. (B) Quantitative affiliation of identified RNA features. (C)(Left) Proportional distribution of intergenic regions or annotated genes with different transcriptional activities within the complete B. licheniformis DSM13 genome. (Middle + Right) Percentage of the total genome covered by the defined RNA classes.
Figure 5.

Length distribution of RNA features. Size range of (A) 1433 identified 5’ untranslated regions, (B) 1365 identified 3’ untranslated regions and (C) 461 identified non-coding RNAs. Please note that the classification scheme corresponds to Figure 4.
Table 1.
Selected 5’untranslated regions (5'UTRs)
| RNA feature | Start | Stop | Length | Downstream gene | Antisense genes | cis -regulatory element | NPKM value* |
|---|---|---|---|---|---|---|---|
| BLi_r0085 |
210580 |
210926 |
347 |
thrZ |
|
T-box |
1966 |
| BLi_r0356 |
542296 |
542520 |
225 |
BLi00536 |
|
ydaO-yuaA leader |
1368 |
| BLi_r0498 |
712264 |
712587 |
324 |
yybP |
|
yybP-ykoY leader |
1144 |
| BLi_r0691 |
942026 |
942290 |
306 |
thiC |
|
TPP |
5030 |
| BLi_r0744 |
998913 |
999033 |
121 |
glpD |
|
|
5693 |
| BLi_r0943 |
1207762 |
1207542 |
180 |
yitJ |
|
SAM |
2782 |
| BLi_r0982 |
1243825 |
1243498 |
328 |
trpS |
|
T-box |
865 |
| BLi_r0983 |
1244045 |
1244232 |
188 |
oppA |
|
|
1465 |
| BLi_r1011 |
1271178 |
1271391 |
255 |
tenA |
|
TPP |
9963 |
| BLi_r1028 |
1291984 |
1292289 |
306 |
metI |
|
SAM |
3605 |
| BLi_r1168 |
1487545 |
1487279 |
226 |
mtnK |
|
SAM |
2207 |
| BLi_r1196 |
1510226 |
1511237 |
1012 |
BLi05023 |
BLi01539, BLi01540 |
|
651 |
| BLi_r1485 |
1973018 |
1973209 |
192 |
BLi02027 |
|
|
1035 |
| BLi_r1609 |
2118173 |
2117083 |
1091 |
gltA |
gltC |
|
34 |
| BLi_r1634 |
2146236 |
2145933 |
304 |
expZ |
|
|
1225 |
| BLi_r1709 |
2204969 |
2205111 |
143 |
dhaS |
|
|
2816 |
| BLi_r1801 |
2295779 |
2295571 |
168 |
xpt |
|
Purine |
1540 |
| BLi_r1835 |
2356768 |
2356478 |
291 |
hbs |
|
|
3478 |
| BLi_r1850 |
2382265 |
2381914 |
352 |
ribU |
|
FMN |
2367 |
| BLi_r1871 |
2409288 |
2409010 |
238 |
ribD |
|
FMN |
2512 |
| BLi_r2045 |
2616129 |
2615830 |
300 |
glyQ |
|
T-box |
787 |
| BLi_r2142 |
2742406 |
2742106 |
301 |
yrzI |
|
|
860 |
| BLi_r2241 |
2878256 |
2877902 |
313 |
lysC |
|
Lysine |
751 |
| BLi_r2286 |
2949768 |
2949565 |
204 |
citZ |
|
|
1563 |
| BLi_r2389 |
3060789 |
3060456 |
292 |
leuS |
|
T-box |
998 |
| BLi_r2510 |
3188213 |
3185988 |
2226 |
kapD |
yuxJ, pbpD |
|
13 |
| BLi_r2628 |
3302655 |
3302393 |
221 |
metN2 |
|
SAM |
2661 |
| BLi_r3184 |
4014316 |
4014539 |
224 |
yxjG |
|
SAM |
5296 |
| BLi_r3195 |
4037045 |
4036819 |
185 |
BLi04205 |
|
TPP |
9303 |
| BLi_r3196 | 4037110 | 4037236 | 127 | BLi04206 | 2693 |
*(pooled RNA-Seq data).
At sampling points I, II, and III, the gene of the sporulation inhibitor KapD (BLi03329) reveals a 5’UTR of 113 nt (Figure 6A), whereas an alternative, dRNA-Seq-supported 5’UTR (BLi_r2510) with a length of 2226 nt is present at the later stages of the fermentation process. The TSS of both 5’UTRs seem to be preceded by a σA recognition site. At sampling point IV, the transcriptional activity of the gene is higher than the activity of the 5’UTR region. In B. subtilis, growth phase-dependent differentiation into subpopulations of distinct cell types with different gene expression patterns is well described [40,41]. The divergent expression levels of kapD and the long 5’UTR in B. licheniformis might therefore result from different usage of promoter sites dependent on the respective cell type. However, the observed effect could also derive from slow decay rates of the short form of the kapD mRNA transcribed earlier. 52 further 5’UTRs exhibited antisense activity towards upstream genes (A 5’UTR ; Figure 4), as shown for the untranslated region BLi_r1609 upstream of the glutamate synthase operon gltAB (BLi02161/62; Figure 6B). The observed 5’UTR is completely antisense to the gene of the corresponding transcriptional activator GltC (BLi02163). The dRNA-Seq data suggest the presence of only one TSS. This finding might be an example for a regulatory linkage between adjacent genes localized on different strands. This concept has recently been termed the excludon by Sesto et al. [20], who demonstrated that long 5’UTRs can act negatively on the transcription of the opposite gene. Following this idea in the case of the glutamate synthase operon, the preceding 5’UTR might establish a negative feedback regulation of the transcriptional activator GltC. A corresponding elongated UTR of the gltAB operon has not been found in B. subtilis[42,43], which indicates different regulations of glutamate homeostasis in the two species.
Figure 6.

Untranslated regions (UTRs). Transcriptional activities of UTR regions. Black arrows indicate genes and green arrows the identified UTRs. (A) 5’UTR of kapD at sampling point II (left) and sampling point IV (right). (B) 5’UTR BLi_t1609 at sampling point IV. (C) 3’UTR BLi_r2654 (pooled RNA-Seq data). Predicted terminator sequences are marked orange.
Next to regulatory effects based on antisense orientation, 5’UTRs can bear intrinsic, so-called cis-regulatory elements [44]. At the time of this study, 62 cis-regulatory elements have been predicted for B. licheniformis DSM13 by covariance models [45,46]. All elements have been shown to be transcriptionally active during the fermentation process (Additional file 2: Table S11), although some are not located in 5’UTRs but in intergenic read-through regions. Three new T-boxes, located upstream of the serine acetyltransferase gene cysE and the tRNA ligase subunit genes glyQ and pheS, could be identified by comparison to the Rfam database. In B. subtilis, 92 cis-regulatory elements have been described [43], comprising RNA switches as well as protein-binding RNAs. For 76 of these instances, transcription could be shown at orthologous loci in B. licheniformis (Additional file 2: Table S12).
1365 3’UTRs (Additional file 2: Table S6) with an average length of 276 nt have been identified according to Figure 4, the most strongly transcribed 3’UTRs ≥150 nt and the 3’UTR discussed in this chapter are listed in Table 2. Of the identified 3’UTRs, 42% exceed 100 nt and 16% even exceed 500 nt in length (Figure 5B). In total, 338 3’UTRs are localized antisense to adjacent genes (A 3’UTR ; Figure 4). A detailed manual inspection revealed that all 3’untranslated regions longer than 1000 nt seem to be protruding after incomplete termination [18,32]. Altogether, 684 3’UTRs with internal termination sites could be determined, whereas 511 3’UTRs end at predicted termination sites. These findings suggest that the effect of fading-out at the end of operons due to imperfect termination might be a common effect in B. licheniformis. An example is the 3965 nt 3’UTR (BLi_r2654) downstream of the cell envelope stress response operon liaIHGFSR (BLi03492-97; Figure 6C). The mRNA transcript of this operon protrudes beyond a termination signal, which is located directly behind the stop codon of liaR. This protruding mRNA sequence is antisense to the next four genes which comprise the germination receptor operon gerAAABAC (BLi03488-90) and a hypothetical protein (BLi03491). A second terminator structure can be found 370 nt upstream of the end of the transcript.
Table 2.
Selected 3’untranslated regions (3'UTRs)
| RNA feature | Start | Stop | Length | Upstream gene | Antisense genes | NPKM value* |
|---|---|---|---|---|---|---|
| BLi_r0075 |
198675 |
198433 |
243 |
citM |
|
127 |
| BLi_r0671 |
919040 |
918595 |
446 |
ygzB |
perR1 |
50 |
| BLi_r0688 |
938151 |
938591 |
441 |
BLi00936 |
|
496 |
| BLi_r0817 |
1054694 |
1054523 |
172 |
msmX |
BLi01051 |
75 |
| BLi_r0859 |
1099760 |
1099445 |
316 |
ynzH |
yhfE |
2310 |
| BLi_r0949 |
1209837 |
1210136 |
300 |
BLi01196 |
|
188 |
| BLi_r1013 |
1278149 |
1277370 |
780 |
cotZ |
fabI, cotO |
116 |
| BLi_r1145 |
1465905 |
1468238 |
2334 |
ykoM |
ykoU, ykoV |
264 |
| BLi_r1333 |
1655028 |
1654895 |
134 |
ylaL |
|
195 |
| BLi_r1357 |
1680492 |
1679988 |
505 |
ylbP |
gerR |
62 |
| BLi_r1521 |
2008172 |
2008031 |
142 |
BLi02067 |
|
224 |
| BLi_r1720 |
2216049 |
2215342 |
708 |
odhB |
yocS |
41 |
| BLi_r1750 |
2244661 |
2244519 |
143 |
yodL |
yoyE |
16 |
| BLi_r1797 |
2291386 |
2291184 |
203 |
ypbQ |
|
268 |
| BLi_r1927 |
2463046 |
2465026 |
1981 |
BLi02544 |
BLi02545,ymaC |
213 |
| BLi_r1985 |
2537707 |
2537586 |
122 |
mntR |
|
151 |
| BLi_r1995 |
2550986 |
2550862 |
125 |
tasA |
|
482 |
| BLi_r2041 |
2606579 |
2606461 |
119 |
cccA |
|
431 |
| BLi_r2067 |
2662102 |
2661914 |
189 |
BLi02768 |
yrhD |
87 |
| BLi_r2141 |
2741955 |
2741415 |
541 |
yrzI |
|
348 |
| BLi_r2152 |
2754987 |
2754859 |
129 |
yrzB |
|
140 |
| BLi_r2178 |
2787395 |
2787201 |
195 |
yrbF |
|
1520 |
| BLi_r2292 |
2952407 |
2952263 |
145 |
pyk |
|
520 |
| BLi_r2582 |
3255915 |
3256420 |
506 |
yutI |
yuxL |
324 |
| BLi_r2620 |
3292613 |
3292481 |
133 |
sufB |
|
405 |
| BLi_r2654 |
3332662 |
3328698 |
3965 |
liaR |
gerAA, gerAB, gerAC, BLi03491 |
11 |
| BLi_r2700 |
3398622 |
3398373 |
250 |
copA |
|
166 |
| BLi_r2729 |
3427607 |
3428159 |
553 |
BLi05033 |
|
345 |
| BLi_r2752 |
3464447 |
3464713 |
267 |
BLi03635 |
|
286 |
| BLi_r2855 | 3591778 | 3591637 | 142 | cccB | 80 |
*(pooled RNA-Seq data).
Non-coding RNA features
Non-coding RNAs were identified in non-coding regions of the chromosome, for example in intergenic regions or localized in antisense direction to protein genes (see Methods). The boundaries of the identified transcripts were determined by upshifts or downshifts of transcriptional activity. All identified RNA features were checked for similarities to complete protein genes as well as protein domains to ensure that they indeed represent non-coding RNAs.
To extract the basic types of ncRNA expression profiles during the examined fermentation process, cluster analysis based on the k-means algorithm [47] was applied to those 273 ncRNAs with highly reliable replicates. In total, 15 clusters of divergent expression profiles were generated (Figure 7, Additional file 2: Table S13). Cluster 1 contains 36% of the applied ncRNAs and 50% of all ncRNAs >1000 nt. It displays a strong up-shift of transcriptional activity at sampling point IV followed by a decrease at sampling point V. The high portion of transcripts in this cluster prompts the conclusion that RNA-based regulation is especially important during the later stages of the fermentation process. Other ncRNAs exhibiting up-shifts of transcriptional activity are displayed in clusters 2 to 4, whereas clusters 5 to 8 include transcripts with activity down-shifts. The further clusters comprise ncRNAs with expression shifts during the early fermentation process, as well as an activity up-shift at sampling point V in clusters 10 to 12.
Figure 7.
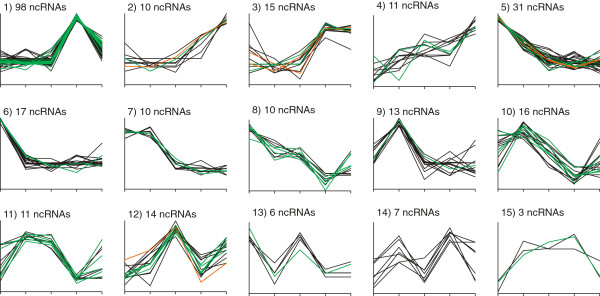
Cluster analysis of ncRNA expression profiles. Expression profiles of ncRNAs after k-means clustering (Additional file 2: Table S13). The x-axis shows sampling points I to V from left to right and the y-axis gives the expression strength in z-score transformed mean NPKM values of each replicate. Clusters are numbered and captioned with the count of included ncRNAs. Transcripts with a maximal NPKM value >100,000 are marked in orange and transcripts with a maximal NPKM value >1000 are marked in green.
All assigned non-coding RNAs were categorized according to the scheme displayed in Figure 4 and subdivided into the classes A 5 , A 3 , A I , A misc and indep. Selected ncRNAs are listed in Table 3, whereas an overview of all identified features is given in Additional file 2: Table S6. Several ncRNAs have been selected for validation by Northern blotting (Additional file 1: Figure S3). The analyzed ncRNAs were chosen as they are exemplarily for their respective class. The occurrence of eight ncRNAs could be verified, especially ncRNAs <500 nt are in good accordance with the results gained by RNA-Seq. The results for transcripts >2000 nt are indicative for RNA degradation or processing and leaky transcription termination. However, three ncRNAs could not be validated, which is most probably due to their low expression levels.
Table 3.
Selected non-coding RNAs (ncRNAs)
| RNA feature | Start | Stop | Length | Class | Rfam | Upstream gene | Downstream gene | Antisense genes | NPKM value* |
|---|---|---|---|---|---|---|---|---|---|
| BLi_r0016 |
30440 |
30837 |
398 |
indep |
Scr |
tadA |
dnaX |
|
223117 |
| BLi_r0026 |
54282 |
49490 |
4793 |
A
misc
|
RnaA |
|
|
metS, yabD, yabE, rnmV |
18 |
| BLi_r0086 |
212998 |
213153 |
156 |
indep |
|
thrZ |
BLi00235 |
|
242952 |
| BLi_r0253 |
413569 |
412086 |
1484 |
A
misc
|
|
|
|
BLi00412, BLi00413 |
14 |
| BLi_r0415 |
617488 |
617027 |
462 |
A
misc
|
|
|
|
thiL |
6 |
| BLi_r0451 |
653178 |
652888 |
291 |
A
misc
|
|
|
|
BLi00649 |
15139 |
| BLi_r0844 |
1082413 |
1082091 |
323 |
indep |
|
yhaA1 |
hit |
|
8039 |
| BLi_r0872 |
1108968 |
1109111 |
144 |
A
I
|
|
|
|
apr |
25689 |
| BLi_r1000 |
1262387 |
1262504 |
118 |
indep |
RsaE |
pepF |
yjbL |
|
5791 |
| BLi_r1034 |
1300088 |
1300311 |
224 |
indep |
|
pbpE1 |
BLi01297 |
|
1902 |
| BLi_r1306 |
1639742 |
1639946 |
205 |
A
misc
|
SR1 |
|
|
speA |
502 |
| BLi_r1347 |
1673741 |
1673635 |
107 |
A
misc
|
CsfG |
|
|
ylbG, ylbH |
5366 |
| BLi_r1424 |
1898847 |
1898597 |
251 |
indep |
|
yqeD |
BLi01936 |
|
2023 |
| BLi_r1454 |
1929530 |
1929709 |
180 |
indep |
BsrB |
|
|
|
26604 |
| BLi_r1474 |
1960434 |
1960112 |
323 |
indep |
|
BLi02008 |
|
|
61592 |
| BLi_r1596 |
2101575 |
2102388 |
814 |
A
I
|
|
|
|
cysP2 |
12 |
| BLi_r1645 |
2156680 |
2156597 |
84 |
indep |
|
yobS |
yndG |
|
13545 |
| BLi_r1808 |
2302203 |
2301803 |
401 |
indep |
RnpB |
gpsB |
ypsC |
|
55930 |
| BLi_r1834 |
2355791 |
2356137 |
347 |
A
misc
|
|
|
|
folE |
1 |
| BLi_r2049 |
2634028 |
2634231 |
204 |
A
misc
|
SurC |
|
|
dnaK |
29 |
| BLi_r2163 |
2770133 |
2769931 |
203 |
indep |
BsrA |
aspS |
yrvM |
|
389442 |
| BLi_r2390 |
3060847 |
3062662 |
1816 |
A
misc
|
|
|
|
ytvB, yttB |
11 |
| BLi_r2624 |
3299320 |
3299025 |
296 |
indep |
BsrI |
yurZ |
BLi03452 |
|
514 |
| BLi_r2645 |
3325524 |
3323636 |
1889 |
A
3'UTR
|
|
yirB |
|
cssR, cssS |
112 |
| BLi_r2758 |
3469610 |
3469009 |
602 |
indep |
SsrA |
smpB |
BLi03638 |
|
78287 |
| BLi_r2780 |
3485163 |
3485306 |
144 |
indep |
|
BLi03658 |
BLi03670 |
|
3747 |
| BLi_r2828 |
3552518 |
3552467 |
52 |
indep |
|
trxB |
yvcI |
|
15924 |
| BLi_r2863 |
3611192 |
3612078 |
887 |
A
misc
|
|
|
|
degU, degS |
5 |
| BLi_r2925 |
3692599 |
3692663 |
65 |
A
I
|
|
|
|
BLi03865 |
14110 |
| BLi_r3203 | 4050815 | 4050900 | 86 | A misc | bglP | 35711 |
*(pooled RNA-Seq data).
Indep ncRNAs
As depicted in Figure 4, indep transcripts are defined as non-coding RNAs not localized antisense to any mRNA. Instead they can be found in intergenic regions or any other position of the chromosome. In total, 53 indep RNAs with sizes between 51 and 602 nt have been identified, of which 40 have TSS verified by dRNA-Seq. Within this group five housekeeping sRNAs could be annotated: the tmRNA SsrA (BLi_r2758), the 6S RNAs BsrA (BLi_r2163) and BsrB (BLi_r1454), the RNA component of RNase P RnpB (BLi_r1808) and the signal recognition particle Scr (BLi_r0016) [13]. 87% of the indep transcripts exhibited NPKM values ≥100 in at least three samples, reflecting a strong transcriptional activity of the encoding genomics regions. For example the sRNA Scr, an essential part of the protein secretion system [48], reaches a maximal NPKM value of almost 400,000. This is in perfect accordance with the fact that the cells are derived from a fermentation process optimized for protein secretion. Interestingly, 39 indep ncRNAs seem to be transcribed constitutively under the examined conditions (Figure 8A), whereas only thirteen indep RNAs show differential expression (likelihood value ≥0,99) [49]), as illustrated exemplarily for BLi_r1424 (Figure 8B).
Figure 8.

Non-coding RNAs (ncRNAs).(Left) Sum of transcriptional activities from all 15 replicates (pooled RNA-Seq data). Black arrows indicate genes and green arrows the identified ncRNAs. (Right) Log-transformed NPKM values of ncRNAs and adjacent genes for single samples. (A)Indep RNA BLi_r0086 is transcribed constitutively with a length of 156 nt and located between the genes of threonyl-tRNA synthetase (thrZ, BLi00234) and a hypothetical protein (BLi00235). Both adjacent genes are also transcribed constitutively, but are less abundant by four and three orders of magnitude, respectively. (B) The differentially expressed indep transcript BLi_r1424 is located between the gene of a hypothetical protein (BLi01936) and a pseudogene (yobN, BLi01938) with a length of 251 nt. The TSS could be confirmed by dRNA-Seq. In the three early conditions the BLi_r1424 transcription level is low, but NPKM values of more than 12,000 were recorded during the productive stages of the fermentation process. A direct transcriptional connection to the adjacent BLi01936 is not visible from the shown NPKM values. (C) BLi_r2390 antisense to ytvB (BLi03176) and yttB (BLi03177) is an example for long antisense ncRNAs. The Amisc RNA occurs only in the later stages of the fermentation process, parallel to a distinct increase in transcriptional activity of ytvB, but does not exceed it regarding the NPKM value. (D) One example suggesting a regulatory function of Amisc RNAs is BLi_r0253, oriented antisense to BLi00413. In the earliest stage the asRNA shows stronger transcription than the corresponding gene, but in all later stages the asRNA is only weakly transcribed. This might indicate a silencing effect in the exponential stage. (E)AI RNA BLi_r1596 localized antisense to the gene of the sulfate permease CysP2 (BLi02153). The transcription of both, the ncRNA and the protein-coding gene, starts during the late stages of the fermentation process.
Antisense ncRNAs
In contrast to the class of indep ncRNAs, the antisense ncRNAs (asRNAs) A I , A 3 , A 5 , and A misc comprise non-coding transcripts localized antisense to annotated protein-coding genes. They either target the protein-coding region of a gene (A I ) or the 5’ and 3’ untranslated regions (A 5 and A 3 ). Furthermore, ncRNAs that target more than one gene or that are only partially antisense are classified (A misc ). In this study, 242 A misc RNAs (Figure 8C/D) could be identified. Approximately 150 of them (and also all A 5 ncRNAs) are located opposite to ribosome binding sites and could therefore function as inhibitors of translation, a very common mechanism of cis-encoded asRNAs [50]. The length distribution of the non-coding RNA features is shown in Figure 5C, and illustrates that 42% of the A misc RNAs are less than 400 nt in length. Twenty-seven of these short A misc RNAs reach maximal NPKM values ≥100, suggesting putative sRNA mechanisms. However, some A misc RNAs are much longer, i.e. BLi_r2246 is 6348 nt in length and spans six genes. The occurrence of antisense transcripts of such length is not unexpected, as asRNAs with very diverse sizes, reaching more than 7000 nt, have been described for several species [18]. Furthermore, 146 A I transcripts (Figure 8E) could be assigned, ranging in size from 54 to 1572 nt. Over 95% of the A I transcripts exhibit maximal NPKM values ≤100, 68% even ≤20, due to the low coverage only 20 TSS could be verified by dRNA-Seq for these asRNAs. In total, 408 non-coding asRNAs were determined, comprising 89% of all identified non-coding RNA transcripts and targeting 15% of all genes. The number of identified antisense ncRNAs is in accordance to previous studies which assume that antisense transcription concerns ~10 to 20% of the bacterial genes [22].
Antisense transcripts with putative impact on productivity
The general aim of our group is the identification of productivity related features. Thus, a special focus has been set on the identification of antisense transcripts with a putative impact on protease production as targets for strain improvement.
The alkaline serine protease Subtilisin Carlsberg (apr, BLi01109) represents the major secreted feeding protease of B. licheniformis. Thus, it competes for energetic and secretory resources with the production protease. We identified a cis-encoded 144 nt A I asRNA (BLi_r0872) which is located at the 3’end of the apr mRNA (Figure 9). A highly active TSS determined from the dRNA-Seq data and a terminator structure downstream of the adjacent gene yhfN confirm the characterization of the transcript as independently transcribed asRNA. BLi_r0872 is highly expressed at all fermentation stages, whereas the transcriptional activity of the Subtilisin Carlsberg gene increases at the productive stages of the process. The presence of the cis-encoded asRNA opposite to the 3’end of the target mRNA resembles the B. subtilis RatA/txpA toxin/antitoxin system or the Escherichia coli GadY/gadX system in which an antisense RNA promotes either mRNA degradation or stability [19]. To elucidate the impact of the detected asRNA, further analyses will be necessary, especially as a corresponding transcript is absent in the transcriptome of B. subtilis[43].
Figure 9.
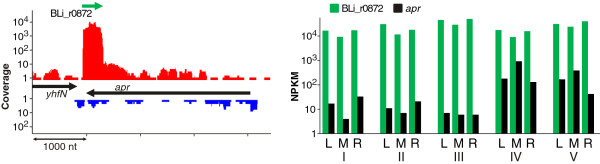
Antisense RNA against Subtilisin Carlsberg.(Left) Transcriptional activities (sample L-III) of apr, the gene which encodes Subtilisin Carlsberg, and the AI RNA BLi_r0872 (green), which is antisense to the 3’UTR of apr. (Right) Log-transformed NPKM values of BLi_r0872 and apr.
Further antisense transcripts against genes involved in cell differentiation, cell stress response, and thiamine and folate biosynthesis could be observed and are presented in Additional file 1: Figure S4.
It is exciting to think about a regulatory impact of the mentioned ncRNAs, but there are also some noteworthy limitations to putative effects. (i) The completed RNA-Seq experiments cannot discern if the sense and the antisense transcript are transcribed in the same type of differentiated cells, which especially challenges stoichiometric estimations of asRNAs and their mRNA targets. Whether they can influence each other or fulfill different purposes in different cell types has to be a topic of single cell targeted investigations. (ii) It has been reported that functional sRNAs are produced in excess amounts over the targeted mRNA [16,51]. Therefore, a regulatory mechanism of poorly transcribed antisense RNA cannot be assumed bona fide, but has to be evaluated carefully. Nonetheless, our data implicate that there might be a biological function assignable to the RNA features, especially when they are conserved within related species as B. subtilis. (iii) At last, it has to be experimentally excluded, especially for low abundant instances, that the found ncRNAs originate from spurious transcriptional events, for instance driven by alternative sigma factors [43].
Comparative transcriptomics
In total, we determined 461 candidate non-coding RNA transcripts, including antisense, as well as indep ncRNAs (see Non-coding RNA features). For Synechocystis sp. PCC6803, Sinorhizobium meliloti and the archaea Sulfolobus solfataricus P2 and Methanosarcina mazei Gö1 between 50 and 107 non-coding RNAs per Mb were identified [31,52-54], matching our result of 109 ncRNAs/Mb. For B. subtilis, the close relative of B. licheniformis, Nicolas et al. [43] have found 472 non-coding RNA features in a tiling array-based, condition-dependent transcriptome study. The majority (68%) of these features are intergenic transcripts determined by promoter analysis, whereas only 32% are derived from independently transcribed (antisense) RNAs. In contrast, the majority of ncRNAs identified in B. licheniformis are antisense RNAs (89%), transcribed independently from protein-coding genes. The identification of more antisense transcripts in B. licheniformis might be accounted to the reduced background noise in RNA-Seq in comparison to tiling arrays, which allows a better detection of low abundant transcripts [28]. 167 of the B. licheniformis ncRNAs are located in regions with high sequence similarity to B. subtilis[55] and 126 ncRNAs are encoded at the frontiers of conserved and not conserved regions of the two genomes. Based on sequence similarity, only 43 (Additional file 2: Table S14) out of the, in total, 293 ncRNAs located in these regions seem to occur in the B. subtilis transcriptome [43], emphasizing the differences of the two closely related species. Comparisons to two earlier B. subtilis transcriptome studies show similar low levels of accordance [56,57]. However, as mentioned above, it is also possible that the identified antisense ncRNAs partly derive from spurious transcription events [43], and hence do not introduce a species-specific effect.
For B. subtilis, 22 sRNAs have been validated experimentally [43,58]. Comparison to Rfam and/or comparison of genomic locations facilitated the detection of eleven of these sRNAs in the transcriptome of B. licheniformis (Additional file 2: Table S15). These include, in addition to the mentioned five housekeeping sRNAs [13], two regulatory RNAs with well-known function in B. subtilis: SR1 and RnaA [59,60]. The other RNAs found in B. licheniformis are BsrI, CsfG, SurC and RsaE [61-64]. The B. subtilis sRNAs which could not be confirmed in B. licheniformis originate from loci with no conserved gene pattern in this organism and thus may contribute to the differences between the two species. Jahn et al. [65] described the toxin-antitoxin system BsrG/SR4 located in the SPβ prophage region of B. subtilis. Although B. licheniformis does not harbor a homolog of the SPβ prophage, two distinct transcripts were found to encode peptides similar to the BsrG toxin (Additional file 1: Figure S5). Additionally, the transcriptional activity of the corresponding loci revealed pairs of overlapping transcripts from both strands (Figure 3 and Additional file 1: Figure S5) as shown for the BsrG/SR4 type toxin-antitoxin system. Therefore both newly identified ORFs were annotated as BsrG-like peptides (BLi05015 and BLi05038). Furthermore, the antisense transcripts (indep RNAs BLi_r1034 and BLi_r2780) resemble the SR4 antitoxin, especially in stem loops SL3, SL4 and TSL [65] directly antisense to the BsrG-encoding mRNA.
Conclusions
The presented study generated substantial data on the transcriptional activity of B. licheniformis within five relevant growth stages of an industrial-oriented fermentation process. A detailed analysis of the transcriptome data enabled us to accomplish a high quality functional genome reannotation of B. licheniformis DSM13 (Figure 10, Ring 4). The integration of the reannotation and the transcriptionally active regions (Figure 10, Ring 1&2) resulted in the identification and quantification of hundreds of RNA based regulatory elements as well as protein encoding genes.
Figure 10.

Circular plot of transcriptional activity and identified RNA features. Combined depiction of reannotated genes and transcriptional activity of B. licheniformis. Unmappable regions, GC skew, transcription start sites, non-coding RNAs, untranslated regions and antisense transcripts are also shown. 5’UTRs and 3’UTRs are evenly distributed over the whole chromosome of B. licheniformis, except for regions a – h: these regions contain long operon structures (a: ribosomal superoperon, b: lch operon, e: fla/che operon, f: trp operon, h: eps operon) or prophage regions with low transcriptional activity (c, d and g). The classification scheme corresponds to Figure 4. (indep) ncRNA, not antisense to any mRNA; (Amisc) ncRNA partially antisense to an mRNA transcript or antisense to more than one gene; (Ai) ncRNA completely antisense to a protein-coding gene; (A5) ncRNA exclusively antisense to the 5'UTR of an mRNA; (A3) ncRNA exclusively antisense to the 5'UTR of an mRNA; (5'UTR) 5'untranslated region of an mRNA; (A5'UTR) 5'UTRs completely or partially antisense to a protein-coding region; (3'UTR) 3'untranslated region of an mRNA transcript; (A3'UTR) 3'UTRs completely or partially antisense to a protein-coding region.
In total, 3314 RNA features have been sorted into ten functional classes (Figure 4). 1433 5’UTRs and 1365 3’UTRs (Figure 10, Ring 8) as well as 461 ncRNAs (Figure 10, Ring 7) and 55 antisense intergenic read-through (A rt ) transcripts have been identified. A striking observation was the identification of 855 RNA features, which mapped antisense to annotated genomic features (Figure 10, Ring 6). Notably antisense RNA features have been found in each of the functional classes and include transcripts of a size range from 38 to 6348 base pairs in length. We have identified both: constitutively as well as growth phase dependently expressed RNA features.
Our data represent a solid amount of knowledge on regulatory elements which orchestrate the cellular activities of B. licheniformis during the succession of growth phases within a productive fermentation. To generate an overview of the functional diversity of the identified RNA features, all instances have been screened against the Rfam database. This approach resulted in hits to experimentally well characterized RNA features known from B. subtilis and other relatives, as well as in a multitude of so far unknown RNA features without any Rfam hit. The knowledge on genes and regulatory RNA features which are transcriptionally active during an industrial-oriented fermentation enables an excellent access to a rational strain design approach for the optimization of B. licheniformis as industrial workhorse. Especially the regulatory features which represent differences to the model organism B. subtilis give new insights to the still open question what makes strains of the species B. licheniformis superior to B. subtilis strains in terms of protease production capacity in industrial applications [2]. In the future it may be promising to correlate the transcriptional activity of the RNA features to the corresponding protein expression patterns.
Methods
Bacterial strain and fermentation conditions
Bacillus licheniformis MW3Δspo (kindly provided by F. Meinhardt and St. Wemhoff, University of Münster) was used for the fermentation experiments. B. licheniformis MW3Δspo is a derivate of the B. licheniformis wild type strain DSM13, bearing three deletions: ΔhsdR and ΔhsdR2 coding for restriction endonucleases [10] and ΔyqfD[66] to prevent the production of viable spores and thus the long-term contamination of the used fermenters [67].
Fermentation was carried out for 46 h in aerated 16 L fermenters with a culture volume of 6 L at 39°C. Medium contained 12% w/v of a complex nitrogen source, 57 mM KH2PO4, 21 mM (NH4)2SO4, 0.53 mM Mn(II)SO4, 0.17 mM Fe(II)SO4, 2.0 mM CaCl2 * 2 H2O, 5.7 mM MgSO4, 0.4% v/v PPG200, 0.03 mM tetracycline and 3% w/v glucose. The pH value was regulated to a set point of 7.9 with sodium hydroxide solution. Glucose-feed was started after exceeding the point of biphasic growth.
RNA isolation and preparation
5 mL of the harvested cells were mixed with 5 mL of RNAprotect Bacteria Reagent (Qiagen) directly upon sampling. After 10 min incubation at room temperature the samples were centrifuged at 4500× g, the supernatant was removed, the sample was snap-frozen in liquid nitrogen and finally stored at -80°C. The cells were separated from the remainders of the fermentation broth by washing repeatedly with Buffer RLT (Qiagen). Subsequent RNA isolation was carried out with a modified protocol of the RNeasy Midi Kit (Purification of RNA including small RNAs using the RNeasy Midi Kit RY39 Apr-09, Qiagen) to retain short RNAs. The cells were disintegrated with the ball mill Mikro-Dismembrator U (B. Braun Biotech) in 400 μL Buffer RLT and afterwards resuspended in 1.4 mL Buffer RLT and 2.7 mL pure ethanol. The initial washing step of the column was done using 4 mL Buffer RWT (Qiagen). The DNA was digested successively with two different DNases (TURBO™ DNase, Ambion and DNase I recombinant, Roche), with a purification step after the first treatment. Purification was performed with a protocol adapted for small RNA purification of the RNeasy MinElute Cleanup Kit (Qiagen). Instead of 250 μL, 675 μL pure ethanol were added to the RNA before binding to the column to shift the binding capacity of the column. A control PCR with 35 cycles was conducted to confirm complete DNA removal. Depletion of rRNA was obtained using the MICROBExpress™ Bacterial mRNA Enrichment Kit (Ambion) according to manufacturer’s instructions. The following purification step was also carried out with the described adaption to the RNeasy MinElute Cleanup Kit.
Library construction and sequencing
cDNA libraries were prepared by vertis Biotechnologie AG, Germany (http://www.vertis-biotech.com). For whole transcriptome libraries [28,33] the RNA samples were fragmented by ultrasound and dephosphorylated with Antarctic phosphatase. After polynucleotide kinase treatment the RNA was poly(A)tailed and an RNA adapter was ligated to the 5’phosphate. cDNA synthesis was accomplished by the use of poly(T) adapters and M-MLV reverse transcriptase. The subsequent PCR was carried out with cycle numbers between nine and twelve. The construction of the libraries for the dRNA-Seq was performed as described by Sharma et al. [32], supplemented by an additional treatment with polynucleotide kinase after the fragmentation step to allow removal of fragments previously not phosphorylated. The samples were incubated with Terminator™ 5’-Phosphate-Dependent Exonuclease (Epicentre) and a poly(A) tail was ligated to the 3’end of the transcripts. Hereafter an incubation step with tobacco acid pyrophosphatase and the ligation of an RNA adapter to the 5’end was conducted. Reverse transcription was processed as described above, the cycle numbers of the following PCR were 14 or 15. The RNA-Seq libraries as well as the libraries for dRNA-Seq were size fractioned in the range of 200 to 400 nt on agarose gels and then sequenced on an Illumina HiSeq 2000 machine with a read length of 50 nt.
In silico sequence read processing
Initially, all sequence reads mapping to B. licheniformis rRNA and tRNA genes according to BLAST analysis were removed. The remaining reads were processed in a multi-step procedure to ensure the reliability of the read mappings used for the analysis of the transcriptional activity of the genome and to estimate the quality of the RNA-Seq data. All reads which mapped over the full read length of 50 bases with 98% or sequence identity were used for further analyses. Additionally, a distinct bit score was required to ensure an unambiguous assignment to one locus. All discarded reads were screened with relaxed similarity quality criteria vs. the B. licheniformis genome. 75% of these reads generated hits and were therefore assigned as bad quality B. licheniformis reads. The remaining reads (approximately 3% of the total generated sequence) cannot be mapped on the genome. A detailed sequence analysis of these unmappable reads revealed that they mainly contain poly(A) tails or concatenated adapters and therefore represent methodic artifacts. All datasets were depleted for plasmid-mapping reads and have been deposited in NCBI Sequence Read Archive database under accession number SRP018744 (Additional file 2: Table S16).
To obtain the maximal number of features, a dataset containing all reads of the 15 samples was prepared and is referred to as pooled RNA-Seq data. To reduce putative background noise, all reads with coverage of one and no intersecting or adjacent reads were omitted prior to combination of the datasets. This is done to reduce transcriptional activity that was not replicated within a dataset in order to avoid incorrect extension of predicted features (e.g. transcriptional activity from leaky termination masking or extending 5’UTR extensions due to overlap). The generation of five datasets describing each sampling point was processed accordingly.
Expression strength values
The analytical methods used to process the 15 generated RNA-Seq datasets require the use of single nucleotide activities instead of read mappings. This makes RPKMs [68] inapplicable as a measure of transcriptional activity. Instead, we defined the nucleotide activity per kilobase of exon model per million mapped reads (NPKM) value. An NPKM is defined as:
Where n and m are the start and stop of the region of interest, f(i) is the base activity of base i on a specific strand and g(i) is the sum of the activities of base i of positive and negative strands.
NPKM values are a derivate of RPKMs [68], adapted to per base nucleotide activities. They are designed to be functionally equivalent to RPKMs, albeit they are more accurate due to the single base-resolution. We are aware that RPKMs and therefore NPKMs do not account for sequencing-based bias [69]. Although sequencing-based bias produces some local errors, the overall comparability of active genomic regions is still possible.
Untranslated regions
5’ and 3’ UTRs were considered as regions of continuous, non-interrupted transcriptional activity upstream or downstream of annotated genomic features, respectively. The boundary of an identified 5’UTRs was set at the point of the rising of the continuous transcript from zero transcriptional activity. The boundary of a 3’ UTRs was accordingly set at the point of the downshift of the continuous transcript to zero transcriptional activity.
The analysis of 5’ and 3’untranslated regions was aimed to find the longest UTR, as the longest transcript should cover all possible alternative UTRs and contain all transcribed regulatory elements. Therefore, the computational analysis was based on the pooled RNA-Seq data. Few 5’ and 3’UTRs were manually extended on account of adjacent transcripts which are only separated from the UTR by a very short downshift and potentially are part of the UTR. To exclude that the resulting UTRs correspond to previously not annotated protein genes, searches versus the InterPro and the UniProtKB/Swiss-Prot databases were performed [70,71].
5’ and 3’UTRs which are antisense to an adjacent gene on the opposite strand were classified as A 5’UTR and A 3’UTR . The respective UTRs were computationally examined and assigned to be antisense when their overlap to an opposite gene exceeded 100 nt in length.
Intergenic read-through transcripts localized antisense to an opposite gene were determined manually and classified as A rt .
Non-coding RNA features
The RNA-Seq data were scanned for transcriptionally active regions that were clearly separated from the transcripts corresponding to any annotated gene or its untranslated regions. This primary computational search identified transcripts which were either located on the opposite strand of a protein-coding gene, in intergenic regions or any other region of the chromosome. The boundaries of the identified transcripts were set to those nucleotides with the first and last occurrence of transcriptional activity higher than zero of the corresponding transcriptional unit. NPKM values for the resulting loci were generated from each of the 15 datasets. Subsequently, all results from the computational search were evaluated as depicted in Additional file 1: Figure S6A to approve the reliability of the identified ncRNAs. Searches vs. the InterPro and the UniProtKB/Swiss-Prot databases were performed to exclude the possibility that the resulting non-coding RNA features correspond to non-annotated protein genes [70,71]. Subsequently, the non-coding transcripts were subdivided into the ncRNA classes described in Figure 4 (A 3 , A 5 , A I , A misc , and indep).
The class indep comprises all identified ncRNAs that are not located antisense to any protein-coding gene or its respective untranslated regions. Several transcripts of this category were added manually as this class comprises RNA transcripts which could not clearly be distinguished from surrounding mRNAs by complete down-shifts of transcriptional activity, but were detected by their remarkably higher abundance.
The categories A 3 , A 5 , A I and A misc comprise ncRNAs which are localized antisense to protein-coding genes or their respective untranslated regions. The class A I contains all ncRNAs with an antisense localization solely towards a protein-coding gene. The class A 5 contains all ncRNAs with an antisense localization solely towards the 5’UTR of an opposite mRNA. The class A 3 contains all ncRNAs with an antisense localization solely towards the 3’UTR of an opposite mRNA. The class A misc contains all ncRNAs with an antisense localization towards more than one protein-coding gene and all ncRNAs which are only partially antisense to an mRNA transcript.
Analysis of dRNA-Seq reads
Transcriptional start sites were determined by the identification of significant increases of the log-scaled expression strength of the dRNA-Seq data from succeeding bases greater than ln 4. The reference value of ln 4 was empirically determined based on the observation that ln 4 represents the smallest expression strength increase for TSS present across all samples of one sampling point. In a second step, all TSS in promoter regions of rRNA or tRNA genes and all TSS being apart less than 20 bp were excluded. TSS matching the boundaries of RNA-Seq predicted 5’UTRs or ncRNAs were determined accordingly to the flow chart depicted in Additional file 1: Figure S6B.
Transcriptome Viewer
Additionally, the gained RNA-Seq data were used to generate logarithmic scaled, color coded graphs representing strand-specific transcription.
Operon prediction
Operon predictions based on whole transcriptome sequencing, dRNA-Seq transcription start sites, and operon and transcription terminator site determination with DOOR [72], OperonDB [73], and TransTermHP [74]. Operon predictions were curated manually as described by Sharma et al. [32], regarding especially level shifts in transcriptional activity.
Reannotation
Functional reannotation was carried out using the ERGO software tool (Integrated Genomics, Chicago, USA) [75] and the IMG/ER (Integrated Microbial Genomes/Expert Review) system [45]. Subsequent manual curation was based on the results of a bidirectional BLAST analysis comprising B. subtilis, B. pumilus and related, manually annotated organisms, the comparisons to UniProtKB/Swiss-Prot and UniProtKB/TrEMBL databases [71] and the analysis of functional domains with InterProScan [70]. The annotation of new genes and the correction of reading frames was based on transcriptional activity and was performed upon analysis of GC frame plots, ribosome-binding sites and -10 and -35 promoter regions using Artemis v12 [76] and comparisons to UniProtKB/Swiss-Prot, UniProtKB/TrEMBL, and InterProScan [70,71]. The removal of gene annotations relied on the combined evaluation of GC frame plots, ribosome-binding sites and -10 and -35 promoter regions using Artemis v12 [76] and comparisons to UniProtKB/Swiss-Prot, UniProtKB/TrEMBL, and InterProScan [70,71]. The absence of transcriptional activity was not used to support the removal of gene annotations. Prophage regions have been annotated by an initial bioinformatic search using Prophagefinder [77] followed by manual evaluation of the candidate regions. Based on the existence of GC content deviations, genes in these regions with significant similarities to known prophages and the identification of insertion repeats, genomic regions were assigned as prophages. The annotation followed the principles of prophage annotation outlined by Casjens [78]. The reannotated data set has been used to update the B. licheniformis DSM13 genome data initially submitted by Veith et al. [1] and is now available at NCBI under accession number AE017333.1.
Clustering of ncRNAs
Cluster analysis to elucidate the fundamental types of ncRNA expression profiles was performed based on the respective NPKM values (Additional file 2: Table S2). To ensure that the data of each replicate are sufficiently reliable, t-tests were performed with MeV [79]. For at least three out of the five samples, the respective ncRNA had to have a P value <0.15 to be taken into further analysis, as described by Koburger et al. [80]. Furthermore, all ncRNAs taken into analysis had to have a minimal NPKM value >10. Means of the replicates of each sampling point were built and z-score transformation was performed. The number of clusters was determined by Figure of merit (FOM) analysis, which basically is an estimate of the predictive power of a clustering algorithm [81]. Clusters were generated by employing k-means clustering [47] with Euclidian distances in the MeV software [79] and subsequent manual curation.
Utilized software and databases
ACT and Mauve
The comparison of RNA features from B. licheniformis with the reference genome B. subtilis was based on sequence similarity analyzed with ACT v11, the Artemis comparison tool [82]. Quantification of ncRNAs located in conserved or not-conserved loci, was done employing the progressive Mauve alignment tool [83].
baySeq
Determination of constitutive or differential expression of the RNA features was employed with baySeq [49], which uses an empirical Bayes approach assuming a negative binomial distribution and is capable of dealing with multi-group experimental designs. Input data were generated by counting the reads referring to every gene.
DOOR and OperonDB
Predictions for operons were thankfully downloaded from the DOOR Database of prOkaryotic OpeRons [72] and OperonDB [73].
Gem mappability
The determination of the genome mappability was calculated for a read length of 50 nt with the Gem mappability program [84].
MeV
Cluster analysis was performed using the Multiexperiment Viewer v4.8 [79].
Rfam
Annotation of cis-regulatory elements and small RNAs was carried out by Infernal searches [85] of RNA features versus the Rfam database [46].
TransTermHP
Transcription terminators pre-computed with TransTermHP v2.07 were gratefully downloaded from transterm.cbcb.umd.edu [74]. 3’UTRs were checked for terminators as described by Martin et al. [86]. Terminators were considered as internal if they were located at least 50 nt upstream of the end of the transcript.
Northern blot analysis
B. licheniformis DSM13 was cultivated at 37°C and 160 rpm in a 5 L Erlenmeyer flask on defined minimal medium [87]. Cells were harvested at OD600 1 and 4.5 and after having reached the stationary phase for at least 2 h. Escherichia coli DH5α was cultivated in Luria broth at 37°C and 180 rpm to an OD600 of 2. RNA was isolated as described in RNA isolation and preparation. Digoxigenin-labeled RNA probes were prepared by in vitro transcription with T7 RNA polymerase (DIG Northern Starter Kit, Roche). Templates for in vitro transcription were generated by PCR using primer pairs (Additional file 2: Table S17) containing a primer flanked with the T7 promoter sequence. Gel electrophoresis of the RNA was carried out using a 1% agarose formaldehyde MOPS gel [88] with 100 V applied for 2,5 h. RNA was transferred to the membrane (Nylon Membranes, positively charged, Roche) via vacuum blotting with the Amersham VacuGene XL Vacuum Blotting System (GE Healthcare) using the recommended protocol. The RNA probe hybridization pro-cedure was performed following the manufacturer’s instructions (DIG Northern Starter Kit, Roche). Detection was accomplished with ChemoCam Imager (Intes). RiboRuler High Range RNA Ladder (Thermo Scientific) ranging from 200 to 6000 nt was used as RNA marker.
Abbreviations
°C: Degrees Celsius; μL: Microliter; A: Adenine; asRNA: Antisense RNA; bp: Base pairs; C: Cytosine; cDNA: Complementary DNA; dRNA-Seq: Differential RNA sequencing; g: Gram; g: Gravitational constant; G: Guanine; h: Hours; L: Liter; ln: Natural logarithm; Mb: Megabase pairs; min: Minute; mL: Milliliter; mM: Millimolar; mRNA: Messenger RNA; ncRNA: Non-coding RNA; NPKM: Nucleotide activity per kilobase of exon model per million mapped reads; nt: Nucleotides; OD: Optical density; ORF: Open reading frame; PCR: Polymerase chain reaction; RNA-Seq: RNA sequencing; rpm: Revolutions per minute; rRNA: Ribosomal RNA; T: Thymine; TAP: Tobacco Acid Pyrophosphatase; TEX: Terminator™ 5′-Phosphate-Dependent Exonuclease; tmRNA: Transfer-messenger RNA; tRNA: Transfer RNA; TSS: Transcription start sites; UTR: Untranslated region; V: Volt; v/v: Volume per volume; w/v: Weight per volume.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SW performed the experiments, analyzed data and wrote paper, SD developed the analysis tools, RH performed northern blots, JB and SE provided industrial fermentation facilities and performed the fermentation, SV prepared submission of genome and transcriptome data, RD wrote paper and provided research facilities, HL wrote paper, designed research and analyzed data. All authors read and approved the final version of the manuscript.
Supplementary Material
Distribution of whole transcriptome sequencing reads. Figure S2: Comparative operon prediction. Figure S3: Northern blot confirmation of non-coding RNAs. Figure S4: Antisense RNAs with putative impact on productivity. Figure S5: BsrG/SR4-like loci in B. licheniformis. Figure S6: Work flow charts.
Whole transcriptome sequencing reads. Table S2: NPKM values of RNA features. Table S3: NPKM values of all genes. Table S4: Differential RNA-Seq reads. Table S5: Transcription start sites. Table S6: Identified RNA features. Table S7: Predicted operons. Table S8: Corrected genes. Table S9: New genes. Table S10: Removed genes. Table S11: Predicted cis-regulatory elements. Table S12: Comparison of cis-regulatory elements known from B. subtilis. Table S13: Cluster analysis of ncRNA expression profiles. Table S14: Comparison of ncRNAs to B. subtilis. Table S15: Comparison of small RNAs from B. subtilis to identified ncRNAs. Table S16: Sequence Read Archive accession. Table S17: Primer pairs for Northern blots.
Contributor Information
Sandra Wiegand, Email: swiegan@gwdg.de.
Sascha Dietrich, Email: sdietri@gwdg.de.
Robert Hertel, Email: rhertel@gwdg.de.
Johannes Bongaerts, Email: bongaerts@t-online.de.
Stefan Evers, Email: stefan.evers@henkel.com.
Sonja Volland, Email: svollan@gwdg.de.
Rolf Daniel, Email: rdaniel@gwdg.de.
Heiko Liesegang, Email: hlieseg@gwdg.de.
Acknowledgements
This study was funded by the Bundesministerium für Bildung und Forschung (FKZ-0315387).
The authors would like to thank the Henkel Company for kind access to their fermentation facility. We are grateful for expert technical assistance by Ayhan Aydemir and Maik Schlieper.
References
- Veith B, Herzberg C, Steckel S, Feesche J, Maurer K-H, Ehrenreich P, Bäumer S, Henne A, Liesegang H, Merkl R, Ehrenreich A, Gottschalk G. The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J Mol Microbiol Biotechnol. 2004;7:204–211. doi: 10.1159/000079829. [DOI] [PubMed] [Google Scholar]
- Schallmey M, Singh A, Ward O. Developments in the use of Bacillus species for industrial production. Can J Microbiol. 2004;50:1–17. doi: 10.1139/w03-076. [DOI] [PubMed] [Google Scholar]
- Maurer K-H. Detergent proteases. Curr Opin Biotechnol. 2004;15:330–334. doi: 10.1016/j.copbio.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Çalık P, Takac S, Çalık G, Özdamar T. Serine alkaline protease overproduction capacity of Bacillus licheniformis. Enzyme Microb Tech. 2000;26:45–60. doi: 10.1016/S0141-0229(99)00126-X. [DOI] [Google Scholar]
- Çalık P, Çalık G, Takaç S, Ozdamar TH. Metabolic flux analysis for serine alkaline protease fermentation by Bacillus licheniformis in a defined medium: effects of the oxygen transfer rate. Biotechnol Bioeng. 1999;64:151–167. doi: 10.1002/(SICI)1097-0290(19990720)64:2<151::AID-BIT4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Degering C, Eggert T, Puls M, Bongaerts J, Evers S, Maurer K-H, Jaeger K-E. Optimization of protease secretion in Bacillus subtilis and Bacillus licheniformis by screening of homologous and heterologous signal peptides. Appl Environ Microbiol. 2010;76:6370–6376. doi: 10.1128/AEM.01146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Beg QK, Lorenz P. Bacterial alkaline proteases: molecular approaches and industrial applications. Appl Microbiol Biotechnol. 2002;59:15–32. doi: 10.1007/s00253-002-0975-y. [DOI] [PubMed] [Google Scholar]
- Nielsen AK, Breüner A, Krzystanek M, Andersen JT, Poulsen TA, Olsen PB, Mijakovic I, Rasmussen MD. Global transcriptional analysis of Bacillus licheniformis reveals an overlap between heat shock and iron limitation stimulon. J Mol Microbiol Biotechnol. 2010;18:162–173. doi: 10.1159/000315457. [DOI] [PubMed] [Google Scholar]
- Rey M, Ramaiya P, Nelson N, Brody-Karpin S. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 2004;5:R77. doi: 10.1186/gb-2004-5-10-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschkau B, Waldeck J, Wieland S, Eichstädt R, Meinhardt F. Generation of readily transformable Bacillus licheniformis mutants. Appl Microbiol Biotechnol. 2008;78:181–188. doi: 10.1007/s00253-007-1278-0. [DOI] [PubMed] [Google Scholar]
- Nahrstedt H, Waldeck J, Gröne M, Eichstädt R, Feesche J, Meinhardt F. Strain development in Bacillus licheniformis: Construction of biologically contained mutants deficient in sporulation and DNA repair. J Biotechnol. 2005;119:245–254. doi: 10.1016/j.jbiotec.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Madan Babu M, Teichmann SA, Aravind L. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J Mol Biol. 2006;358:614–633. doi: 10.1016/j.jmb.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Romby P, Charpentier E. An overview of RNAs with regulatory functions in gram-positive bacteria. Cell Mol Life Sci. 2010;67:217–237. doi: 10.1007/s00018-009-0162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, Storz G. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol. 2011;3:a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters L, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel JM, Axmann IM, Legewie S. Multi-target regulation by small RNAs synchronizes gene expression thresholds and may enhance ultrasensitive behavior. PLoS One. 2012;7:e42296. doi: 10.1371/journal.pone.0042296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georg J, Hess WR. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol R. 2011;75:286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantl S. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr Opin Microbiol. 2007;10:102–109. doi: 10.1016/j.mib.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Sesto N, Wurtzel O, Archambaud C, Sorek R, Cossart P. The excludon: a new concept in bacterial antisense RNA-mediated gene regulation. Nat Rev Microbiol. 2013;11:75–82. doi: 10.1038/nrmicro2934. [DOI] [PubMed] [Google Scholar]
- Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet. 2010;11:9–16. doi: 10.1038/nrg2695. [DOI] [PubMed] [Google Scholar]
- Güell M, Yus E, Lluch-Senar M, Serrano L. Bacterial transcriptomics: what is beyond the RNA horiz-ome? Nat Rev Microbiol. 2011;9:658–669. doi: 10.1038/nrmicro2620. [DOI] [PubMed] [Google Scholar]
- Breaker RR. Prospects for riboswitch discovery and analysis. Mol Cell. 2011;43:867–879. doi: 10.1016/j.molcel.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo PR, Yoon K, Ko D, Smith AD, Qiao M, Suresh U, Burns SC, Penalva LOF. Before It Gets Started: Regulating Translation at the 5’ UTR. Comp Funct Genom. 2012;2012:475731. doi: 10.1155/2012/475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoll J, Heus H, van Kuppeveld F, Melchers W. The structure-function relationship of the enterovirus 3’-UTR. Virus Res. 2009;139:209–216. doi: 10.1016/j.virusres.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Kaberdin VR, Bläsi U. Translation initiation and the fate of bacterial mRNAs. FEMS Microbiol Rev. 2006;30:967–979. doi: 10.1111/j.1574-6976.2006.00043.x. [DOI] [PubMed] [Google Scholar]
- Evguenieva-Hackenberg E, Klug G. New aspects of RNA processing in prokaryotes. Curr Opin Microbiol. 2011;14:587–592. doi: 10.1016/j.mib.2011.07.025. [DOI] [PubMed] [Google Scholar]
- Vivancos AP, Güell M, Dohm JC, Serrano L, Himmelbauer H. Strand-specific deep sequencing of the transcriptome. Genome Res. 2010;20:989–999. doi: 10.1101/gr.094318.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnvig KB, Comas I, Thomson NR, Houghton J, Boshoff HI, Croucher NJ, Rose G, Perkins TT, Parkhill J, Dougan G, Young DB. Sequence-based analysis uncovers an abundance of non-coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 2011;7:e1002342. doi: 10.1371/journal.ppat.1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockenhuber M-P, Sharma CM, Statt MG, Schmidt D, Xu Z, Dietrich S, Liesegang H, Mathews DH, Suess B. Deep sequencing-based identification of small non-coding RNAs in Streptomyces coelicolor. RNA Biol. 2011;8:468–477. doi: 10.4161/rna.8.3.14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter J-P, Reinkensmeier J, Daschkey S, Evguenieva-Hackenberg E, Janssen S, Jänicke S, Becker JD, Giegerich R, Becker A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics. 2010;11:245. doi: 10.1186/1471-2164-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, Stadler PF, Vogel J. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- Passalacqua KD, Varadarajan A, Ondov BD, Okou DT, Zwick ME, Bergman NH. Structure and complexity of a bacterial transcriptome. J Bacteriol. 2009;191:3203–3211. doi: 10.1128/JB.00122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dötsch A, Eckweiler D, Schniederjans M, Zimmermann A, Jensen V, Scharfe M, Geffers R, Häussler S. The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS One. 2012;7:e31092. doi: 10.1371/journal.pone.0031092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristoffersen SM, Haase C, Weil MR, Passalacqua KD, Niazi F, Hutchison SK, Desany B, Kolsto A-B, Tourasse NJ, Read TD, Okstad OA. Global mRNA decay analysis at single nucleotide resolution reveals segmental and positional degradation patterns in a Gram-positive bacterium. Genome Biol. 2012;13:R30. doi: 10.1186/gb-2012-13-4-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel O, Sesto N, Mellin JR, Karunker I, Edelheit S, Bécavin C, Archambaud C, Cossart P, Sorek R. Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol Syst Biol. 2012;8:1–14. doi: 10.1038/msb.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire S, Figueroa-Bossi N, Bossi L. Bacteriophage crosstalk: coordination of prophage induction by trans-acting antirepressors. PLoS Genet. 2011;7:e1002149. doi: 10.1371/journal.pgen.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Hu G-Q, She Z-S, Zhu H. Leaderless genes in bacteria: clue to the evolution of translation initiation mechanisms in prokaryotes. BMC Genomics. 2011;12:361. doi: 10.1186/1471-2164-12-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Niimura Y, Miura K, Gojobori T. Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc Natl Acad Sci U S A. 2010;107:6382–6387. doi: 10.1073/pnas.1002036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening J-W, Igoshin OA, Eijlander RT, Nijland R, Hamoen LW, Kuipers OP. Transient heterogeneity in extracellular protease production by Bacillus subtilis. Mol Syst Biol. 2008;4:184. doi: 10.1038/msb.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank EA, Kolter R. Extracellular signaling and multicellularity in Bacillus subtilis. Curr Opin Microbiol. 2011;14:741–747. doi: 10.1016/j.mib.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunka K, Commichau FM. Control of glutamate homeostasis in Bacillus subtilis: a complex interplay between ammonium assimilation, glutamate biosynthesis and degradation. Mol Microbiol. 2012;85:213–224. doi: 10.1111/j.1365-2958.2012.08105.x. [DOI] [PubMed] [Google Scholar]
- Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisicchia P, Botella E, Delumeau O, Doherty G, Denham EL, Fogg MJ, Fromion V, Goelzer A, Hansen A, Hartig E, Harwood CR, Homuth G, Jarmer H, Jules M, Klipp E, Le Chat L, Lecointe F, Lewis P, Liebermeister W. et al. Condition-Dependent Transcriptome Reveals High-Level Regulatory Architecture in Bacillus subtilis. Science. 2012;335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- Brantl S. Acting antisense: plasmid- and chromosome-encoded sRNAs from Gram-positive bacteria. Future Microbiol. 2012;7:853–871. doi: 10.2217/fmb.12.59. [DOI] [PubMed] [Google Scholar]
- Markowitz VM, Mavromatis K, Ivanova NN, Chen I-MA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, Bateman A. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 2011;39:D141–D145. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock G. Analysis of large-scale gene expression data. Curr Opin Immunol. 2000;12:201–205. doi: 10.1016/S0952-7915(99)00074-6. [DOI] [PubMed] [Google Scholar]
- Pool MR. Signal recognition particles in chloroplasts, bacteria, yeast and mammals (Review) Mol Membr Biol. 2005;22:3–15. doi: 10.1080/09687860400026348. [DOI] [PubMed] [Google Scholar]
- Hardcastle TJ, Kelly KA. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiley Thomason M, Storz G. Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet. 2010;44:167–188. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine E, Zhang Z, Kuhlman T, Hwa T. Quantitative characteristics of gene regulation by small RNA. PLoS Biol. 2007;5:e229. doi: 10.1371/journal.pbio.0050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke J, Georg J, Scholz I, Sharma CM, Dienst D, Bantscheff J, Voß B, Steglich C, Wilde A, Vogel J, Hess WR. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci U S A. 2011;108:1–6. doi: 10.1073/pnas.1015154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons B, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger D, Sharma C, Thomsen J, Ehlers C, Vogel J, Schmitz R. Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A. 2009;106:21878–21882. doi: 10.1073/pnas.0909051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbe V, Cruveiller S, Kunst F, Lenoble P, Meurice G, Sekowska A, Vallenet D, Wang T, Moszer I, Médigue C, Danchin A. From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology. 2009;155:1758–1775. doi: 10.1099/mic.0.027839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S, Nielsen H, Jarmer H. The Transcriptionally Active Regions in the Genome of Bacillus subtilis. Mol Microbiol. 2009;73:1043–1057. doi: 10.1111/j.1365-2958.2009.06830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irnov I, Sharma C, Vogel J, Winkler W. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010;38:6637–6651. doi: 10.1093/nar/gkq454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäder U, Schmeisky AG, Flórez LA, Stülke J. SubtiWiki–a comprehensive community resource for the model organism Bacillus subtilis. Nucleic Acids Res. 2012;40:D1278–D1287. doi: 10.1093/nar/gkr923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiamphungporn W, Helmann JD. Extracytoplasmic function sigma factors regulate expression of the Bacillus subtilis yabE gene via a cis-acting antisense RNA. J Bacteriol. 2009;191:1101–1105. doi: 10.1128/JB.01530-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpel M, Heidrich N, Mäder U, Krügel H, Brantl S. A dual-function sRNA from B. subtilis: SR1 acts as a peptide encoding mRNA on the gapA operon. Mol Microbiol. 2010;76:990–1009. doi: 10.1111/j.1365-2958.2010.07158.x. [DOI] [PubMed] [Google Scholar]
- Geissmann T, Chevalier C, Cros M-J, Boisset S, Fechter P, Noirot C, Schrenzel J, François P, Vandenesch F, Gaspin C, Romby P. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 2009;37:7239–7257. doi: 10.1093/nar/gkp668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchais A, Duperrier S, Durand S, Gautheret D, Stragier P. CsfG, a sporulation-specific, small non-coding RNA highly conserved in endospore formers. RNA Biol. 2011;8:358–364. doi: 10.4161/rna.8.3.14998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Kakeshita H, Nakamura K. Novel small RNA-encoding genes in the intergenic regions of Bacillus subtilis. Gene. 2009;428:2–8. doi: 10.1016/j.gene.2008.09.024. [DOI] [PubMed] [Google Scholar]
- Silvaggi J, Perkins J, Losick R. Genes for Small, Noncoding RNAs under Sporulation Control in Bacillus subtilis. J Bacteriol. 2006;188:532–541. doi: 10.1128/JB.188.2.532-541.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn N, Preis H, Wiedemann C, Brantl S. BsrG/SR4 from Bacillus subtilis–the first temperature-dependent type I toxin-antitoxin system. Mol Microbiol. 2012;83:579–598. doi: 10.1111/j.1365-2958.2011.07952.x. [DOI] [PubMed] [Google Scholar]
- Feucht A, Evans L, Errington J. Identification of sporulation genes by genome-wide analysis of the σE regulon of Bacillus subtilis. J Mol Biol. 2003;149:3023–3034. doi: 10.1099/mic.0.26413-0. [DOI] [PubMed] [Google Scholar]
- Wemhoff S. Deletionsmutagenese des yqfCD/phoH-Operons in Bacillus licheniformis - Untersuchungen zur Auswirkung auf die Sporulation. Westfälische Wilhelms-Universität Münster, Institut für Molekulare Mikrobiologie und Biotechnologie; 2008. (Master thesis). [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium. The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 2009;37:D169–D174. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 2009;37:D459–D463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Ayanbule K, Smedinghoff M, Salzberg SL. OperonDB: a comprehensive database of predicted operons in microbial genomes. Nucleic Acids Res. 2009;37:D479–D482. doi: 10.1093/nar/gkn784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007;8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R. The ERGOTM genome analysis and discovery system. Nucleic Acids Res. 2003;31:164–171. doi: 10.1093/nar/gkg148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Bose M, Barber RD. Prophage Finder: a prophage loci prediction tool for prokaryotic genome sequences. In Silico Biol. 2006;6:223–227. [PubMed] [Google Scholar]
- Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- Saeed A, Bhagabati N, Braisted J, Liang W, Sharov V, Howe E, Li J, Thiagarajan M, White J, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Koburger T, Weibezahn J, Bernhardt J, Homuth G, Hecker M. Genome-wide mRNA profiling in glucose starved Bacillus subtilis cells. Mol Genet Genomics. 2005;274:1–12. doi: 10.1007/s00438-005-1119-8. [DOI] [PubMed] [Google Scholar]
- Hahne H, Mäder U, Otto A, Bonn F, Steil L, Bremer E, Hecker M, Becher D. A comprehensive proteomics and transcriptomics analysis of Bacillus subtilis salt stress adaptation. J Bacteriol. 2010;192:870–882. doi: 10.1128/JB.01106-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Sola S, Sammeth M, Guigó R, Ribeca P. The Gem mapper: fast, accurate and versatile alignment by filtration. Nat Methods. 2012;9:1186–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, Zhu W, Passalacqua K, Bergman N, Borodovsky M. Bacillus anthracis genome organization in light of whole transcriptome sequencing. BMC Bioinformatics. 2010;11:S10. doi: 10.1186/1471-2105-11-S3-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer M. Physiologische Untersuchungen zur Regulation des Aminosäure-Stoffwechsels von Bacillus licheniformis DSM13. Georg-August-Universität Göttingen, Institut für Mikrobiologie und Genetik; 2010. (PhD thesis). [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning - A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of whole transcriptome sequencing reads. Figure S2: Comparative operon prediction. Figure S3: Northern blot confirmation of non-coding RNAs. Figure S4: Antisense RNAs with putative impact on productivity. Figure S5: BsrG/SR4-like loci in B. licheniformis. Figure S6: Work flow charts.
Whole transcriptome sequencing reads. Table S2: NPKM values of RNA features. Table S3: NPKM values of all genes. Table S4: Differential RNA-Seq reads. Table S5: Transcription start sites. Table S6: Identified RNA features. Table S7: Predicted operons. Table S8: Corrected genes. Table S9: New genes. Table S10: Removed genes. Table S11: Predicted cis-regulatory elements. Table S12: Comparison of cis-regulatory elements known from B. subtilis. Table S13: Cluster analysis of ncRNA expression profiles. Table S14: Comparison of ncRNAs to B. subtilis. Table S15: Comparison of small RNAs from B. subtilis to identified ncRNAs. Table S16: Sequence Read Archive accession. Table S17: Primer pairs for Northern blots.