

Single molecule Förster resonance energy transfer (smFRET) allows the distance between two fluorophores to be determined, and when these probes are attached to biomolecules such as DNA and protein, both static and dynamic structural features can be quantified.[1–8] However, when measuring smFRET, photons from each single fluorophore can only be collected for a finite period of time, resulting in a fairly small window of time to observe structural changes.[6] Longer smFRET traces are desirable because they improve information content in both time and space domains.[9, 10]
The primary cause for truncated photon collection times is deleterious fluorophore photophysics and/or photochemistry. In some cases, the fluorophore quantum yield fluctuates due to transient interactions with residues on the biomolecule to which it is attached.[11–14] Much more commonly, temporary photoblinking and irreversible photobleaching restrict photon collection. Photoblinking describes a temporary dark state for a fluorophore and is usually due to the molecule entering a long-lived, low-emissive triplet state.[3, 15–18] While in the dark state there is an increased risk of irreversible photobleaching by reaction with singlet oxygen or other highly reactive species present in solution.[3, 15]
Ameliorating unwanted photophysical and photochemical processes has been pursued by designing new fluorophores and introducing chemical extensions and blockers to biomolecules in order to reduce unwanted interactions.[8, 11, 19, 20] The still ubiquitous photoblinking and photobleaching must be addressed, thus motivating the development of a variety of photoprotection systems (PSs), involving different combinations of photostabilizing solutions and oxygen scavenging solutions to optimize the relaxation pathways and ultimately to extend the photobleaching lifetime.[15, 21, 22]
Typically, photostabilization solutions reduce the time the fluorophore spends in the triplet state.[15, 16, 22–25] An oxygen scavenging solution’s primary function is to react with aqueous oxygen preventing it from forming singlet oxygen, which will react with the fluorophores, and therefore allowing for maximum fluorescent cycles.[21, 26, 27] The appropriate choice for PS elements depends on the fluorophore species and the surrounding environment.[11] smFRET measurements, in particular, are even more complex, as there are at least two separate fluorophores that both need to respond well to a PS without the PS interfering with the energy transfer between the fluorophores. In PSs, the most commonly used oxygen scavenger is glucose/glucose oxidase (GGO) in conjunction with catalase.[26] GGO provides a preferential binding site for oxygen, reducing the overall oxygen concentration present in the solution available for the photobleaching reaction. More recently, alternative options for singlet oxygen removal have included protocatechuric acid /protocatechuric-3,4-dioxygenase (PCA), which possesses a higher binding affinity for oxygen than GGO, and pyranose oxidase with catalase, which is a scavenger designed for pH sensitive systems.[21, 28]
Early on, β-mercaptoethanol was employed as a photostabilizing solution in order to quench the triplet state.[29] Although it helped to limit photoblinking in some cases, it failed to adequately limit photoblinking for Cy5, one of the most commonly used smFRET fluorophores.[30, 31] Trolox was later introduced as an alternative to β-mercaptoethanol and is a commonly used photostabilizing system.[32, 33] The reducing and oxidizing system (ROXS) and the triplet state quenching system (TSQ) are two more recently developed photostabilizing solutions.[15, 23] ROXS operates by introducing a reducing agent and oxidizing agent to the solution, which opens up faster alternate pathways from the triplet state to the ground state.[15, 34] TSQ is a combination of Trolox, nitrobenzyl alcohol, and cyclooctatetraene and works similarly to the ROXS system by incorporating triplet quenching by energy transfer with cyclooctatetraene.[23] The PCA solution can also act as a reducing agent and can serve as a partial photostabilizer.[21] Because of this, the PCA system is known to interfere with photostabilization solutions designed around reducing and oxidizing the fluorophore, such as ROXS or TSQ.[34] Thus, one should expect wide variability of photoblink/photobleach improvement as a function of both fluorophore and biomolecule chemistry.
In order to demonstrate variability in PS effectiveness on a model smFRET system and to show that optimizing PS chemistry can result in dramatic improvements in smFRET observation time, we compared smFRET photostability on a model DNA hairpin labeled with an smFRET pair in the presence of PSs comprised of two commonly used oxygen scavenging solutions (GGO and PCA) alone and in combination with three commonly used photostabilizing solutions (Trolox, ROXS, and TSQ).[15, 21–23, 26, 32] The chosen fluorophore pair was Cy3/Cy5, as the pair is commonly used in smFRET studies, and with appropriate attention to position and spacer chemistry,[15–18, 21–25, 35] photophysical interactions aside from photoblinking and photobleaching are minimal.
We selected a mutant version of transactivation response element (TAR) DNA hairpin (Figure 1), because it has been well-studied by smFRET [7, 36–40] and has been shown to be conformationally stable and static. This is important because, for the purposes of evaluating the variability of photostability of smFRET fluorophores, it is necessary to keep all other control parameters constant. The ensemble smFRET distribution shown in Figure 1B is from a series of single molecule traces under GGO ROXS conditions and confirms that, as shown previously, the overall smFRET efficiency is tightly grouped near an efficiency of 1, demonstrating that we can consider the hairpin, and the expected smFRET values, to be static throughout the measurement. The smFRET efficiency distribution did not change with different PS. Thus, the current experiment reports only the effects of PS on smFRET via Cy5 emission, with none of the complications of relating photocycles with hairpin conformational changes.
Figure 1.

A) The structure of the mutant TAR DNA. The structure has been modified to remove intermediate bulges, add thymine spacers to reduce fluorophore-base interactions, attach the Cy3 and Cy5 to the ends of the hairpin, and place a biotin molecule on the loop for the immobilization procedure. B) Ensemble histogram of the smFRET efficiency response as accumulated from 51 single molecule traces using the GGO ROXS system.
An additional complication arises from interactions between PS and the fluorophores that alter photophysics or hinder free rotation. Such interactions would distort the calculated decay rate. Although unwanted photophysical interactions have not been reported for this fluorophore/hairpin system,[7, 37, 40] in order to rule out hindered rotation of Cy5 as a source of any smFRET efficiency fluctuations, we investigated the rotational lifetime of the fluorophore in the various oxygen scavenging solutions, the results of which are shown in the Supporting Information. In all cases, measured lifetimes were in the range of tens to hundreds of picoseconds. The PCA solution in combination with the TSQ and ROXS, proved much more difficult to fit with traditional multi-exponential kinetics. This could be a sign of complex interaction kinetics or alternate excitation relaxation pathways and warrants further study. However, all rotational lifetimes are at least seven orders of magnitude faster than the bin time used for the smFRET experiments, and therefore we are confident that rotation of the fluorophore is not a significant contributor to smFRET efficiency fluctuations.
Some PSs can induce rapid pH changes upon being introduced to the system and there has been recent research on oxygen scavenging solutions that do not affect the pH.[28, 41] To reduce the effects of changing pH in these experiments, a HEPES buffer solution was used, as discussed in the Experimental section. Also, each sample was equilibrated for 30 min after the introduction of the PS before beginning measurments, and overall photon counts were compared to identify any changes in quantum yield. No significant changes in pH (or in pH-induced quantum yield changes) were observed over the course of the experimental measurements (data not shown).
Next, as summarized in Figure 2, statistical benchmarks were established for identifying single Cy3/Cy5 labeled hairpins and for distinguishing photoblink and photobleach criteria. We used an automated routine to determine molecule positions from the initial frame at time = 0, and corrected for drift in subsequent frames as described in previous work.[42] Once located, the background corrected sum total photons for each molecule’s 350 × 350 nm region (i.e. 3 × 3 pixels) were fit to a 2 D normal distribution and screened with a 99% confidence level to remove false positives and closely clustered molecules, as shown in the Figure 2 inset for the GGO ROXS solution. The photons from identified molecules were collected over the course of the experiment from all subsequent frames and the resulting distribution was fit to a full normal plus a half-normal cumulative distribution function corresponding to an ‘on’ distribution and an ‘off’ distribution (Figure 2). A threshold for determining the on/off status of a molecule was set based on the 95% confidence level for the off state and all of the molecules in each frame assigned to the appropriate state based on whether the sum photon counts fell inside or outside the confidence level.
Figure 2.
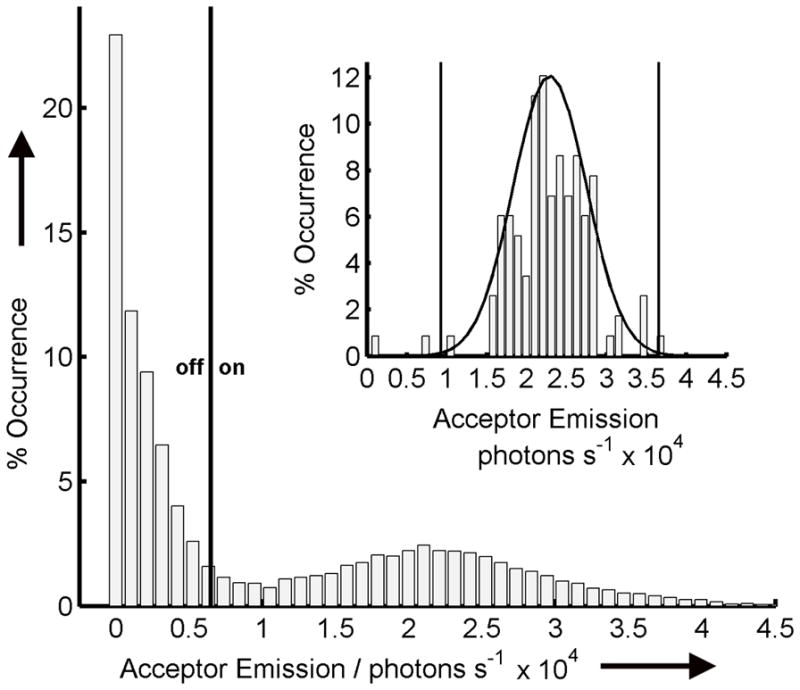
Example emission intensity distribution for the acceptor channel for all identified molecules in all frames for GGO ROXS solution. The threshold value for determining on/off states was determined to be approximately 6500 photons/s with values above it being considered to be in the on state and values below to be in the off state. The inset shows the intensity distribution for molecules in the first frame before molecule selection process with the Gaussian fit with the two threshold points.
After the identification of ‘on‘ and ‘off‘ molecules in initial and subsequent frames, it was necessary to classify ‘off‘ molecules as either photoblinks, if reversible, or photobleaches, if irreversible, as shown in Figure 3. When a molecule‘s signal dropped below the threshold discussed earlier, it’s status was termed a photoblink if the emission recovered for at least one frame subsequent to the first ‘off‘ occasion, or a photobleach if the emission did not recover. Both cases are shown in Figure 3 in sample frames, with identified molecules and cases indicated.
Figure 3.

Example initial and subsequent donor and acceptor frames for GGO ROXS solution. Identified molecules are marked as being either ‘on’ (yellow triangle), ‘photoblinking’ (green circle), or ‘photobleached’ (blue square) for the shown duration based on their status in frames 2 and 8.
The decay in the percentage of smFRET pairs remaining in the ‘on‘ state over the course of photoexcitation is shown in Figure 4 for the range of PSs compared, demonstrating that the photostability of the smFRET pair/DNA hairpin used in the present study varied considerably as a function PS conditions. Shown are all of the combinations of the two different oxygen scavenging systems, GGO and PCA, and three different photostabili-zation systems, Trolox, TSQ, and ROXS, tested in order to determine which combination of oxygen scavenger and photostabilizer form the best solution for prolonging fluorophore lifetimes in single molecule smFRET experiments for the Cy3/Cy5 smFRET pair on the TAR DNA hairpin. The variability in photostability as a function of PS was quantified by a single exponential fit of the decay data to yield the relative photobleach rate constants included in the first two columns of Table 1 and the solid lines in Figure 4. The values are plotted against the calculated exposure time for each molecule based on the beam size, effective cross sectional area of the molecule, and scan rate.
Figure 4.

Percentage of on state molecules as a function of excitation time for each combination of oxygen scavenger and photostabilizer. A single exponential decay curve was fit to each series in order to extract the rate constant for each scavenging solution. PSs were listed in order of effectiveness from most effective to least effective.
Table 1.
Photobleaching and photoblinking for each PS combination[a]
| Solution | Photobleaching Rate Constant | Photoblink Dark % | |
|---|---|---|---|
| (1/s) | R2 | ||
|
| |||
| Buffer | 24.7 | 0.99 | NA |
| GGO | 5.04 | 0.96 | 16 ± 4 |
| GGO Trolox | 1.33 | 0.99 | 15 ± 4 |
| GGO TSQ | 0.86 | 0.99 | 26 ± 6 |
| GGO ROXS | 1.06 | 0.98 | 24 ± 6 |
| PCA | 1.62 | 0.94 | 22 ± 6 |
| PCA Trolox | 1.23 | 0.98 | 18 ± 6 |
| PCA TSQ | 4.35 | 0.94 | 20 ± 4 |
The combination of PCA ROXS solution resulted in a vigorous reaction and was not measured for photobleaching and photoblink analysis
It is important to note that, although the photostability was highly variable as a function of the specific PS, as shown in Figure 4, all of the PSs offer an improvement in comparison to a buffer alone (buffer data/curve shown in black). By introducing a PS, the photobleaching decay rate is reduced, yielding an improvement factor of 4.9 for the standard solution GGO, and 15.2 for PCA. When both an oxygen scavenging solution and a photostabilizing solution are present the decay rate of the acceptor is reduced even further than just using an oxygen scavenging solution, up to 28.7 times for the GGO ROXS combination. For the GGO systems, adding a photostabilization solution decreased the decay rate over simply using an oxygen scavenging solution by itself, whereas the PCA systems showed less consistent results for combinations with a photostabilizer with only the PCA Trolox combination yielding improved lifetimes over PCA alone.
TSQs and ROXS, the two more complicated photostabilization solutions, were highly dependent on the oxygen scavenging solution used. When used with GGO, both resulted in decreased photobleaching decay rates compared to either oxygen scavenging solution paired with Trolox. However when paired with the PCA solution the results were very different. PCA caused a visible reaction with the ROXS system (data not shown), likely a redox reaction between protocatechuric acid and methyl viologen, resulting in the inability to measure a decay rate for that combination. The PCA TSQ pairing resulted in faster bleaching of the fluorophores than PCA by itself and just slightly shorter times than the GGO system by itself.
As discussed earlier, reduction in photoblinking is another important property of PSs. The photoblink dark fraction, calculated as described below in the experimental section, is included in Table 1. Unlike the large variability observed in photobleaching rate constants, the photoblink dark fraction statistics were very similar for the PSs studied. Because the smFRET pair photobleached so quickly in buffer, it was not possible to calculate reliable photoblink statistics for this system. It is important to note that the observed lack of variability in photoblink dark fraction as a function of chosen PS likely results from the relatively long observation times and low time resolution necessary in the present study to collect adequate photobleach statistics. Because photoblinking occurs on faster time scales, a comparison at kHz collection frequency, outside the scope of the current study, might resolve differences in photoblink rates among the PSs.
In conclusion, we have demonstrated that PS selection can have a drastic effect on the photobleaching lifetime. Selecting the proper combination of oxygen scavenging solution and photostabilization solution can significantly increase the observable lifetime of a smFRET pair, even for a relatively simple biomolecule and a well-characterized smFRET pair. We determined which PS had the biggest effect for Cy3/Cy5 labeled DNA in single molecule smFRET environment. Our results showed that using the GGO oxygen scavenging solution in combination with the TSQ photostabilization solution yielded the longest average photobleaching lifetimes and that significant differences in photoblinking were not observed under our measurement conditions. All of the PSs, except for the combination of PCA/ROXS, were effective at increasing the observation time for smFRET with the Cy3/Cy5 system, but PS optimization is worthwhile given the strong variability.
Experimental Section
Sample preparation
22 × 22 mm # 1 thick glass slides were plasma cleaned and functionalized via Vectabond treatment (Vector Laboratories, Burlingame, CA).[7, 32] A 100:1 mixture of 5 kDa, methoxy-terminated, N-succinimidyl polyethylene glycol (Fluka, 33% w/w PEG in MB water) and 5 kDa biotin-terminated PEG (NOF Corporation, 2.5% w/w in MB water) in sodium bicarbonate (1% v/v, pH 8.0) buffer was added to the slide surface and incubated for 5hrs. A custom hybriwell chamber (Grace bio-labs) with dual silicon press fit tubing connectors (Grace bio-labs) was placed on top of the biotin-PEG glass slide. The biotin-PEG chamber was filled with streptavidin (40 μL, 20% w/w, Invitrogen) in HEPES buffer solution, comprised of HEPES buffer (0.025 M, Sigma) and NaHCO3 (0.04 M), before being incubated in the dark for 10 min. A mutant version of TAR DNA (2 nM, Trilink) was diluted in HEPES buffer solution and then heated to 80 °C then 60 °C before cooling at 0 °C for 2:30min apiece. The TAR DNA solution was added in two 17 μL increments into the chamber and incubated for 20 min. After incubation excess DNA was flushed out via HEPES buffer solution. The sample was placed over the objective and the PS for that trial was flowed through the sample for 30 minutes to allow for the system to equilibrate. The outlet flow was monitored for pH changes using pH-indicator strips (colorpHast pH 5.0 – 10.0).
Oxygen Scavenger System preparation
A PCA stock solution was prepared by making a solution of 3–4 dihydroxybenzoic acid (0.2 M, Aldrich), by adding NaOH (1 M, Sigma-Aldrich) until all of the acid had dissolved and the pH of the solution was approximately 9. An entire bottle of protocatechuric-3,4-dioxygenase (~9 mg, Sigma) was dissolved in a 0.45 mL buffer solution containing Glycerol (10 %, Sigma-Aldrich), Tris HCl pH8 (100 mM, Sigma), KCl (50 mM, FisherChemical), and EDTA (1mM, Sigma). Volumes were adjusted so that the final PCA oxygen scavenger solution contained 10 mM protocatechuric acid and 60 nM protocatechuric-3,4-dioxygenase. The initial pH for the PCA solution was ~7.5 at the beginning of data collection and remained there throughout the experiment.
Stock solutions of glucose were prepared by adding glucose (3 g, Sigma) to water (7 mL). The solution was then vortexed and filtered before adding to the final solution cocktail. Glucose oxidase (3 mg, Sigma) was added to water (3 mL) to create the glucose-oxidase stock solution. Catalase (20 μL Sigma) was added to HEPES buffer (3980 μL, 1 M, Sigma). The final GGO solution concentrations were 3.3% w/w β-D-(+)-glucose, 1% w/w glucose oxidase, and 0.1% v/v catalase. The GGO solutions had a pH value of ~ 7.5 after the equilibrating period and pH slowly dropped to between 7.5 and 70 throughout the experiment.
Trolox solution was prepared by adding 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (50 mg, Aldrich) to water (2 mL) immediately before use in PS. The solution was vortexed vigorously and filtered. The filtered Trolox solution was used in lieu of water as the dilution agent for PS preparation.
To prepare the three solutions that comprise the TSQ system, nitrobenzyl alcohol (6.2 mg, Aldrich) was dissolved in DMSO (1 mL, Sigma), Trolox (10 mg) was dissolved in DMSO (1 mL) and cyclooctatetraene (5 μL, Sigma-Aldrich) was added to DMSO (495 μL). The nitrobenzyl alcohol solution (50 μL), Trolox solutions (50 μL), and cyclooctatetraene solution (22 μL) were added to the oxygen scavenger cocktail and after the oxygen scavenging system was diluted to a total volume of 1 mL.
The ascorbic acid and methyl viologen redox pair was selected for use in the ROXS solution. Ascorbic acid (100 μL,10 mM, Sigma) was mixed with methyl viologen (100 μL, 10 mM, Aldrich). This mixture was added to the oxygen scavenger and diluted with water to the 1 mL mark.
Microscope set-up
All single-molecule fluorescence measurements were performed using a custom built confocal microscope.[7] The sample was excited with a 532-nm diode- pumped solid-state laser (Coherent, Compass 315M-100 SL). The light was expanded to overfill the back aperture of a Fluar 100× 1.3 NA oil immersion microscope objective lens (Carl Zeiss, GmBH) resulting in a beam radius of ~ 250 nm and height of ~ 1 μm. A closed-loop-x-y-z piezo stage (P-517.3CL; Physik Instrumente) with 100 × 100 × 20 μm travel range and 1-nm specificity (SPM 1000; RHK Technology) was used to locate individual molecules and perform the scanning routine. The power of the laser was set to approximately 2 μW at the sample for all experiments. Fluorescence emission was collected back through the objective and was passed through a notch filter (zet532nf, Chroma Technology) and towards the detector box. The emitted light was separated by a second dichroic mirror (640-nm high pass filter) (Chroma 640 DCXR) to split donor emission and acceptor emission by wavelength, and these fluorescence signals were collected by two avalanche photodiodes (SPCM-AQR-15; Perkin-Elmer) fitted with an additional filters (NHPF-532.0, Kaiser Optical and ET585, Chroma Technology) selected to further increase the selectivity of each detector.
Data collection
A 30 um × 30 um sample region was raster scanned until a majority of the molecules present initially no longer appeared in the acceptor channel or the donor channel. The images were then processed by an in-house MATLAB script that identified molecules in the first frame of the acceptor channel and tracked them throughout the experiment to account for sample drift.[42] Molecules were screened based on their relative intensities and size in order to remove false positives due to clustering, with identified positions closer than 600 nm center to center being removed. Photon counts at the remaining identified positions were stored and then summed up in a 350 nm × 350 nm area for both the donor and the acceptor channels and then fit to a normal Gaussian distribution where a 95% confidence interval was set and molecules whose sum counts existing outside the tolerance being removed (see Figure 2 for sample distribution). After screening, the sum counts for each molecule in every frame were collected in order to determine a threshold for on/off state from the resulting full-Gaussian/half-Gaussian distribution and set at three times the sigma for the half-Gaussian representing off state molecules. For each molecule the on/off state was determined in every frame and the photoblinking and photobleaching rates determined. For each PS, the percentage of acceptor on state molecules vs. the total number of identified molecules was plotted for every frame and fit to a single exponential decay, where the decay rate constant was extracted along with the R2 for the fit. To determine the photoblinking rate, the number of times that a molecule returned to the on state from the off state was divided by the total length of time that a molecule was determined to be on. To determine the photoblinking dark state percentage the number of off frames were divided by the sum of both the on and off frames before a molecule was determined to have photobleached. For both the photoblink rate and photoblink dark percentage bootstrapping was used to determine the error of the averages with a 95% confidence limit.
Anisotropy
Lifetime and anisotropy data were acquired using a Fluotime 300 (PicoQuant, Germany) with a pulsed diode laser excitation source 640 nm PDL-D-C 640 (PicoQuant, Germany) driven by a PDL 820 (PicoQuant, Germany) laser driver. The emission monochromator was set to 665 nm. Photons were detected using a PMT model PMA-C 182-N-M (PicoQuant, Germany) DNA solutions, 100 nM, were placed in 1 cm polystyrene cuvettes for analysis. Data analysis was conducted using Fluofit software (PicoQuant, Germany). Fluorescence decay curves measured with the polarizer set to the magic angle were analyzed using reconvolution analysis to extract the fluorescence lifetimes. Fluorescence decay curves were then measured with the emission polarizer set parallel and perpendicular to the excitation polarizer. These curves were analyzed using reconvolution analysis and the previously measured florescence decay times to extract the rotational lifetimes.
Supplementary Material
Acknowledgments
We would like to thank the Welch Foundation (Grant C-1787), the National Science Foundation(Grants CBET-1133965 and CHE-1151647), and the National Institute of Health (Grant GM94246-01A1) for providing funding. We would also like to thank PicoQuant for loaning the use of the Fluotime 300 instrument. Special thanks also goes to Dr. Jixin Chen, Lydia Kisley, and Chad Byers for their thoughts and discussions as well as Prof. Stephan Link and his research group for their feedback on this project.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author
References
- 1.Roy R, Hohng S, Ha T. Nature Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landes CF, Rambhadran A, Taylor JN, Salatan F, Jayaraman V. Nat Chem Biol. 2011;7:168–173. doi: 10.1038/nchembio.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakowicz JR. Principles of fluorescence spectroscopy. Springer; New York: 2006. p. 443. [Google Scholar]
- 4.Weiss S. Science. 1999;283:1676–1683. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- 5.Weiss S. Nature Structural Biology. 2000;7:724–729. doi: 10.1038/78941. [DOI] [PubMed] [Google Scholar]
- 6.Ha TJ, Ting AY, Liang J, Deniz AA, Chemla DS, Schultz PG, Weiss S. Chemical Physics. 1999;247:107–118. [Google Scholar]
- 7.Darugar Q, Kim H, Gorelick RJ, Landes C. J Virol. 2008;82:12164–12171. doi: 10.1128/JVI.01158-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preus S, Wilhelmsson LM. Chembiochem. 2012;13:1990–2001. doi: 10.1002/cbic.201200400. [DOI] [PubMed] [Google Scholar]
- 9.Blanco M, Walter NG. Method Enzymol. 2010;472:153–178. doi: 10.1016/S0076-6879(10)72011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watkins LP, Yang H. Biophysical Journal. 2004;86:4015–4029. doi: 10.1529/biophysj.103.037739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilderbrand SA. In: Labels and Probes for Live Cell Imaging: Overview and Selection Guide. Papkovsky DB, editor. Vol. 591. Humana Press; 2010. pp. 17–45. [DOI] [PubMed] [Google Scholar]
- 12.Dolghih E, Roitberg AE, Krause JL. Journal of Photochemistry and Photobiology a-Chemistry. 2007;190:321–327. [Google Scholar]
- 13.Ranjit S, Gurunathan K, Levitus M. Journal of Physical Chemistry B. 2009;113:7861–7866. doi: 10.1021/jp810842u. [DOI] [PubMed] [Google Scholar]
- 14.Sanborn ME, Connolly BK, Gurunathan K, Levitus M. Journal of Physical Chemistry B. 2007;111:11064–11074. doi: 10.1021/jp072912u. [DOI] [PubMed] [Google Scholar]
- 15.Vogelsang J, Kasper R, Steinhauer C, Person B, Heilemann M, Sauer M, Tinnefeld P. Angewandte Chemie-International Edition. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 16.Cordes T, Vogelsang J, Tinnefeld P. Journal of the American Chemical Society. 2009;131:5018. doi: 10.1021/ja809117z. [DOI] [PubMed] [Google Scholar]
- 17.Misik V, Miyoshi N, Riesz P. Free Radical Biology and Medicine. 1999;26:961–967. doi: 10.1016/s0891-5849(98)00279-2. [DOI] [PubMed] [Google Scholar]
- 18.Stein IH, Capone S, Smit JH, Baumann F, Cordes T, Tinnefeld P. Chemphyschem. 2012;13:931–937. doi: 10.1002/cphc.201100820. [DOI] [PubMed] [Google Scholar]
- 19.Wang HX, Nakata E, Hamachi I. Chembiochem. 2009;10:2560–2577. doi: 10.1002/cbic.200900249. [DOI] [PubMed] [Google Scholar]
- 20.Patterson GH, Lippincott-Schwartz J. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 21.Aitken CE, Marshall RA, Puglisi JD. Biophysical Journal. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campos LA, Liu JW, Wang XA, Ramanathan R, English DS, Munoz V. Nature Methods. 2011;8:143–U163. doi: 10.1038/nmeth.1553. [DOI] [PubMed] [Google Scholar]
- 23.Dave R, Terry DS, Munro JB, Blanchard SC. Biophysical Journal. 2009;96:2371–2381. doi: 10.1016/j.bpj.2008.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Q, Jockusch S, Zhou Z, Altman RB, Warren JD, Turro NJ, Blanchard SC. The Journal of Physical Chemistry Letters. 2012;3:2200–2203. doi: 10.1021/jz300670p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cordes T, Maiser A, Steinhauer C, Schermelleh L, Tinnefeld P. Phys Chem Chem Phys. 2011;13:6699–6709. doi: 10.1039/c0cp01919d. [DOI] [PubMed] [Google Scholar]
- 26.Benesch RE, Benesch R. Science. 1953;118:447–448. doi: 10.1126/science.118.3068.447. [DOI] [PubMed] [Google Scholar]
- 27.Gruebele M. Nature Methods. 2011;8:213–215. doi: 10.1038/nmeth0311-213. [DOI] [PubMed] [Google Scholar]
- 28.Swoboda M, Henig J, Cheng HM, Brugger D, Haltrich D, Plumere N, Schlierf M. Acs Nano. 2012;6:6364–6369. doi: 10.1021/nn301895c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yanagida T, Nakase M, Nishiyama K, Oosawa F. Nature. 1984;307:58–60. doi: 10.1038/307058a0. [DOI] [PubMed] [Google Scholar]
- 30.Ha T, Tinnefeld P. Annu Rev Phys Chem. 2012;63:595–617. doi: 10.1146/annurev-physchem-032210-103340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heilemann M, Margeat E, Kasper R, Sauer M, Tinnefeld P. Journal of the American Chemical Society. 2005;127:3801–3806. doi: 10.1021/ja044686x. [DOI] [PubMed] [Google Scholar]
- 32.Rasnik I, McKinney SA, Ha T. Nature Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 33.Grunwell JR, Glass JL, Lacoste TD, Deniz AA, Chemla DS, Schultz PG. Journal of the American Chemical Society. 2001;123:4295–4303. doi: 10.1021/ja0027620. [DOI] [PubMed] [Google Scholar]
- 34.Le Gall A, Dulin D, Clavier G, Meallet-Renault R, Bouyer P, Perronet K, Westbrook N. Chemphyschem. 2011;12:1657–1660. doi: 10.1002/cphc.201100085. [DOI] [PubMed] [Google Scholar]
- 35.Di Fiori N, Meller A. Biophysical Journal. 2010;98:2265–2272. doi: 10.1016/j.bpj.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor JN, Darugar Q, Kourentzi K, Willson RC, Landes CF. Biochemical and Biophysical Research Communications. 2008;373:213–218. doi: 10.1016/j.bbrc.2008.05.191. [DOI] [PubMed] [Google Scholar]
- 37.Landes CF, Zeng YN, Liu HW, Musier-Forsyth K, Barbara PF. Journal of the American Chemical Society. 2007;129:10181–10188. doi: 10.1021/ja071491r. [DOI] [PubMed] [Google Scholar]
- 38.Liu HW, Cosa G, Landes CF, Zeng YN, Kovaleski BJ, Mullen DG, Barany G, Musier-Forsyth K, Barbara PF. Biophysical Journal. 2005;89:3470–3479. doi: 10.1529/biophysj.105.065326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu HW, Zeng YN, Landes CF, Kim YJ, Zhu YJ, Ma XJ, Vo MN, Musier-Forsyth K, Barbara PF. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5261–5267. doi: 10.1073/pnas.0700166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeng YN, Liu HW, Landes CF, Kim YJ, Ma XJ, Zhu YJ, Musier-Forsyth K, Barbara PF. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12651–12656. doi: 10.1073/pnas.0700350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi XH, Lim J, Ha T. Analytical Chemistry. 2010;82:6132–6138. doi: 10.1021/ac1008749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Claytor K, Khatua S, Guerrero JM, Tcherniak A, Tour JM, Link S. Journal of Chemical Physics. 2009;130:164710. doi: 10.1063/1.3118982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.