

Abstract
Rationale
Nox2 and Nox4 are major components of the NADPH oxidase (Nox) family, which purposefully produce reactive oxidative species (ROS), namely O2− and H2O2, in the heart. The isoform-specific contribution of Nox2 and Nox4 to ischemia/reperfusion (I/R) injury is poorly understood.
Objective
We investigated the role of Nox2 and Nox4 in mediating oxidative stress and myocardial injury during I/R using loss of function mouse models.
Methods and Results
Systemic (s) Nox2 KO, sNox4 KO, and cardiac-specific (c) Nox4 KO mice were subjected to I (30 min)/R (24 h). Both myocardial infarct size/area at risk (MI/AAR) and O2− production were lower in sNox2 KO, sNox4 KO, and cNox4 KO than in wild-type (WT) mice. Unexpectedly, however, the MI/AAR was greater, despite less O2− production, in sNox2 KO+cNox4 KO (DKO) mice and transgenic mice with cardiac-specific expression of dominant-negative Nox (Tg-DN-Nox), which suppresses both Nox2 and Nox4, than in WT or single KO mice. Hypoxia-inducible factor-1α (HIF-1α) was downregulated while peroxisome proliferator-activated receptor-alpha (PPARα) was upregulated in Tg-DN-Nox mice. A cross with mice deficient in prolyl hydroxylase 2, which hydroxylates HIF-1α, rescued the I/R injury and prevented upregulation of PPARα in Tg-DN-Nox mice. A cross with PPARα KO mice also attenuated the injury in Tg-DN-Nox mice.
Conclusions
nBoth Nox2 and Nox4 contribute to the increase in ROS and injury by I/R. However, low levels of ROS produced by either Nox2 or Nox4 regulate HIF-1α and PPARα, thereby protecting the heart against I/R, suggesting that Noxs also act as a physiological sensor for myocardial adaptation.
Keywords: Reactive oxygen species, oxidative stress, free radicals, ischemia/reperfusion
INTRODUCTION
Reactive oxygen species (ROS), such as superoxide (O2−) and hydrogen peroxide (H2O2), play an important role in regulating the cell growth and death of cardiac myocytes.1 The family of NADPH oxidases (Noxs) represents the only known enzyme system whose sole biological function is to purposefully produce O2− or H2O2,2 by transferring electrons from NADPH to molecular oxygen. NADPH oxidases 2 and 4 (Nox2 and Nox4) are the major Nox isoforms in the heart.3 Nox2 is involved in cardiac remodeling after myocardial infarction,4 whereas Nox4 is a critical mediator of mitochondrial oxidative stress and mitochondrial dysfunction during heart failure.5
Nox2 and Nox4 are localized at distinct subcellular locations and appear to have distinct roles in the heart.3 Although it has been speculated that Nox isoforms play an important role in mediating increases in oxidative stress during ischemia/reperfusion (I/R), the role of Nox4 in mediating I/R injury has not been evaluated with genetically altered mouse models. The role of Nox2 in mediating I/R injury has been investigated indirectly with p47phox KO mice.6 but has only been examined directly with Nox2 KO mice in the context of preconditioning.7 Directly examining the roles of the Nox isoforms in mediating I/R injury would provide important clues for the development of an effective intervention to inhibit myocardial I/R injury.
Hypoxia-inducible factors (HIFs) are master regulators of hypoxia-regulated gene expression 8 that mediate adaptive responses to low oxygen (O2) levels and/or oxidative stress. HIF-1 transcriptionally activates genes associated with angiogenesis, energy metabolism, nutrient transport, the cell cycle, and cell migration.8 Activation of glycolytic genes by HIF-1 is considered critical for metabolic adaptation to hypoxia through increased conversion of glucose to pyruvate and, subsequently, to lactate. A hypoxia-induced metabolic switch shunts glucose metabolites from mitochondria to glycolysis to maintain ATP production and prevent toxic ROS production.9 Therefore, HIFs play a protective role in I/R injury through regulation of the cardiac metabolism.10 Although it has been reported that ROS activate HIF-1α,11, 12 the role of Noxs in regulating the expression of HIF-1 during I/R remains to be elucidated.
Thus, the major goal of this study was to elucidate the functions of Nox2 and Nox4 during I/R in the heart. To this end, we investigated the effects of I/R in the context of Nox2 and Nox4 loss-of-function mouse models. Our results suggest that both Nox2 and Nox4 play critical roles in mediating ROS production and myocardial injury in response to I/R. Interestingly, a low level of ROS produced by either Nox2 or Nox4 is required for the heart to activate adaptive mechanisms, including regulation of HIF-1α and PPARα. Thus, Nox2 and Nox4 have both pathological and adaptive roles in the heart during myocardial I/R.
METHODS
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Genetically altered mouse models
Tg-Nox4 and Tg-DN-Nox mice were generated with the use of the α-myosin heavy chain promoter 3 on a C57BL/6J background. The baseline cardiac phenotypes of Tg-Nox4 and Tg-DN-Nox mice have been described 3. Nox4 flox/flox mice were generated as described previously 5. Cardiac-specific Nox4 KO mice were generated by crossing Nox4 flox/flox mice with a C57BL/6J background with α-myosin heavy chain promoter-driven Cre mice (αMHC-Cre, courtesy of Dr M. Schneider, Imperial College, London, UK). Systemic Nox4 KO mice were generated by crossing Nox4 flox/flox mice with a C57BL/6J background with cytomegalovirus promoter-driven Cre mice purchased from Jackson Laboratory. Systemic Nox2 KO mice were purchased from Jackson Laboratory. Cardiac-specific PHD2 KO mice were generated by crossing PHD2 flox/flox mice with a C57BL/6J background with αMHC-Cre mice. We generated a genetic cross between Tg-DN-Nox and cardiac-specific PHD2+/− mice 13. We also generated a genetic cross between Tg-DN-Nox and PPARα−/− mice. We used only male mice in these experiments. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Medicine and Dentistry of New Jersey.
Statistical analysis
Data are expressed as mean±SEM. The between-group comparisons of means were performed by one-way ANOVA, followed by t-tests. The Bonferroni’s correction was done for multiple comparisons of means. P<0.05 was considered to be statistically significant.
RESULTS
Individual downregulation of Nox2 or Nox4 attenuates I/R injury in the heart
We investigated the involvement of the different Nox isoforms in I/R injury in the heart. We first characterized O2− production in the heart after I/R using superoxide dismutase (SOD)-inhibitable lucigenin chemiluminescence. Wild-type (WT) mice were subjected to 30 min of ischemia and 24 h of reperfusion. O2− production from the heart homogenates 1, 6 and 24 hours after reperfusion was significantly higher than that at baseline (Figure 1A). In order to investigate the role that Nox4 in cardiomyocytes plays in mediating I/R injury, we applied I/R to cardiac-specific Nox4 knockout (cNox4 KO) and WT mice. Nox4 expression was increased by I/R in WT mice, but was decreased in cNox4 KO mice at baseline and after I/R (Figure 1B). Downregulation of Nox4 led to a significant decrease in the infarct size/area at risk (AAR) after I/R, as evaluated with triphenyltetrazolium chloride (TTC) and Alcian blue staining, compared to in WT mice (Figure 1C). We also examined the role of Nox2 in the heart in I/R injury by subjecting systemic Nox2 knockout (Nox2 KO) mice to I/R. I/R increased Nox2 expression in WT mice, an effect that was abolished in Nox2 KO mice (Figure 1D). Knockdown of Nox2 also led to a significant decrease in the infarct size after I/R compared to in WT mice (Figure 1E). Apoptotic cell death in the AAR, as evaluated by TUNEL staining, was significantly decreased in both cNox4 KO and Nox2 KO mice compared to in WT mice (Figure 1F). Consistent with the TUNEL staining, cleaved caspase-3 was decreased in both cNox4 KO and Nox2 KO mice compared to in WT mice (Figure 1G). These results suggest that individual downregulation of either Nox2 or Nox4 reduces I/R injury, accompanied by decreases in apoptosis in the myocardium.
Figure 1. The role of Nox isoforms in I/R injury.


A. Superoxide-producing activity of LV homogenates at the indicated time points was evaluated with lucigenin chemiluminescence assays. *P<0.05, **P<0.01. B. Expression levels of Nox4 and tubulin in the LV in WT and cNox4 KO mice with or without I/R were evaluated by immunoblotting. *P<0.05. C. WT and cNox4 KO mice were subjected to I/R. Infarct size was evaluated by TTC and Alcian blue staining. *P<0.05. Bar=1mm. D. Expression levels of Nox2 and tubulin in the LV in WT and Nox2 KO mice with or without I/R were evaluated by immunoblotting. N.D. not detectable. E. WT and Nox2 KO mice were subjected to I/R. Infarct size was evaluated by TTC and Alcian blue staining. *P<0.05. Bar=1mm. F. Apoptotic cells in the ischemic area were evaluated by TUNEL staining 24 h after reperfusion. The number of TUNEL-positive myocytes was decreased in LVs from cNox4 KO and Nox2 KO mice compared to in those of WT mice. *P<0.05. Bar=10 m. G. Expression levels of cleaved caspase-3 and tubulin in the ischemic area were evaluated by immunoblotting. *P<0.05. H. Mitochondrial function in the indicated mice was evaluated by the mitochondrial swelling assay. *P<0.05, N.S. not significant. I. The expression levels of cleaved caspase-12 and tubulin were evaluated by immunoblotting.
To investigate the mechanism by which apoptosis is attenuated in cNox4 KO and Nox2 KO mice, we evaluated mitochondrial function in the heart. Mitochondria were challenged with calcium overload and the rate of mitochondrial swelling was evaluated by light scattering, where decreases in the absorbance reflect passive swelling of the mitochondrial matrix. I/R-induced decreases in absorbance were significantly inhibited in cNox4 KO and Nox2 KO mice compared to in WT mice (Figure 1H), suggesting that both Nox4 and Nox2 contribute to I/R-induced mitochondrial permeability transition pore opening.
The expression of cleaved caspase-12, a mediator of ER stress-induced apoptosis, in the ischemic area was significantly increased by I/R in WT mice. This increase was reduced in both cNox4 KO and Nox2 KO mice (Figure 1I). These data indicate that both Nox4 and Nox2 are involved in ER stress during I/R injury.
Since Nox4 is expressed not only in myocytes but also in non-myocytes, in order to investigate the role of Nox4 in all cell types in the heart during I/R, we generated Nox4 systemic KO (sNox4 KO) mice by crossing Nox4 flox/flox mice with CMV-Cre on a C57 background. The expression of Nox4 in the left ventricle (LV) was completely abolished in sNox4 KO mice (Online Figure IA). sNox4 KO mice showed a decrease in I/R injury compared to WT mice (Online Figure IB). The extent of I/R injury in sNox4 KO mice was similar to that in cNox4 KO mice (Online Figure IB) and systemic Nox2 KO mice (not shown). We also evaluated the O2− production in the ischemic area. The extent of O2− production in sNox4 KO was similar to that in cNox4 KO mice (Online Figure IC).
To elucidate the role of inflammatory responses in mediating I/R injury, the extent of cell infiltration was evaluated with hematoxylin-eosin (HE) staining. Although I/R increased the total cell density in the myocardium in WT, cNox4 KO, and Nox2 KO mice, there was no significant difference between the three groups (Online Figure IIA, B).
Overexpression of DN-Nox causes broad suppression of Nox activity in the heart
In order to further investigate the role of Nox in the heart, we next examined transgenic mice with cardiac-specific overexpression of dominant-negative Nox4 (DN-Nox) 3. Although we initially named these mice Tg-DN-Nox4 mice, we have recently re-designated them as Tg-DN-Nox because we have discovered that DN-Nox inhibits other Nox isoforms besides Nox4 as well. For example, when cardiomyocytes were transduced with Ad-Nox2 and Ad-DN-Nox, DN-Nox efficiently inhibited Nox2-induced O2− production, evaluated as the SOD-inhibitable component of lucigenin chemiluminescence, in cardiomyocytes (Figure 2A). In addition, in sNox4 KO mice, O2− production in the LV, as evaluated with dihydroethidium (DHE) staining, was significantly decreased after injection of Ad-DN-Nox (Figure 2B). Moreover, O2− production, evaluated as the SOD-inhibitable component of lucigenin chemiluminescence, in the mitochondrial fraction, where Nox4 is mainly expressed, and in the microsomal fraction, where Nox2 is mainly expressed, were both suppressed in myocytes transduced with Ad-DN-Nox (Figure 2C, D). These results indicate that DN-Nox inhibits both Nox4 and Nox2 in cardiomyocytes.
Figure 2. DN-Nox inhibits Nox isoforms.

A. Cardiomyocytes were transduced with either Ad-LacZ or Ad-Nox2 in the presence or absence of Ad-DN-Nox, and the cellular level of O2− was evaluated with SOD-inhibitable lucigenin chemiluminescence. *P<0.05, N.S. not significant. B. O2− production in the LV in systemic Nox4 KO mice, as evaluated with dihydroethidium (DHE) staining, was significantly decreased after injection of Ad-DN-Nox adenovirus. *P<0.05. C and D. O2−-producing activity of mitochondrial and microsomal fractions from cardiomyocytes transduced with the indicated adenoviruses was evaluated with lucigenin chemiluminescence assays. *P<0.05 vs. LacZ, N.S. not significant. E. Lysates from myoctytes transduced with Ad-LacZ or Ad-DN-Nox-HA were used for immunoprecipitation with HA antibody. Immunoblots for p22phox, Nox4, and HA were performed. Immunoblot of remaining lysate after incubation with HA-agarose beads is also shown. F. NADP/NADPH ratio was measured in the indicataed mouse hearts. * P<0.05 vs. WT, #P<0.05 vs. cNox4 KO, # # P<0.05 vs. Nox2 KO.
To elucidate the mechanism of suppression of Nox isoforms by DN-Nox, we evaluated the interaction between DN-Nox and p22 phox in myocytes. p22phox was effectively immunoprecipitated with anti-HA antibody from DN-Nox-HA-expressing cardiomyocytes. Importantly, the p22phox level in the supernatant after immunoprecipitation with anti-HA agarose was markedly decreased in myocytes transduced with Ad-DN-Nox-HA (Figure 2E), suggesting that DN-Nox effectively sequestrates p22phox. In addition, we evaluated the NADP/NADPH ratio in the heart. The NADP/NADPH ratio of the heart homogenates was in the order WT > cNox4 KO, Nox2 KO > Tg-DN-Nox (Figure 2F), suggesting that the consumption of NADPH is reduced markedly in myocytes in the presence of DN-Nox. Taken together, the data suggest that DN-Nox broadly suppresses Nox isoforms by sequestrating p22phox from endogenous Noxs.
Tg-DN-Nox and double-knockout of Nox2 and Nox4 exhibit exacerbated I/R injury in the heart
To further understand the role of ROS produced by Nox isoforms, we conducted I/R experiments in transgenic mice with cardiac-specific overexpression of Nox4 (Tg-Nox4) and Tg-DN-Nox mice. Interestingly, the infarct size/AAR in Tg-Nox4 was not significantly different than in non-transgenic (NTg) mice (Online Figure IIIA). Apoptosis in the ischemic area, as evaluated by TUNEL staining, was also similar in Tg-Nox4 and WT mice (Online Figure IIIB), in spite of an increase in O2− production (Online Figure IIIC). On the other hand, unexpectedly, Tg-DN-Nox exhibited a significant increase in the infarct size/AAR (Figure 3A), which was accompanied by increases in apoptosis, as evaluated by the number of TUNEL positive myocytes and the level of cleaved caspase-3 in the AAR after I/R (Figure 3B,C). Tg-DN-Nox also exhibited an increase in necrosis compared to NTg mice as evaluated with hairpin2 staining (Online Figure IV).
Figure 3. DN-Nox and double-knockout of Nox2 and Nox4 exacerbates I/R injury.

A. NTg and Tg-DN-Nox mice were subjected to ischemia (30 min) and reperfusion (24 h). Infarct size was evaluated by TTC and Alcian blue staining. *P<0.05. Bar=1 mm. B. Apoptotic cells in the ischemic area were evaluated by TUNEL staining 24 h after reperfusion. The number of TUNEL-positive myocytes was increased in the LVs of Tg-DN-Nox mice compared to in those of NTg mice. *P<0.05. Bar=10 m. C. Expression levels of cleaved caspase-3 and tubulin in the indicated mice were evaluated by immunoblotting. *P<0.05. D. Cardiac function in NTg and Tg-DN-Nox mice was evaluated using the isolated perfused heart (Langendorff) system. *P<0.05 vs. NTg at same time point. LVEDP: left ventricular end-diastolic pressure. E. WT and DKO mice were subjected to ischemia (30 min) and reperfusion (24 h). Infarct size was evaluated by TTC and Alcian blue staining. *P<0.05. Bar=1 mm. F. Apoptotic cells in the ischemic area were evaluated by TUNEL staining 24 h after reperfusion. The number of TUNEL-positive myocytes was increased in the LVs of DKO mice compared to in those of WT mice. *P<0.05, Bar=10 μm.
We also evaluated cardiac function in Tg-DN-Nox mice using the Langendorff system. Although there was no difference in LVP or dP/dtmax, indexes of cardiac systolic function, or in dP/dtmin, an index of cardiac diastolic function, between NTg and Tg-DN-Nox at baseline, Tg-DN-Nox showed a dramatic decrease in these parameters after I/R in glucose-rich substrates (Figure 3D, Online Figure V). LVEDP was significantly elevated in Tg-DN-Nox mouse hearts compared to in NTg hearts after I/R (Figure 3D). Although heart rate decreased rapidly in Tg-DN-Nox mice after reperfusion, it recovered within 30 min (Online Figure V). Taken together, these data show that suppression of multiple Nox isoforms with DN-Nox exacerbated I/R injury and suppressed the recovery of LV function after reperfusion.
Since Nox2 and Nox4 are the major NADPH oxidases in the heart, the Tg-DN-Nox data raised the possibility that combined downregulation of Nox2 and Nox4 may exacerbate I/R injury. To address these issues, we generated a double-knockout of Nox2 and Nox4 (DKO) by crossing cNox4 KO mice with systemic Nox2 KO mice. We confirmed that the expression levels of both Nox2 and Nox4 in the LV were decreased in DKO mice compared to in WT mice (Online Figure VIA). There was no compensatory upregulation of Nox1 or Nox3 in DKO mice. The expression level of other sources of ROS, such as nitric oxide synthase (NOS) and xanthine oxidase (XO), and the volume of mitochondria, as determined by the level of mitochondrial complex IV (COX IV), in Tg-DN-Nox and DKO mice were not significantly different from those in WT mice (Online Figure VIA). The expression levels of the antioxidants SOD1, Trx1, and catalase were also not altered in Tg-DN-Nox or DKO mice. Only SOD2, an enzyme that dismutates O2− in mitochondria, was decreased in Tg-DN-Nox and DKO mice compared to in WT mice (Online Figure VIB), possibly due to the decreased production of O2−. Interestingly, the antioxidant capacity was significantly increased in Tg-DN-Nox and DKO mice (Online Figure VIC). In addition, the NADP/NADPH ratio was significantly lower in DKO mouse hearts than in control hearts (Online Figure VID), consistent with the results obtained in Tg-DN-Nox mouse hearts. These data indicate that Tg-DN-Nox and DKO mice exhibit a similar phenotype regarding the redox state in the heart. Echocardiographically determined fractional shortening, LV chamber size and wall thickness in DKO mice were all similar to those in WT and Tg-DN-Nox mice at baseline (Online Figure VII). Importantly, DKO mice exhibited a significantly greater infarct size/AAR (Figure 3E) and a greater increase in cardiomyocyte apoptosis in response to I/R (Figure 3F) than WT mice.
Tg-DN-Nox and DKO mice exhibit marked downregulation of ROS in the heart
To elucidate the mechanism of exacerbation of I/R injury in Tg-DN-Nox and DKO mice, we evaluated ROS in the heart. Myocardial O2− production at baseline, as evaluated with DHE staining, was lower in Tg-DN-Nox and DKO mice than in WT, cNox4 KO, and Nox2 KO mice (Online Figure VIIIA). In addition, O2− production was significantly lower in Tg-DN-Nox and DKO than in WT, cNox4 KO, and Nox2 KO mice after I/R (Figure 4A, B). Consistent with these results, the O2− and H2O2 producing activity of cardiac homogenates after I/R, evaluated with lucigenin chemiluminescence assays and Amplex Red assays, was in the order WT > cNox4 KO, Nox2 KO > Tg-DN-Nox, DKO (Figure 4C, Online Figure VIIIB). cNox4 KO and Tg-DN-Nox4 showed an equivalent decrease in O2−-producing activity in the mitochondrial fraction after I/R (Online Figure VIIIC). These data indicate that DN-Nox inhibits not only Nox4 but also other Nox isoforms.
Figure 4. Noxs are major sources of oxidative stress during I/R.

A and B. O2− production in LV sections was evaluated with DHE staining 24 h after reperfusion. *P<0.05 vs. WT, #P<0.05 vs. cNox4 KO, ##P<0.05 vs. Nox2 KO. C. O2−-producing activity of LV homogenates was evaluated with lucigenin chemiluminescence assays. *P<0.05. D. H2O2 production in LV sections was evaluated with the Amplex Red Assay. *P<0.05. E. MDA+4HAE contents in LV sections were evaluated with the LPO-586 assay. *P<0.05.
We also evaluated H2O2 production and malondialdehyde (MDA) + 4-hydroxyalkenals (4HAE) content in LV blocks from NTg and Tg-DN-Nox at baseline, and 30 min and 24 h after reperfusion using the Amplex Red assay and LPO-586 assay, respectively. H2O2 production was significantly lower in Tg-DN-Nox than in NTg mice at all time points (Figure 4D). MDA+4HAE content was also lower in Tg-DN-Nox than in NTg mice after I/R (Figure 4E). Taken together, these results suggest that both Nox2 and Nox4 contribute to oxidative stress in the heart at baseline and in response to I/R, whereas I/R-induced increases in oxidative stress were markedly suppressed in Tg-DN-Nox and DKO hearts after I/R.
Marked downregulation of oxidative stress increases cardiomyocyte death in response to hypoxia/reoxygenation in vitro
To analyze the role of ROS in mediating the cell survival response to I/R at the cellular level, cultured neonatal rat ventricular myocytes were subjected to hypoxia and reoxygenation. O2− production was evaluated after 12 h of hypoxia followed by 24 h of reoxygenation. Pretreatment with either Ad-sh-Nox4 or Ad-sh-Nox2 decreased O2− production in myocytes (Figure 5A). Pretreatment with Ad-DN-Nox or combined Ad-sh-Nox4 and Ad-sh-Nox2 markedly decreased O2− production in myocytes even further (Figure 5A). Cell survival, as evaluated by Cell Titer-Blue assays, was significantly increased in myocytes pretreated with Ad-sh-Nox4 or Ad-sh-Nox2 compared to in those with control adenovirus (Figure 5B, C). In contrast, pretreatment with Ad-DN-Nox or Ad-sh-Nox4 plus Ad-sh-Nox2 decreased cell survival in myocytes compared to in those with control adenovirus (Figure 5D, E). To further elucidate the mechanism of cell survival, we evaluated MPTP opening, an indicator of mitochondrial membrane potential, with JC1 staining. Although pretreatment with Ad-sh-Nox2 or Ad-sh-Nox4 prevented hypoxia/reoxygenation-induced depolarization of the mitochondrial membrane potential, pretreatment with Ad-DN-Nox or Ad-sh-Nox4 plus Ad-sh-Nox2 failed to prevent it (Online Figure IXA). The mRNA level of PGC-1α and the protein level of TFAM were preserved by downregulation of Nox4 or Nox2. On the other hand, combined downregulation of the Nox isoforms showed further reduction of those levels (Online Figure IXB and C). These results indicate that, whereas mild downregulation of oxidative stress is protective, marked downregulation of oxidative stress increases cardiomyocyte death in response to hypoxia/reoxygenation in vitro through stimulation of MPTP opening and suppresses mitochondrial biogenesis.
Figure 5. Combined downregulation of Nox2 and Nox4 exacerbates cell death caused by hypoxia/reoxygenation.

A. O2−-producing activity of myocytes treated with the indicated adenoviruses was evaluated with lucigenin chemiluminescence assays after 12 h of hypoxia and 24 h of reoxygenation. *P<0.05, N.S. not significant. B–E. Cell survival in myocytes treated with the indicated adenoviruses was evaluated by Cell Titer Blue assay at baseline and 1, 6, and 24 h after reoxygenation. *P<0.05 vs. sh-Scramble or sh-LacZ at same time point.
Normalization of HIF-1α expression rescues the increased I/R injury in Tg-DN-Nox mice
The Langendorff experiments and in vitro experiments indicate that the altered cell metabolism may be responsible for the enhancement of I/R injury in the presence of marked suppression of ROS. HIF-1α plays an important role in regulating cell metabolism during I/R. Thus, to elucidate the role of Noxs in mediating the adaptive response to I/R injury, we investigated the role of Noxs in mediating HIF-1α expression. Time course experiments showed that I/R increases the HIF-1α protein level in NTg hearts 1 h and 6 h after reperfusion (Figure 6A). HIF-1α was downregulated at baseline in Tg-DN-Nox (Figure 6B, C). HIF-1α remained downregulated in Tg-DN-Nox after reperfusion, whereas it was upregulated after I/R in Tg-Nox4 (Figure 6B, C). The expression level of HIF-1α was also significantly decreased in DKO mice heart after I/R, whereas it was preserved in cNox4 KO and Nox2 KO mice hearts (Figure 6D). These results are consistent with the notion that Nox-derived ROS play an important role in inducing HIF-1α expression at baseline and during I/R in the heart.
Figure 6. Normalization of HIF-1α mediates exacerbation of I/R injury in Tg-DN-Nox mice.


A. Expression levels of HIF-1α and tubulin in the ischemic area at the indicated time points were evaluated by immunoblotting. *P<0.05, N.S. not significant. B. Expression levels of HIF-1α and tubulin in the LV in NTg, Tg-Nox4, and Tg-DN-Nox mice with or without I/R were evaluated by immunoblotting. *P<0.05. C. Expression levels of HIF-1α and tubulin in the ischemic area in the LV in NTg and Tg-DN-Nox mice 6 h after reperfusion were evaluated by immunoblotting. *P<0.05. D. Expression levels of HIF-1α and tubulin in the ischemic area in the indicated mouse hearts were evaluated by immunoblotting. N.S. not significant. *P<0.05. E. WT, PHD2+/−, Tg-DN-Nox and Tg-DN-Nox/PHD2+/− mice were subjected to ischemia (30 min) and reperfusion (24 h). Infarct size was evaluated by TTC and Alcian blue staining. *P<0.05, **P<0.01. Bar=1 mm. F. The expression levels of cleaved caspase-3 and tubulin in the indicated mice were evaluated by immunoblotting. *P<0.05. G. Expression levels of HIF-1α, GLUT4, Hexokinase, and PPARα in the LVs were evaluated by immunoblotting. *P<0.05.
To investigate the role of HIF-1α downregulation in mediating the exaggerated myocardial injury in Tg-DN-Nox after I/R, we crossed Tg-DN-Nox with PHD2 knockout mice, in which prolyl hydroxylase 2, an enzyme known to induce hydroxylation and downregulation of HIF-1α, is deleted in a cardiomyocyte-specific manner,13 and generated PHD2 heterozygous knockout (PHD2+/−) mice and Tg-DN-Nox/PHD2+/− mice. We then conducted I/R experiments in WT, PHD2+/−, Tg-DN-Nox, and Tg-DN-Nox/PHD2+/− mice. Whereas PHD2+/− mice did not exhibit a significant difference in I/R injury compared to WT mice, the increase in I/R injury observed in Tg-DN-Nox mice was significantly alleviated in Tg-DN-Nox/PHD2+/− mice (Figure 6E). The expression level of cleaved caspase-3 was also significantly lower in Tg-DN-Nox/PHD2+/− mice than in Tg-DN-Nox mice (Figure 6F). HIF-1α expression was downregulated in Tg-DN-Nox hearts after I/R, but was normalized in Tg-DN-Nox4/PHD2+/− hearts (Figure 6G).
HIF-1α regulates glycolysis, fatty acid oxidation (FAO) and angiogenesis in the heart. Although GLUT4 and hexokinase, key regulators of glycolysis, were downregulated in the Tg-DN-Nox mouse heart, expression of these molecules was normalized in the Tg-DN-Nox/PHD2+/− mouse heart (Figure 6G). On the other hand, peroxisome proliferator-activated receptor-alpha (PPARα), a key regulator of FAO, was upregulated in the Tg-DN-Nox mouse heart, but was also normalized in the Tg-DN-Nox/PHD2+/− mouse heart (Figure 6G). The cardiac ATP content 6 h after reperfusion was preserved after downregulation of either Nox4 or Nox2. On the other hand, combined downregulation of the Nox isoforms induced significant reduction of it (Online Figure X). Importantly, the decrease in the ATP content observed in Tg-DN-Nox mice was significantly reversed in Tg-DN-Nox/PHD2+/− mice. These data indicate that both ROS and HIF-1α are involved in ATP production after I/R. To determine whether angiogenesis is affected in Tg-DN-Nox mice, we evaluated capillary density in the LV in NTg and Tg-DN-Nox mice at baseline. There was no significant difference in capillary density between groups (Online Figure XI). Taken together, these data suggest that normalization of HIF-1α expression rescues the increased I/R injury observed in Tg-DN-Nox mice, and is accompanied by a normalization of genes involved in glycolysis and FAO.
Broad suppression of Nox activity upregulates PPARα and induces TG deposition in the heart
Inadvertent activation of PPARα may be detrimental during I/R,14–16 I/R decreased endogenous PPARα in NTg hearts in a time-dependent manner in vivo (Figure 7A). However, both Tg-DN-Nox and DKO mice exhibited greater PPARα expression following I/R than control and single KO mice (Figure 7B, Online Figure XIIA). On the other hand, PPARα was further downregulated after I/R in Tg-Nox4 mice compared to in NTg (Online Figure XIIB).
Figure 7. Broad suppression of Nox activity upregulates PPARα and induces TG deposition.

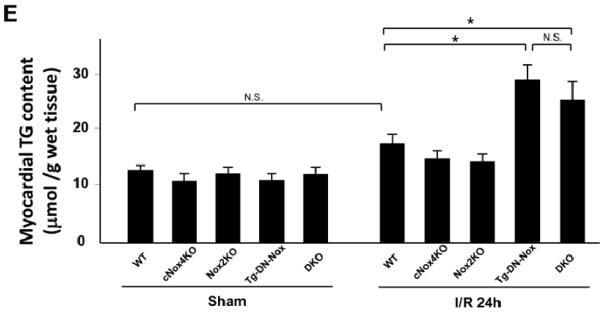
A. Expression levels of PPARα and tubulin in the ischemic area at baseline and after 1, 6 and 24 h of reperfusion were evaluated by immunoblotting. N.S. not significant. *P<0.05. B. Expression levels of PPARα and tubulin in the ischemic area in the indicated mouse hearts were evaluated by immunoblotting. *P<0.05, N.S. not significant. C. Expression levels of PPARα and MCAD in myocytes transduced with the indicated adenoviruses for 48 h were evaluated by immunoblotting. N.S. not significant. *P<0.05. D. PPARα luciferase activity was measured in cardiac myocytes transduced with the indicated adenovirus for 48 h. *P<0.05. E. Myocardial TG content was measured in the indicated mice after either sham or I/R operation. *P<0.05, N.S. not significant.
Knockdown of either Nox4 or Nox2 in myocytes using a short hairpin adenovirus did not change the expression level of either PPARα or medium-chain acyl-CoA dehydrogenase (MCAD), a gene known to be regulated by PPARα, compared to control, whereas overexpression of DN-Nox or combined downregulation of Nox2 and Nox4 upregulated both PPARα and MCAD (Figure 7C). In addition, the activity of a PPARα- luciferase reporter gene was increased in myocytes transduced with Ad-DN-Nox or both Ad-sh-Nox4 and Ad-sh-Nox2 (Figure 7D).
Although the expression of PPARα was downregulated in response to hypoxia/reoxygenation in control myocytes, it was not downregulated after hypoxia/reoxygenation in myocytes treated with Ad-DN-Nox or Ad-sh-Nox4 plus Ad-sh-Nox2 (Online Figure XIIC), suggesting that suppression of both Nox2 and Nox4 prevents hypoxia/reoxygenation-induced downregulation of PPARα in a cell autonomous manner. Tg-DN-Nox and DKO mice showed equivalent decreases in HIF-1α and increases in PPARα and MCAD protein expression (Online Figure XIII).
The circulating level of triglyceride (TG) increases during I/R. In order to test whether upregulation of PPARα during I/R leads to increased accumulation of TG in the myocardium, we measured the TG content in the heart. Cardiac TG content was increased in Tg-DN-Nox and DKO mice compared to in WT, cNox4, and Nox2 KO mice after I/R (Figure 7E). These results suggest that inadvertent upregulation of PPARα caused by combined suppression/downregulation of Noxs induces lipotoxicity in response to I/R.
Downregulation of PPARα attenuates the increased I/R injury in Tg-DN-Nox mice
To investigate the role of PPARα in mediating the enhancement of I/R injury observed in Tg-DN-Nox mouse hearts, we crossed Tg-DN-Nox with PPARα knockout mice and generated Tg-DN-Nox/PPARα−/− mice. We then conducted I/R experiments in WT, PPARα−/−, Tg-DN-Nox, and Tg-DN-Nox/PPARα−/− mice. Protein levels of PPARα and MCAD were upregulated in the Tg-DN-Nox mouse heart after I/R, but the upregulation was abolished in the Tg-DN-Nox/PPARα−/− mouse heart (Figure 8A). Consistent with the protein levels, the mRNA levels of target genes of PPARα, such as PDK4, AcoxI, CD36, CPT1, and MCAD, were upregulated in Tg-DN-Nox but were normalized in Tg-DN-Nox/PPARα−/− mice after I/R (Online Figure XIVA). Interestingly, PPARα−/− mice showed an increase in the LV HIF-1α protein level compared to WT mice (Online Figure XIVB). Thus, HIF-1α and PPARα negatively regulate one another.
Figure 8. Downregulation of PPARα attenuates the increased I/R injury in Tg-DN-Nox mice.

A. Expression levels of PPARα, MCAD and tubulin in the LVs of the indicated groups were evaluated by immunoblotting. *P<0.05. B. WT, PPARα−/−, Tg-DN-Nox and Tg-DN-Nox/PPARα−/− mice were subjected to ischemia (30 min) and reperfusion (24 h). Infarct size was evaluated by TTC and Alcian blue staining. *P<0.05, **P<0.01. Bar=1 mm. C. Intramyocardial TG deposition was evaluated by oil red O staining. Bar=10 μm D. Myocardial TG content was measured in the indicated mice. *P<0.05. E. Expression levels of cleaved caspase-3 and tubulin in the indicated mice were evaluated by immunoblotting. *P<0.05. F. A schematic representation of the hypothesis.
Whereas PPARα−/− mice did not exhibit a significant difference in the extent of I/R injury compared to WT mice, Tg-DN-Nox/PPARα−/− mice exhibited an attenuation in the level of I/R injury compared to Tg-DN-Nox mice (Figure 8B). Myocardial TG content was significantly increased in Tg-DN-Nox mice after I/R, but this increase was attenuated in Tg-DN-Nox/PPARα−/− mice (Figure 8C, D). We also evaluated ATP content during reperfusion in each group. Although ATP content in the ischemic area was similar in the four groups 24 h after reperfusion, Tg-DN-Nox exhibited a decrease in ATP content 6 h after reperfusion that was not apparent in Tg-DN-Nox/PPARα−/− mice (Online Figure XVA). Furthermore, the expression level of C/EBP homologous protein (CHOP), an ER stress marker, was increased in Tg-DN-Nox mice, an effect that was attenuated in Tg-DN-Nox/PPARα−/− mice (Online Figure XVB). The expression level of cleaved caspase-3 in Tg-DN-Nox/PPARα−/− mice was also significantly decreased compared to in Tg-DN-Nox mice (Figure 8E). Taken together, these results show that upregulation of PPARα plays an important role in mediating the increased I/R injury observed in Tg-DN-Nox mice.
DISCUSSION
Our results suggest that both Nox2 and Nox4 are major sources of ROS production in the heart in response to I/R and that oxidative stress induced by Nox2 and Nox4 plays an important role in mediating myocardial I/R injury. However, low levels of oxidative stress produced by either endogenous Nox2 or Nox4 mediate essential adaptive mechanisms for myocardial survival against I/R through activation of HIF-1α and consequent suppression of the PPARα pathway.
Although O2− is rapidly dismutated to H2O2, which is diffusible and membrane permeable, since Nox2 and Nox4 are localized in distinct subcellular spaces,2 they may have distinct local targets promoting I/R injury. However, thus far, we have found that both Nox4 and Nox2 affect common cellular responses, namely, mitochondrial dysfunction and ER stress. Thus, unique targets of Nox2 and Nox4 in the context of I/R injury remain to be identified. Interestingly, downregulation of a single Nox is sufficient to achieve a more than 50% reduction of ROS. Furthermore, upregulation of Nox4 alone failed to enhance I/R injury in Tg-Nox4 mice compared to in control mice. Thus, ROS produced by Nox2 and Nox4 in distinct subcellular localizations may cooperatively enhance further ROS production by both isoforms, which in turn promotes myocardial injury. In addition, although the reduction in superoxide production at the level of 25- 50% is beneficial to I/R injury, more than 75% is detrimental in WT. These data indicate a possibility that there is a threshold effect for ROS in terms of I/R injury.
Perhaps one of the most surprising findings in this work is the fact that a significant enhancement of infarct size/AAR after I/R occurs when both Nox2 and Nox4 are downregulated or inhibited. Importantly, ROS production in Tg-DN-Nox and DKO mice was markedly downregulated in the ischemic area, to an extent significantly greater than in either Nox2 KO or cNox4 KO mice. This indicates that a certain level of ROS is important for cell survival in response to I/R. Purposeful production of ROS in mitochondria could mediate upregulation of cell-protective mechanisms, such as preconditioning effects.17 ROS generated by Nox may act as a second messenger to alert cells and activate adaptive mechanisms.18 The protective mechanism activated by the low level of ROS appears quite powerful, because, in the absence of such a mechanism, the myocardial injury caused by I/R is even greater than usual, despite the fact that the cell death mechanism activated by excessive ROS is no longer active under these conditions (Fig. 8F).
Previous reports have suggested that Nox isoforms play some beneficial roles in the heart. For example, upregulation of angiogenic growth factors in response to I/R is inhibited in p47phox−/− mouse hearts.8 Stretch-induced production of ROS (X-ROS) from Nox2 increases Ca2+-sensitivity by stimulating ryanodine receptors in healthy cardiomyocytes.19 Stimulation of angiogenesis in pressure overload heart is attenuated in systemic Nox4 −/−mice, which was accompanied by downregulation of HIF-1α.20 It should be noted, however, that c-Nox4 −/− mice are protected from pressure overload5. Thus, the protective action of Nox4 may originate from non-myocytes during pressure overload. The molecular mechanism mediating the protective actions of Nox isoforms and how they are regulated by cellular levels of ROS appear stimulus- and cell type-dependent.
As discussed below, since PHD2, one of the critical targets of ROS mediating the hypoxic adaptation during I/R, is localized primarily in cytosol, ROS produced by either Nox2 or Nox4 may be sufficient to inactivate this enzyme, whereas the absence of both Noxs appears to prevent this inactivation, an essential step for stabilizing HIF-1α. Consistent with the in vivo data, whereas cell death in cultured cardiomyocytes was attenuated by single downregulation of Nox2 or Nox4 after hypoxia/reoxygenation, it was exacerbated by expression of DN-Nox or combined downregulation of Nox4 and Nox2. Thus, ROS produced by either Nox2 or Nox4 act as a second messenger to promote cell survival under stress in a cell-autonomous fashion.
HIF-1α is activated by low oxygen conditions during ischemia or by increased oxidative stress during I/R, and activation of HIF-1α reduces reperfusion injury in the heart.21, 22 Although HIF-1α protects the heart through multiple mechanisms, one important mechanism particularly relevant for protecting the heart against acute I/R may be its effect upon cardiac metabolism.10 Activation of endogenous HIF-1α stimulates the glycolytic pathway 23 and inhibits the TCA cycle 9 and fatty acid oxidation (FAO),24, 25 thereby preserving ATP while reducing O2 consumption during I/R.26 The preservation of ATP production induced by stimulation of the glycolytic mechanism is protective against ischemia and facilitates restoration of the high energy phosphate content after reperfusion.27 Although it has been speculated that Nox may be involved in the stabilization of HIF-1α,28–31 a definitive link between Nox and HIF-1α during I/R has not yet been established in vivo. We show here that combined inhibition/downregulation of Nox2 and Nox4 decreases HIF-1α at baseline and in response to I/R in the heart, suggesting that endogenous Noxs play an important role in HIF-1α regulation in response to I/R.
Hydroxylation of prolyl residues in HIF-1α promotes proteolytic degradation of HIF-1α by the ubiquitin-proteasome pathway. ROS increase the stability and transcription of HIF-1α through suppression of prolyl hydroxylase (PHD).11, 12 In response to ROS, PHD is inactivated and proline hydroxylation of HIF-1α is reduced, which in turn reduces ubiquitin-proteasomal degradation of HIF-1α. PHD1 knockout mice exhibit attenuation of myocardial I/R injury.32 However PHD2 was found to be the most active isoform in a number of cell lines. PHD2 has a preference for HIF-1α, whereas PHD1 and PHD3 primarily hydroxylate HIF-2α.33 Here, downregulation of PHD2 prevented the increase in myocardial infarct size after I/R in Tg-DN-Nox mice, suggesting that Noxs play an important role in regulating the activation of HIF-1α through inactivation of PHD2 during I/R.
Although overexpression of Nox4 was sufficient to enhance I/R-induced upregulation of HIF-1α, it did not significantly reduce the infarct size/AAR. Excessive production of ROS by overexpressed Nox4 may counteract the beneficial effect of HIF-1α. Alternatively, the beneficial effect of HIF-1α on I/R injury may have already been saturated at the level of HIF-1α upregulation seen in NTg mice, which would be consistent with the fact that downregulation of PHD2 alone did not reduce the infarct size, despite upregulation of HIF-1α.
HIF-1α not only upregulates proteins involved in glycolysis but also downregulates those involved in FAO, including PPARα.25 Upregulation of HIF-1α, such as by I/R and downregulation of PHD2, rapidly decreased the expression of PPARα, whereas downregulation of HIF-1α increased the PPARα level after hypoxia/reoxygenation, suggesting that HIF-1α negatively regulates PPARα in cardiomyocytes. Consistent with its effect upon HIF-1α, combined inhibition/downregulation of Nox4 and Nox2 significantly upregulated PPARα in the heart at baseline and in response to I/R. Although a previous report suggested that ROS negatively regulate PPARα expression in response to repetitive ischemia,34 underlying mechanisms remain to be elucidated. Our results clearly show that endogenous Noxs negatively regulate PPARα expression through HIF-1α in the heart.
The effect of PPARα activation upon I/R injury is controversial.14–16, 35 In our study, an increase in PPARα expression was correlated with the extent of TG accumulation in the heart after reperfusion in Tg-DN-Nox and DKO mice. Downregulation of PPARα in this condition induced a significant attenuation of infarct size, apoptosis and deposition of TG, indicating the causative involvement of endogenous PPARα in mediating the detrimental phenotype caused by suppression of both Nox2 and Nox4.
The upregulation of PPARα observed in Tg-DN-Nox mice was accompanied by upregulation of PPARα target genes, including CD36. Since myocardial ischemia rapidly increases the plasma free fatty acid (FFA) concentration,36 these data suggest that upregulation of PPARα caused by the lack of Nox-derived ROS could stimulate FFA uptake, which in turn induces TG deposition and consequent lipotoxicity. Stimulation of FAO may also delay the recovery of intracellular pH during reperfusion of ischemic hearts due to the increased production of H+ by glycolysis uncoupled from glucose oxidation.37 Inhibition of Noxs affected HIF-1α/PPARα signaling even at baseline but its effect appears to manifest only in post- I/R hearts, suggesting that the events associated with I/R, such as increases in FFA concentration and acidosis, contribute to the phenotype in Tg-DN-Nox4 and DKO mice.
Interestingly, downregulation of PPARα increased the expression of HIF-1α in the heart, suggesting that PPARα and HIF-1α negatively regulate one another’s functions in the heart. Thus, the salutary effect seen in Tg-DN-Nox/PPARα−/− mice might be due in part to an increase in HIF-1α and activation of glycolysis. Inhibition of glycolysis delays recovery of ATP during I/R in the Langendorff heart model.27 In fact, Tg-DN-Nox/PPARα−/− mice showed an improved recovery in ATP content 6 h after reperfusion compared to Tg-DN-Nox mice, suggesting that endogenous Noxs play an important role in mediating the suppression of PPARα and downstream metabolic effects that are essential for myocyte survival during I/R.
There is an issue to be acknowledged as limitations of this study. In terms of the pathophysiological equivalence of DN and KO Nox4 mice, cross-breeding of Nox4flox/flox-αMHC-Cre-Nox2−/−-PHD2flox/flox (or PPARα −/−) mice was technically challenging and we could not generate them. However, additional lines of evidence that DN-Nox and combined downregulation of Nox2/Nox4 exhibit identical effects upon cardiomyocytes (Online Fig. IXC, X, XI and XIV).
In conclusion, both Nox2 and Nox4 play an essential role in mediating oxidative stress and myocardial injury after I/R. Importantly, however, low levels of ROS produced by either Nox2 or Nox4 are required to activate essential adaptive mechanisms to protect the heart against I/R, including preventing inactivation of HIF-1α and inhibition of PPARα. Unfortunately, these protective mechanisms are easily overcome by cell-death promoting mechanisms activated by the excessive ROS produced by coordinated activation of Nox2 and Nox4 during I/R (Online Figure XVII). Our study suggests that although either Nox2 or Nox4 can be targeted for the treatment of I/R, any such intervention should proceed with extreme caution so as not to impair the physiological function of the Noxs.
Supplementary Material
Novelty and Significance.
What Is Known?
Oxidative stress plays an important role in mediating ischemia/reperfusion (I/R) injury in the heart.
NADPH oxidases produce O2- and H2O2, and it contribute to oxidative stress during I/R in the heart.
Myocardial I/R injury is suppressed in mice with genetic deletion of p47phox, a cytosolic factor positively regulating Nox2.
Hypoxia-inducible factors (HIFs) play a protective role in I/R injury through regulation of the cardiac metabolism.
What New Information Does This Article Contribute?
Both Nox2 and Nox4 contribute to increases in oxidative stress during I/R injury in the heart.
Isoform-specific genetic deletion of either Nox2 or Nox4 significantly attenuates I/R injury in the heart.
Combined deletion of Nox2 and Nox4 or overexpression of dominant negative Nox, which suppresses multiple isoforms of Nox, markedly reduces oxidative stress during I/R, but exacerbates I/R injury.
Physiological levels of reactive oxygen species (ROS) produced by either Nox2 or Nox4 play an essential role in activating HIF-1α, which in turn plays an essential role in mediating protection against I/R in part through downregulation of PPARα.
Oxidative stress plays an important role in mediating myocardial injury during I/R. The molecular source of ROS during I/R and their role in mediating myocardial injury remain unclear. Using genetically-altered mouse models, we found that both Nox2 and Nox4, major isoforms of the NADPH oxidase in the heart, contribute to oxidative stress during I/R and suppression of either of them significantly reduces I/R injury by inhibiting excessive production of ROS. However, combined downregulation of both Nox2 and Nox4 paradoxically exacerbated I/R injury, althought oxidative stress by I/R was markedly suppressed. These findings suggest that during I/R, low levels of ROS production from either Nox2 or Nox4 are required for activation of adaptive mechanisms essential for cardioprotection, which is mediated by the activation of HIF-1α and downregulation of PPARα. Thus ROS production by Nox appears to have dichotomous functions during I/R: excessive production of ROS by Nox2 and Nox4 is detrimental whereas minimum production of ROS by either Nox2 or Nox4 is essential for adaptation. Hence, broad Nox suppression during I/R could be detrimental for the heart.
Acknowledgments
We thank Daniela Zablocki and Christopher D. Brady for critical reading of the manuscript and Yasuhiro Maejima for technical suggestions.
SOURCES OF FUNDING
Dr. Matsushima is the recipient of an AHA FDA Postdoctoral Fellowship (12POST9460007). This work was supported in part by U.S. Public Health Service Grants HL110349 (RT), HL59139, HL67724, HL69020, HL91469, HL102738, and AG27211 (JS), and by the Fondation Leducq Transatlantic Networks of Excellence (JS).
Nonstandard Abbreviations
- AAR
area at risk
- Ad
adenovirus
- DN
dominant-negative
- HIF
hypoxia-inducible factor
- LV
left ventricle (or left ventricular)
- Nox
NADPH oxidase
- NTg
non-transgenic
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- sh-RNA
short hairpin RNA
- TTC
triphenyltetrazolium chloride
- Tg
transgenic
Footnotes
DISCLOSURES
None.
References
- 1.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese zucker rat heart. Diabetes. 2002;51:2587–2595. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 2.Maejima Y, Kuroda J, Matsushima S, Ago T, Sadoshima J. Regulation of myocardial growth and death by nadph oxidase. J Mol Cell Cardiol. 2011;50:408–416. doi: 10.1016/j.yjmcc.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC, Marber M, Monaghan MJ, Shah AM. Involvement of nox2 nadph oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension. 2008;51:319–325. doi: 10.1161/HYPERTENSIONAHA.107.101980. [DOI] [PubMed] [Google Scholar]
- 5.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. Nadph oxidase 4 (nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmeyer MR, Jones SP, Ross CR, Sharp B, Grisham MB, Laroux FS, Stalker TJ, Scalia R, Lefer DJ. Myocardial ischemia/reperfusion injury in nadph oxidase-deficient mice. Circ Res. 2000;87:812–817. doi: 10.1161/01.res.87.9.812. [DOI] [PubMed] [Google Scholar]
- 7.Bell RM, Cave AC, Johar S, Hearse DJ, Shah AM, Shattock MJ. Pivotal role of nox-2-containing nadph oxidase in early ischemic preconditioning. FASEB J. 2005;19:2037–2039. doi: 10.1096/fj.04-2774fje. [DOI] [PubMed] [Google Scholar]
- 8.Chen JX, Zeng H, Tuo QH, Yu H, Meyrick B, Aschner JL. Nadph oxidase modulates myocardial akt, erk1/2 activation, and angiogenesis after hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2007;292:H1664–1674. doi: 10.1152/ajpheart.01138.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Shohet RV, Garcia JA. Keeping the engine primed: Hif factors as key regulators of cardiac metabolism and angiogenesis during ischemia. J Mol Med (Berl) 2007;85:1309–1315. doi: 10.1007/s00109-007-0279-x. [DOI] [PubMed] [Google Scholar]
- 11.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ros but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex iii is required for hypoxia-induced ros production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambandam N, Morabito D, Wagg C, Finck BN, Kelly DP, Lopaschuk GD. Chronic activation of pparalpha is detrimental to cardiac recovery after ischemia. Am J Physiol Heart Circ Physiol. 2006;290:H87–95. doi: 10.1152/ajpheart.00285.2005. [DOI] [PubMed] [Google Scholar]
- 15.Panagia M, Gibbons GF, Radda GK, Clarke K. Ppar-alpha activation required for decreased glucose uptake and increased susceptibility to injury during ischemia. Am J Physiol Heart Circ Physiol. 2005;288:H2677–2683. doi: 10.1152/ajpheart.00200.2004. [DOI] [PubMed] [Google Scholar]
- 16.Hafstad AD, Khalid AM, Hagve M, Lund T, Larsen TS, Severson DL, Clarke K, Berge RK, Aasum E. Cardiac peroxisome proliferator-activated receptor-alpha activation causes increased fatty acid oxidation, reducing efficiency and post-ischaemic functional loss. Cardiovasc Res. 2009;83:519–526. doi: 10.1093/cvr/cvp132. [DOI] [PubMed] [Google Scholar]
- 17.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 18.Acker H. The oxygen sensing signal cascade under the influence of reactive oxygen species. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 2005;360:2201–2210. doi: 10.1098/rstb.2005.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prosser BL, Ward CW, Lederer WJ. X-ros signaling: Rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 20.Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. Nadph oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Natarajan R, Salloum FN, Fisher BJ, Kukreja RC, Fowler AA., 3rd Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ Res. 2006;98:133–140. doi: 10.1161/01.RES.0000197816.63513.27. [DOI] [PubMed] [Google Scholar]
- 22.Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, Thistlethwaite PA. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46:2116–2124. doi: 10.1016/j.jacc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 23.Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K, Johnson RS. Transcription factor hif-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. 2001;21:3436–3444. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belanger AJ, Luo Z, Vincent KA, Akita GY, Cheng SH, Gregory RJ, Jiang C. Hypoxia-inducible factor 1 mediates hypoxia-induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator-activated receptor alpha/retinoid x receptor. Biochem Biophys Res Commun. 2007;364:567–572. doi: 10.1016/j.bbrc.2007.10.062. [DOI] [PubMed] [Google Scholar]
- 25.Narravula S, Colgan SP. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. 2001;166:7543–7548. doi: 10.4049/jimmunol.166.12.7543. [DOI] [PubMed] [Google Scholar]
- 26.Simon MC. Coming up for air: Hif-1 and mitochondrial oxygen consumption. Cell Metab. 2006;3:150–151. doi: 10.1016/j.cmet.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Tian R, Abel ED. Responses of glut4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation. 2001;103:2961–2966. doi: 10.1161/01.cir.103.24.2961. [DOI] [PubMed] [Google Scholar]
- 28.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. Jund reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 29.Block K, Gorin Y, Hoover P, Williams P, Chelmicki T, Clark RA, Yoneda T, Abboud HE. Nad(p)h oxidases regulate hif-2alpha protein expression. J Biol Chem. 2007;282:8019–8026. doi: 10.1074/jbc.M611569200. [DOI] [PubMed] [Google Scholar]
- 30.Goyal P, Weissmann N, Grimminger F, Hegel C, Bader L, Rose F, Fink L, Ghofrani HA, Schermuly RT, Schmidt HH, Seeger W, Hanze J. Upregulation of nad(p)h oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free radical biology & medicine. 2004;36:1279–1288. doi: 10.1016/j.freeradbiomed.2004.02.071. [DOI] [PubMed] [Google Scholar]
- 31.Maranchie JK, Zhan Y. Nox4 is critical for hypoxia-inducible factor 2-alpha transcriptional activity in von hippel-lindau-deficient renal cell carcinoma. Cancer research. 2005;65:9190–9193. doi: 10.1158/0008-5472.CAN-05-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adluri RS, Thirunavukkarasu M, Dunna NR, Zhan L, Oriowo B, Takeda K, Sanchez JA, Otani H, Maulik G, Fong GH, Maulik N. Disruption of hypoxia-inducible transcription factor-prolyl hydroxylase domain-1 (phd-1−/−) attenuates ex vivo myocardial ischemia/reperfusion injury through hypoxia-inducible factor-1alpha transcription factor and its target genes in mice. Antioxid Redox Signal. 2011;15:1789–1797. doi: 10.1089/ars.2010.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuckerman JR, Zhao Y, Hewitson KS, Tian YM, Pugh CW, Ratcliffe PJ, Mole DR. Determination and comparison of specific activity of the hif-prolyl hydroxylases. FEBS Lett. 2004;576:145–150. doi: 10.1016/j.febslet.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Dewald O, Sharma S, Adrogue J, Salazar R, Duerr GD, Crapo JD, Entman ML, Taegtmeyer H. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation. 2005;112:407–415. doi: 10.1161/CIRCULATIONAHA.105.536318. [DOI] [PubMed] [Google Scholar]
- 35.Yue TL, Bao W, Jucker BM, Gu JL, Romanic AM, Brown PJ, Cui J, Thudium DT, Boyce R, Burns-Kurtis CL, Mirabile RC, Aravindhan K, Ohlstein EH. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation. 2003;108:2393–2399. doi: 10.1161/01.CIR.0000093187.42015.6C. [DOI] [PubMed] [Google Scholar]
- 36.Lopaschuk GD, Collins-Nakai R, Olley PM, Montague TJ, McNeil G, Gayle M, Penkoske P, Finegan BA. Plasma fatty acid levels in infants and adults after myocardial ischemia. American heart journal. 1994;128:61–67. doi: 10.1016/0002-8703(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q, Docherty JC, Rendell JC, Clanachan AS, Lopaschuk GD. High levels of fatty acids delay the recovery of intracellular ph and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol. 2002;39:718–725. doi: 10.1016/s0735-1097(01)01803-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.