

Abstract
Analogs of S-adenosyl-L-methionine (SAM) are increasingly applied to the methyltransferase (MT) catalysed modification of biomolecules including proteins, nucleic acids, and small molecules. However, SAM and analogs suffer from an inherent instability, and their chemical synthesis is challenged by low yields and difficulties in stereoisomer isolation and inhibition. Here we report the chemoenzymatic synthesis of a series of SAM analogs using wild-type (wt) and point mutants of two recently identified halogenases, SalL and FDAS. Molecular modelling studies are used to guide the rational design of mutants, and the enzymatic conversion of L-Met and other analogs into SAM analogs is demonstrated. We also apply this in situ enzymatic synthesis to the modification of a small peptide substrate by protein arginine methyltransferase 1 (PRMT1). This technique offers an attractive alternative to chemical synthesis and can be applied in situ to overcome stability and activity issues.
S-adenosyl-L-methionine (SAM, 1a) is the most common enzyme substrate in nature after ATP.1 It serves as a substrate for methylation of proteins, nucleic acids, and small molecules, but is also involved in polyamine biosynthesis and radical reactions.2 There has been considerable interest in using analogs of SAM as selective inhibitors, for exploring methyltransferases (MTases), and for profiling MTase substrates.3 The latter approach is highly interesting as it is speculated that many substrates are still to be identified.2b, 4
Two types of enzymatically active SAM analogs have been reported.5 Aziridinium-based SAM analogs have been employed with DNA and protein MTases resulting in bi-substrate products in which the SAM analog is covalently linked to the MTase substrate.3b,3c,6 However, when using protein arginine MTase 1 (PRMT1) the bi-substrate product has been observed to cause product inhibition indicating that the scope of these analogs is limited.3c Recently, SAM analogs have been applied where the methyl group in L-Met of SAM is exchanged with a new reactive functional group (e.g. terminal alkynyl, keto, or amino group). Similar to the methyl group in SAM, the new chemical moiety is transferred to MTase substrates by the respective MTases. This method offers a compelling strategy, as it allows for subsequent selective bioorthogonal conjugation of the MTase product with functional tags such as fluorescent or affinity probes.3a, 3d–i
Currently, SAM analogs are synthesized chemically from S-adenosyl-L-homocysteine (SAH, 2) with low yields and in approximately a 1:1 ratio of the sulfonium ion diastereomers. Usually only the (S,S) diastereomer is biologically active, whereas the (R,S) diastereomer has been demonstrated to inhibit MTases.7 The impractical separation of these diastereomers typically leads to application of diastereomeric mixtures for the MT transformations.3a, 3f–3i Additionally, caution must be applied when storing and handling SAM and SAM analogs, as these molecules are inherently unstable, particularly at neutral and alkaline pH.8 An in situ enzymatic synthesis of SAM analogs would therefore be of high value if diastereoselective and able to overcome instability problems by producing SAM analogs.
We recently demonstrated the enzymatic synthesis of SAM and the direct coupling to the enzymatic methylation of the antibiotic teicoplanin by MtfA MTase and of DNA by HhaI MTase.9 The applied SAM producing enzyme was a recently discovered chlorinase SalL from Salinispora tropica10 and this enzyme is homologous to a fluorinase FDAS from Streptomyces cattleya.11 In vivo these halogenases catalyse the breakdown of SAM by a nucleophilic attack of chloride or fluoride at C5′ of the ribosyl moiety of SAM producing L-Met (3a) and 5′-chloro or fluoro-5′-deoxyadenosine (ClDA or FDA, 4, X = Cl or F) (Figure 1). FDAS can also utilize chloride as a nucleophile, and for both enzymes it has been shown that in vitro equilibrium favors the synthesis of SAM from ClDA and L-Met.10, 12 Herein, we report the successful use of wt SalL, designed SalL point mutants, and FDAS for the enzymatic diastereoselective synthesis of SAM analogs from ClDA and readily prepared L-Met analogs (3b–j) (Figure 1).
Fig. 1.

The enzymatic synthesis of SAM analogs and its coupling to the modification of MTase substrates
Inspecting molecular interaction field calculations of the SalL (PDB: 2Q6I) and FDAS (PDB: 1RQR) crystal structures containing L-Met,10,11 two water molecules were found tightly bound in the active site of the enzymes where the L-Met analogs are assumed to propagate (Figure 2 and supporting information). This suggests that destabilization of these water molecules might facilitate the accommodation of larger substituents. When analysing refined structures of SalL and FDAS, the following amino acids were found to be involved in an extensive hydrogen bonding network with the two water molecules in SalL (S) and FDAS (F): T128S (T155F), W190S (W217F), Y239S (Y266F), backbone amide of S242S (S269F), and the carboxylate group of L-Met (Figure 2). A series of point mutants were generated for this T, W and Y in both SalL and FDAS with the aim of accommodating larger substituents through introduction of smaller amino acids and destabilizing the hydrogen bonding network (Figure 2).
Fig. 2.
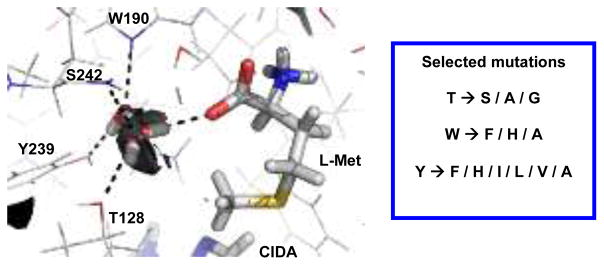
The active site of SalL overlaid with the molecular interaction field of water at −10.0 kcal/mol (black surfaces) and listing of the series of mutations for T128S (T155F), W190S (W217F) and Y239S (Y266F).
For FDAS, all mutants expressed in sufficient yield whereas for SalL, only 6 out of the 12 mutants expressed properly (T128A and Y239H-A) (Supporting Information). The activities of these expressed mutants, as well as wt enzymes, were analyzed for activity with L-Met (3a) and the series of synthetic L-Met analogs (3b–j) of varying size and polarity, as illustrated in Figure 1 (see tables with activity data in Supporting information).
Activities were estimated by HPLC and reported in terms of initial velocities from the ratio of the product area against the total area of product and unreacted substrate (ClDA) relative to the applied enzyme concentrations. Identical concentrations (15 mM) of L-Met and analogs were used except for benzyl and phenethyl analogs (1.5 mM) due to low solubility. Negative control reactions without enzymes were performed in each series to verify enzyme activity. Consistent with earlier studies, the activity of L-Met was approximately 500-fold higher for SalL compared to FDAS.11, 12d In general, activity with L-Met was lower for the mutants compared to wt enzymes, except for Y266FF and T155SF, that displayed a slightly increased activity compared to wt FDAS.
For wt SalL, activity was detected for all L-Met analogs except propargyl (3f), phenethyl (3h) and polar analogs (3i,j) (Figure 3 and Supporting Information). The formation of ethyl-, propyl, butyl-, allyl-, and benzyl-SAM was confirmed by LC MS (Figure 3 and Supporting Information). In general, activity decreased with increasing size of the L-Met analog. The rate of formation of ethyl-, propyl, butyl-, and allyl-SAM compared to SAM, were reduced 10-, 5000-, 15000- and 5000-fold, respectively.
Fig. 3.

A) Enzyme activity of wt SalL and the W190 mutant series of SalL using L-Met analogs. B) HPLC chromatograms illustrating the formation of butyl-SAM using wt and W190A SalL. The synthesis of butyl-SAM was verified by LC MS. Mmono = monoisotopic mass. *1.5 mM S-benzyl-L-Hcy. ∇These peaks are impurities from the enzyme solutions.
For some mutants the decrease in activity relative to L-Met was much less pronounced (Figure 3 and Supporting Information). In particular, the Trp mutant series showed good activity. For W190FS activity to L-Met analogs decreased in a similar manner as observed for wt SalL. For W190HS activity was observed using the propyl and butyl analogs of L-Met, but no activity was detected using either L-Met or the ethyl analog suggesting increased activity towards the larger L-Met analogs. The W190AS mutant accepted the same set of L-Met analogs as wt SalL, but the activities were not reduced as drastically as seen for SalL and the other mutants. Here, the activity of ethyl-, propyl, butyl-, and allyl-SAM formation compared to SAM formation was reduced 20-, 30-, 30-, and 60-fold, respectively. These results indicate that the activity of the ethyl analog of L-Met is reduced with similar magnitude compared to L-Met for both wt and W190A SalL, whereas the reduction in activity when using the larger substrates was 150–500-fold higher for wt SalL. W190AS catalyzed the formation of butyl-SAM with a three-fold higher velocity than when using wt SalL (0.934 ± 0.010 nM/min compared to 0.318 ± 0.006 nM/min). Propyl-SAM was produced with a similar velocity as with wt SalL, whereas the formation of SAM was reduced 160-fold compared to wt SalL. Unlike wt SalL, W190AS showed no reduction in activity when exchanging the propyl analog of L-Met with the butyl analog. Only wt and W190A SalL were tested for activity towards the benzyl and phenethyl analogs of L-Met, and similar rates for the two enzymes were seen for the benzyl analog. This finding is intriguing with respect to further development of enzyme variants in order to discover even broader substrate specificities. From the Tyr mutant series, only Y239FS showed sufficient expression yield to study activity. This mutant showed a similar reduction in activity to wt SalL when increasing the size of the L-Met analogs. Finally, the Thr mutant series showed activity only for L-Met and the ethyl analog of L-Met. Unfortunately, the propargyl analog of L-Met was observed to be unstable, but also propargyl-SAM has been reported to highly unstable (see Supporting Information).3e,3f,3h
These data partially confirm our in silico structural analysis of SalL and FDAS, as we succeeded in modulating the active site to accept some L-Met analogs. Mutants designed to reduce the occupancy of water (W190FS and Y239FS) led to reduced activity of all analogs, presumably due to impaired interaction with the carboxylate group of L-Met and analogs. But for W190AS, this loss was compensated by an increased size of the pocket, leading to activities comparable to wt for propyl, allyl, and benzyl L-Met analogs; and for the butyl analog a significant increase in activity was observed
Wt and mutant FDAS generally displayed much lower activities compared to SalL (Figure 4 and Supporting Information). However, two mutants (T155SF and Y266FF) showed a small but significant increase in the activity towards both L-Met and the ethyl analog of L-Met – ranging from an L-Met activity of 10.6 ± 1.5 nM/min for wt FDAS to 18.5 ± 0.1 and 15.6 ± 0.5 nM/min for the T155SF and Y266FF mutants, respectively. Activities of L-Met and the ethyl analog of L-Met were observed for all FDAS variants and the relative reductions in activity when exchanging L-Met with the ethyl analog were comparable with those found for wt SalL. Furthermore, HPLC analysis indicated formation of trace amounts of propyl-SAM for Y266F, but this could not be confirmed by LC MS because the produced amount was below detection limit.
Fig. 4.

The enzyme activity of wt and mutants of FDAS. Note that one y- axis is for L-Met and one is for the ethyl analog.
To probe the application of these enzymes in an in situ enzymatic synthesis of SAM and analogs coupled to the modification of various substrates by MTs, we applied rat PRMT1 to explore the enzymatic modification of a small peptide sequence, the RGG peptide, that mimics the natural substrate fibrillarin (Figure 5). PRMT1 is the predominant PRMT in mammalian cells, estimated to be responsible for 85% of arginine methylation.13 As a type 1 PRMT, the product of PRMT1 is asymmetrically dimethylated arginine.14 First, we tested the use of the natural substrate SAM as well as the in situ SAM producing coupled assay using L-Met, ClDA, and wt SalL. The peptide was successfully dimethylated at all three arginines under both conditions (Supporting Information). Coupled assays were then performed using the ethyl, propyl, butyl, allyl, and benzyl analogs of L-Met, along with negative controls without rPRMT1.
Fig. 5.

A) A schematic illustration of the coupling of the enzymatic SAM and analog formation and the modification of RGG peptide by rPRMT1. B) LCMS chromatogram of the coupled assay with the allyl analog of L-Met and the spectra of the RGG peptides. L-MetX is an abbreviation for both L-Met and analogs.
The allyl L-Met analog transferred effectively, and both mono- and di-modified peptides were identified by LC MS. While allylation via synthetic analogs has been reported,14 this is the first time di-allylation has been seen using rPRMT1. This finding is likely the result of our technique providing only the biologically active (S,S) diastereomer and that no inhibitory (R,S) diastereomer contaminates the allyl-SAM preparation. Indeed, these findings strongly support the use of this chemoenzymatic method for a stereoselective analog preparation.
Transfer was also observed when using both the ethyl and benzyl analogs of L-Met. It is notable that that the bulky benzyl group is transferred and is probably due to the increased reactivity of this group. For the benzyl analog, MS indicated the formation of only mono-modified peptide, whereas for the ethyl analog both mono-, di- and tri-modified RGG peptides were identified.
However, when employing the larger alkyl-SAM analogs (propyl and butyl), no activity was observed. Given that the rate of propyl- and butyl-SAM formation is approximately 500-fold lower when compared to ethyl-SAM, it remains unclear whether this lack of activity is simply due to reduced specificity or that the activity is abolished.
Conclusions
In conclusion, this study demonstrates the first published method for a chemoenzymatic synthesis of various SAM analogs using both wt and engineered halogenases. This method uses synthetically accessible L-Met analogs as precursors for the diastereoselective formation of SAM analogs. The method is useful for the enzymatic modification by MT’s using in situ generated SAM analogs, hereby overcoming major issues of instability with SAM and related analogs. The utility of in situ coupled assays with other MTs becomes evident when considering the stability of these enzymes and their applicability in a range of conditions. Based on the in silico analysis of SalL and FDAS active sites, it is conceivable that future protein engineering studies can increase activities and further expand substrate specificity for additional SAM analogs. Recently, engineered MAT1 was applied for the production of a SAM analogue in vivo demonstrating the scope of such reactions. However, only one analogue was probed and no enzyme kinetic data was reported.15 Considering the involvement of SAM in metabolic processes and disease states,2d the scope of these applications is highly promising.
Supplementary Material
Acknowledgments
The authors thank Professor Bradley S. Moore and Professor James H. Naismith for the kind donations of the plasmids pAEM7 and pFLA_HT for the expression of SalL and FDAS, respectively. This work was financed by the Drug Research Academy Program and the Novo Nordisk STAR program.
Footnotes
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/b000000x/
Notes and references
- 1.Cantoni GL. Annu Rev Biochem. 1975;44:435–451. doi: 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- 2.a) Wang SC, Frey PA. Trends Biochem Sci. 2007;32:101–110. doi: 10.1016/j.tibs.2007.01.002. [DOI] [PubMed] [Google Scholar]; b) Schubert HL, Blumenthal RM, Cheng X. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pegg AE, Casero RA., Jr Methods Mol Biol. 2011;720:3–35. doi: 10.1007/978-1-61779-034-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Loenen WA. Biochem Soc Trans. 2006;34:330–333. doi: 10.1042/BST20060330. [DOI] [PubMed] [Google Scholar]
- 3.a) Dalhoff C, Lukinavicius G, Klimasauskas S, Weinhold E. Nat Chem Biol. 2006;2:31–32. doi: 10.1038/nchembio754. [DOI] [PubMed] [Google Scholar]; b) Pljevaljcic G, Schmidt F, Scheidig AJ, Lurz R, Weinhold E. Chem bio chem. 2007;8:1516–1519. doi: 10.1002/cbic.200700294. [DOI] [PubMed] [Google Scholar]; c) Osborne T, Roska RL, Rajski SR, Thompson PR. J Am Chem Soc. 2008;130:4574–4575. doi: 10.1021/ja077104v. [DOI] [PubMed] [Google Scholar]; d) Lukinavicius G, Lapiene V, Stasevskij Z, Dalhoff C, Weinhold E, Klimasauskas S. J Am Chem Soc. 2007;129:2758–2759. doi: 10.1021/ja0691876. [DOI] [PubMed] [Google Scholar]; e) Peters W, Willnow S, Duisken M, Kleine H, Macherey T, Duncan KE, Litchfield DW, Luscher B, Weinhold E. Angew Chem Int Ed. 2010;49:5170–5173. doi: 10.1002/anie.201001240. [DOI] [PubMed] [Google Scholar]; f) Islam K, Zheng W, Yu H, Deng H, Luo M. ACS Chem Biol. 2011;6:679–684. doi: 10.1021/cb2000567. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Lee BWK, Sun HG, Zang TZ, Kim BJ, Alfaro JF, Zhou ZS. J Am Chem Soc. 2010;132:3642–3643. doi: 10.1021/ja908995p. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Wang R, Zheng W, Yu H, Deng H, Luo M. J Am Chem Soc. 2011;133:7648–7651. doi: 10.1021/ja2006719. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Binda O, Boyce M, Rush JS, Palaniappan KK, Bertozzi CR, Gozani O. Chem Bio Chem. 2011;12:330–334. doi: 10.1002/cbic.201000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klimasauskas S, Weinhold E. Trends Biotechnol. 2007;25:99–104. doi: 10.1016/j.tibtech.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Luo M. ACS Chem Biol. 2012;7:443–463. doi: 10.1021/cb200519y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Pljevaljcic G, Pignot M, Weinhold E. J Am Chem Soc. 2003;125:3486–3492. doi: 10.1021/ja021106s. [DOI] [PubMed] [Google Scholar]; b) Pljevaljcic G, Schmidt F, Weinhold E. Chem Bio Chem. 2004;5:265–269. doi: 10.1002/cbic.200300739. [DOI] [PubMed] [Google Scholar]
- 7.a) Khani-Oskouee S, Jones JP, Woodard RW. Biochem Biophys Res Commun. 1984;121:181–187. doi: 10.1016/0006-291x(84)90704-6. [DOI] [PubMed] [Google Scholar]; b) Borchardt RT, Wu YS. J Med Chem. 1976;19:1099–1103. doi: 10.1021/jm00231a004. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman JL. Biochemistry. 1986;25:4444–4449. doi: 10.1021/bi00363a041. [DOI] [PubMed] [Google Scholar]
- 9.Lipson JM, Thomsen M, Moore BS, Clausen RP, La Clair JJ, Burkart MD. Chem Bio Chem. doi: 10.1002/cbic.201300221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eustaquio AS, Pojer F, Noel JP, Moore BS. Nat Chem Biol. 2008;4:69–74. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Hagan D, Schaffrath C, Cobb SL, Hamilton JT, Murphy CD. Nature. 2002;416:279. doi: 10.1038/416279a. [DOI] [PubMed] [Google Scholar]
- 12.a) Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]; b) Zhu XF, Robinson DA, McEwan AR, O’Hagan D, Naismith JH. J Am Chem Soc. 2007;129:14597–14604. doi: 10.1021/ja0731569. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Deng H, O’Hagan D. Curr Opin Chem Biol. 2008;12:582–592. doi: 10.1016/j.cbpa.2008.06.036. [DOI] [PubMed] [Google Scholar]; d) Deng H, Cobb SL, McEwan AR, McGlinchey RP, Naismith JH, O’Hagan D, Robinson DA, Spencer JB. Angew Chem Int Ed. 2006;45:759–762. doi: 10.1002/anie.200503582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Gary JD, Lin WJ, Yang MC, Herschman HR, Clarke S. J Biol Chem. 1996;271:12585–12594. doi: 10.1074/jbc.271.21.12585. [DOI] [PubMed] [Google Scholar]; b) Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, Herschman HR. J Biol Chem. 2000;275:7723–7730. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]; c) Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE. Mol Cell Biol. 2000;20:4859–4869. doi: 10.1128/mcb.20.13.4859-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR. J Biol Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Islam K, Liu Y, Zheng W, Tang H, Lailler N, Blum G, Deng H, Luo M. J Am Chem Soc. 2013;135:1048–1056. doi: 10.1021/ja309412s. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cantoni GL. Annu Rev Biochem. 1975;44:435–451. doi: 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.