

Abstract
Evaluation of a series of MetAP inhibitors in an in vitro enzyme activity assay led to the first identification of potent molecules that show significant growth inhibition against Burkholderia pseudomallei. Nitroxoline analogues show excellent inhibition potency in the BpMetAP1 enzyme activity assay with the lowest IC50 of 30 nM and inhibit the growth of B. pseudomallei and B. thailandensis at concentrations ≥31 μM.
Keywords: BpMetAP1, methionine aminopeptidase inhibitor, melioidosis, Burkholderia pseudomallei
Burkholderia pseudomallei is the causative agent of melioidosis, a severe and often fatal infection that manifests as pneumonia or septicemia.1 Melioidosis is endemic to Southeast Asia and Northern Australia, where it is a significant cause of morbidity and mortality and can also be found in other tropical regions.2 The Centers for Disease Control and Prevention consider B. pseudomallei to be a bioterrorism risk and have thus listed the organism as a Tier 1 select agent that could potentially cause a large-scale public health crisis if used in an attack (www.selectagents.gov). Although it is critical to identify antimicrobial agents active against B. pseudomallei, the process of drug discovery has been challenging. The current standard treatment for melioidosis is intensive administration of the third-generation cephalosporin antibiotic ceftazidime followed by a regimen of trimethoprim-sulfamethoxazole or amoxicillin-clavulanic acid for 3–6 months.2 Ceftazidime is highly active in vitro and was shown to be safe for clinical use; however, resistance to this drug correlating with clinical use has been documented.2−4 In addition, B. pseudomallei is intrinsically resistant to several classes of antibiotics, including aminoglycosides and macrolides, due to expression of resistance determinants such as beta-lactamase and multidrug efflux pumps.5 This intrinsic resistance creates an even greater challenge for the scientific community to find alternative treatments that act via novel mechanisms.
Methionine aminopeptidase (MetAP) is a dinuclearmetalloprotease that removes the N-terminal methionine from nascent proteins.6 MetAP is conserved in all life forms from bacteria to humans. Genetic studies have shown that deleting the MetAP-encoding gene in numerous prokaryotes results in either a slow-growth or lethal phenotype.6 These studies reveal the critical role of MetAPs in prokaryote survival and suggest that these enzymes may be promising targets for the discovery of new antibiotics.
Early studies by Lu et al. suggested inhibitors of M. tuberculosis MetAP enzymes (MtMetAP1a and MtMetAP1c) inhibited the growth of mycobacteria in culture.7 Similarly, potent inhibitors of human MetAP2 were shown to exhibit activities against HMVEC proliferation.8 While possessing only limited amino acid sequence similarity to Mt-MetAP1 (37% identity/46% similarity) and human MetAP2 (12% identity/18% similarity), B. pseudomallei MetAP1 contains significant similarity in the active site (Supplemental Figure 1). This observation suggests that inhibitors effective against MetAP1 from both M. tuberculosis and humans might also be effective inhibitors of B. pseudomallei MetAP1.
Several inhibitors of MetAP enzymes have been reported over the past several years. Triazole compounds were reported to be potent MetAP inhibitors, demonstrating activity in many organisms through the interaction of each of the triazole nitrogen atoms in positions 1 and 2 with divalent cobalt ions.9 Furan-type compounds displayed submicromolar inhibitory activity against Escherichia coli methionine aminopeptidase.10 The carboxyl group in these compounds coordinates with the metal ions at the enzyme active site pocket. Derivatives of anthranilic acid sulfonamide were reported to have potent MetAP inhibition activities against human MetAP2.8 Chlorosubstitution at the para-position on the sulfonyl phenyl was observed to enhance inhibition. Nitroxoline, or 5-nitro-8-hydroxyquinoline, exhibited activity against human MetAP2 in vitro in the nanomolar range.11 The phenolic group at the 8-position dissociates to O– at physiological pH allowing the O– and the nitrogen atom at the 1 position to participate in binding to the active site metal ion.12 Here, representative compounds from each chemical series of MetAP inhibitors were generated and characterized to determine their inhibition potencies against BpMetAP1 (Figure 1 and Scheme 1).
Figure 1.

Commercial compounds purchased from Sigma-Aldrich. aBpMetAP1 IC50 values in μM with standard error obtained from nonlinear regression analysis of the average observed activity (in triplicate) versus inhibitor concentration data. bIn cases where analysis is limited by compound solubility, the minimum estimate of IC50 is provided.
Scheme 1. Syntheses of MetAP Inhibitors and BpMetAP1 IC50 Data.

Reagents and conditions: (a) CH2O, EtOH, R7H, 80 °C 24 h. (b) NaOH, EtOH, 70 °C, 20 min. (c) 1 M Na2CO3, pH 8.
BpMetAP1 IC50 values in μM with standard error obtained from nonlinear regression analysis of the average observed activity (in triplicate) versus inhibitor concentration data.
In cases where analysis is limited by compound solubility, the minimum estimate of IC50 is provided.
Seven compounds (1–4, 9, and 22–23) were purchased from Sigma-Aldrich (Figure 1). Compounds 5–8 were synthesized by Mannich reaction of 5-chloroquinolin-8-ol with formaldehyde and secondary amine. The synthesis of each of these compounds is shown in Scheme 1. Compounds 5–8 were produced with yields between 68 and 95% after recrystallization. Compounds 10–15 were synthesized similarly using 5-nitroquinolin-8-ol as the starting material, with similar yields. Compounds 16–21 were synthesized by alkylation of 3-amino-5-thio-1,2,4-triazole with the respective benzyl bromide. Compounds 16–21 were produced with yields between 47 and 92% following recrystallization. Compound 24 was synthesized from 2-amino-5-bromobenzoic acid and p-toluenesulfonyl chloride, providing the product in a 76% yield. Following a similar procedure, compound 25 was synthesized using 4-chlorobenzenesulfonyl chloride as a starting material, with a yield of 92%
The four chemical series of compounds (1–25) were profiled for in vitro BpMetAP1 activity (Figure 1 and Scheme 1) using an established activity inhibition assay.13 Overall, compounds across the four different series exhibited a wide range of inhibition potencies.
It was shown that oxine derivatives (compounds 1–15) generally did not exhibit significant inhibition if the nitro group is replaced in the 5-position (compounds 1–8). The inhibition potencies of compounds 1 through 8 ranged from low micromolar to more than 250 μM (Figure 1 and Scheme 1). The addition of functional groups on the 7-position of the 5-chloroquinolin-8-ol generally did not increase the potency as shown by compounds 6–8. However, the Mannich product 5, bearing a piperidyl group, displayed increased potency (9 μM). Nitroxoline derivatives were found to effectively inhibit BpMetAP1 activity compared to other oxines, with IC50 values ranging from low micromolar to 30 nM. The addition of functional groups on the 5-position was shown to increase the inhibitory potencies. The dimethylamino and pyrrolidyl Mannich products (11 and 12) displayed increased potency, while the piperidyl, dimethylamino, morpholino, thiomorpholino, and N-methylpiperazino groups (10, 13, 14, and 15) displayed decreased potency. The triazole-type compounds 16, 17, and 18 demonstrated inhibition of BpMetAP1 in the low micromolar range. Different substituents at the C2 and C4 position on the phenyl ring did not alter the inhibitory effect of these compounds. Addition of bulky functional groups, such as tert-butyl and isopropyl group at the C4 position, was shown to decrease the potency of the triazole compounds against BpMetAP1. Compounds 19–21 displayed an increase in IC50 values compared to triazole compounds with smaller functional group at the C4 position (16, 17, and 18). Furan-type compounds (22 and 23) and sulfonamide compounds (24 and 25) displayed IC50 values of more than 250 μM, indicating that they are not potent inhibitors of BpMetAP1.
The selectivity of several oxine derivatives (compounds 1–6 and 8–9) was evaluated through comparisons to published IC50 values from human MetAP1 and MetAP2 enzymes (Table 1).14 Overall, most compounds display a lack of apparent selectivity across the three MetAP enzymes. While several compounds display lower potency against BpMetAP1, compounds 5 and 9 possess higher potency toward BpMetAP1 versus hMetAP1, but similar potency when compared to published IC50 values for hMetAP2. These results may indicate similar binding determinants for the nitroxline-based compounds within BpMetAP1 and hMetAP2 enzymes. In addition, the lack of a differentiation between BpMetAP1 and human MetAP (particularly hMetAP2) may reflect the relatively small scaffolding and/or similarities across the three different MetAP active sites.
Table 1. Comparison IC50 Values of Compounds against BpMetAP1 and Human MetAP Enzymes, hMetAP1 and hMetAP2a.
| # | BpMetAP1b | hMetAP1d | hMetAP2d |
|---|---|---|---|
| 1 | 213 ± 118 | >15 | >15 |
| 2 | >250c | >15 | >15 |
| 3 | >250 | >15 | 2.03 ± 0.3 |
| 4 | >250c | 12.9 ± 1.0 | 1.27 ± 0.6 |
| 5 | 9 ± 6 | >50 | 7.68 ± 1.2 |
| 6 | >250c | >50 | 2.66 ± 0.2 |
| 8 | >250c | >50 | 3.81 ± 0.74 |
| 9 | 0.06 ± 0.03 | >15 | 0.055 ± 0.02 |
| 24 | >250 | ND | 0.009e |
| 25 | >250 | ND | 11e |
IC50 values in μM.
IC50 values and standard error obtained from nonlinear regression analysis of the average observed activity (in triplicate) versus inhibitor concentration data.
In cases where analysis is limited by compound solubility, the minimum estimate of IC50 is provided.
Values from Bhat et al.14 unless otherwise noted.
Values from Wang et al.15
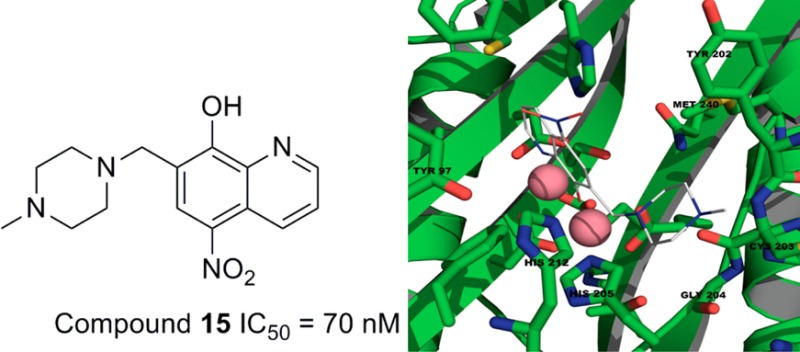
Structural studies were explored to better understand the nature of inhibitor interactions within the BpMetAP1 active site. Because of poorly diffracting BpMetAP1 crystals, a homology model of the BpMetAP1 was constructed from MtMetAP1 (PDB code: 3IU9). Using the Sybyl docking suite, compounds 9–15 and 18–20 were docked into the active site of BpMeAP1. The 8-hydroxyquinoline ligand within the Aeromonasproteolytica aminopeptidase binding pocket (PDB code: 3VH9) served as a structural constraint for docking compounds 9–15. A full negative charge was assigned to the oxygen atom in each compound due to the phenolic group donating its proton at physiological pH. Docking produced ligand poses where the negative charge from the oxygen and the nitrogen atom at the 1 position was centered on the two active site metal ions (Figure 2a). Functional groups extended from position 7, are observed to reside within a binding pocket (Tyr202, Cys203, Gly204, His205, His212, and Met240). Analysis of triazole-type compound 18 suggested that its inhibitory effect is dependent upon the presence of nitrogen atoms at positions 1 and 2 (Figure 2b). These nitrogen atoms coordinate with the metal ions in a manner similar to other triazole-type compounds that bind at the active site of MetAP.9 The phenyl group of compound 18 fits in to a hydrophobic binding pocket (Tyr27, Pro94, Tyr97, Cys105, Thr 133, and Phe211). The presence of bulky functional groups extending from the C4 position (e.g., 20) appear to clash into the side of the pocket, which is consistent with the observed loss of inhibitory potency. There are several differences in the binding pockets of hMetAP2 and BpMetAP1. In particular, Phe366/Met240, Leu328/Tyr202, and Asn329/Cys203 represent several residue differences (hMetAP2/BpMetAP1) that reside within the active site. These different residues may allow for introduction of larger substituents, or introduction of functional groups to hydrogen bond may result in increased selectivity.
Figure 2.
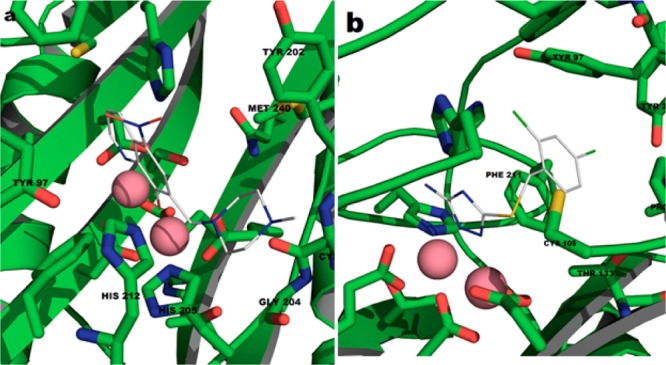
Proposed binding modes for 15 (a) and 18 (b) in the BpMetAP1 binding site.
To determine if inhibitors of BpMetAP1 were effective against Burkholderia in vivo, compounds 1–25 were evaluated for the ability to inhibit the growth of Burkholderia thailandensis. B. thailandensis is closely related to B. pseudomallei, displaying 98% sequence identity at the nucleotide level. However, B. thailandensis is not a select agent pathogen, which allows for its use as a model without the requirement of a BSL-3 facility.16 Cells were grown in the presence of 1 mM inhibitor, 0.5 mM kanamycin (Km), or DMSO alone for approximately 24 h. Treatment with Km at 0.5 mM results in complete killing of both B. thailandensis and B. pseudomallei in vitro and thus serves as a positive control for growth inhibition in our assay.5,17−19 Growth was monitored by reading the absorbance at 600 nm every 15 min, and the area under the curve was calculated for cells grown in each condition (Supplemental Table 1). Treatment with the majority of oxine derivatives (compounds 2–8) resulted in greater than 50% growth inhibition compared to the DMSO control and similar to the Km control, while compound 1 showed minimal inhibition of bacterial growth (approximately 9%; Supplemental Table 1 and Figure 3a). Nitroxoline (compound 9) and three nitroxoline derivatives (compounds 11, 12, and 15) were potent growth inhibitors, while the remaining nitroxoline derivatives exhibited weak to moderate growth inhibition (Supplemental Table 1). None of the triazole compounds exhibited growth inhibition greater than 26%, consistent with the moderate to high IC50 values observed in the activity inhibition assay. It was previously reported that triazole-derived compounds do not exhibit antibacterial activity, and our results support this observation.8 Neither the furan (22 and 23) nor the sulfonamide (24 and 25) derivatives demonstrated significant growth inhibition (Supplemental Table 1), consistent with the low enzyme inhibition potencies. In fact, the furan compounds displayed essentially no ability to inhibit bacterial growth and represent the weakest class of compounds tested.
Figure 3.
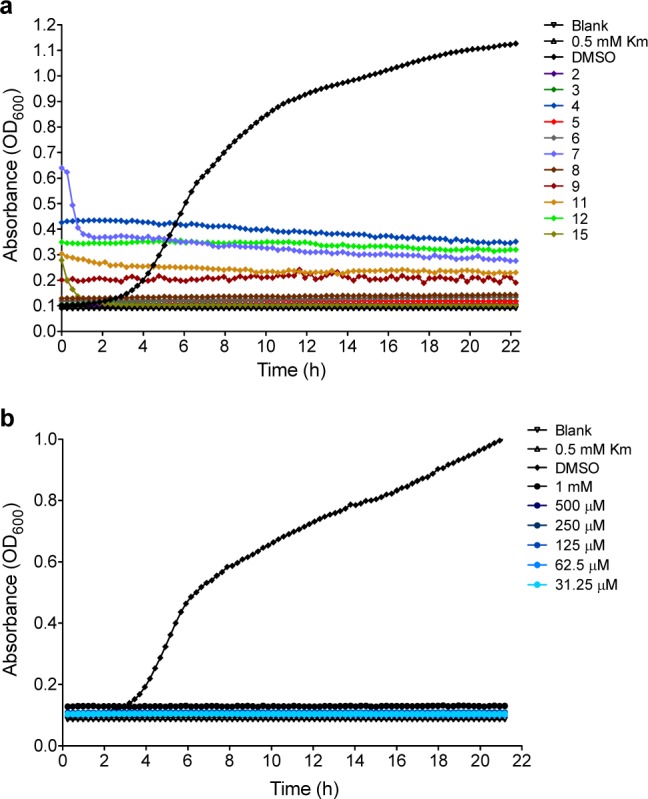
(a) Growth inhibition of B. thailandensis by indicated compounds and (b) growth inhibition of B. pseudomallei by compound 9.
Ten compounds demonstrated activity in the enzyme inhibition assay with IC50 values lower than 60 μM, while 11 compounds showed growth inhibition of B. thailandensis greater than 50%. Only five compounds were active against both purified BpMetAP1 and B. thailandensis, and of these, four are nitroxoline or its derivatives. Compounds 5, 9, 11, 12, and 15 displayed inhibition of cell growth and MetAP inhibition. Six compounds 2, 3, 4, 6, 7, and 8 inhibited cell growth but had IC50 values greater than 250 uM in the MetAP assay. The compounds 4, 6, 7, and 8 had limited solubility in the enzyme inhibition assay, which may explain the discrepancy between the enzyme inhibition data and the antibacterial data. Compounds 4, 7, 9, 11, and 12 showed inhibition of growth but had high background due to solubility limitations.
Compound 10 was inactive in the cell growth assay but had an IC50 of 100 nM in the MetAP inhibition assay. Our results highlight the fact that in vitro and in vivo assays are required to sufficiently evaluate the antimicrobial potential of small molecule inhibitors. The observation that oxine derivatives 2 and 3 were inactive against purified BpMetAP1, yet inhibited growth of B. thailandensis, suggest that these oxine compounds may have a target other than MetAp.
Following this initial assessment of potential inhibitors against B. thailandensis, we evaluated nitroxoline (compound 9) and a nitroxoline derivative (compound 15) at different concentrations to determine the minimum inhibitory concentration (MIC) that would prevent growth and the minimum bactericidal concentration (MBC) that would result in cell death. Using the cell growth inhibition assay, we observed an MIC of 31 μM (approximately 6 μg/mL) for compound 9, which is within the range of MIC values reported for ceftazidime against clinical B. pseudomallei isolates (1–6 μg/mL).4 For compound 15, we found the MIC to be 500 μM. The MBC of compound 9 was 250 μM; however, we did not reach the MBC of compound 15 as growth was observed at the highest concentration tested (1 mM). Growth curves used to determine the MIC values can be found in the Supporting Information.
Although testing these compounds in B. thailandensis is a simple and effective process, it is possible that B. pseudomallei-specific resistance mechanisms may reduce or eliminate the effectiveness of nitroxoline in the growth inhibition assay. Therefore, we tested nitroxoline against the virulent B. pseudomallei strain Bp340, a derivative of the clinical melioidosis isolate 1026b that lacks an aminoglycoside/macrolide-specific multidrug efflux pump (to facilitate antibiotic selection in the laboratory).5 Treatment with nitroxoline resulted in the complete growth inhibition of B. pseudomallei Bp340 at the lowest concentration tested (31.25 μM; Figure 3b). While we did not determine the MIC of nitroxoline against B. pseudomallei, our data indicate that it is at least as low as for B. thailandensis. A subsequent test of nitroxoline against B. pseudomallei 1026b revealed no difference in inhibition compared to Bp340, suggesting that the lack of the multidrug efflux pump does not affect the activity of this compound (data not shown).
In conclusion, we evaluated a set of small molecule inhibitors of B. pseudomallei MetAP1 in an enzyme activity assay. These compounds displayed a range of MetAP1 inhibition. A homology model of the BpMetAP1 was generated from MtMetAP1, and the proposed binding mechanisms of two of the most potent compounds from the in vitro assay were evaluated through molecular docking. Assessment of the antibacterial cell-growth inhibition reveals that five of the compounds that display MetAP1 inhibition had modest to high cell growth inhibitory effects, while others had minimal effects. Compound 15 is a potent inhibitor in the enzyme activity assay and shows nearly complete cell growth inhibition in vivo. The efficiency of this molecule in inhibiting BpMetAP1 activity and in arresting cell growth suggests that nitroxoline-derived compounds may be useful candidates for potential melioidosis therapeutics. While there was little selectivity observed between BpMetAP1 and hMetAP2, homology modeling suggests there are several difference in the binding pocket that may be targeted in future design efforts.
Acknowledgments
We appreciate comments and advice regarding homology model and docking studies from Dr. Michael Clare. We also thank Mr. Sriram Jakkaraju for technical assistance regarding surface plasmon resonance work. Dr. Herbert Schweizer kindly provided Burkholderia strains.
Glossary
Abbreviations
- BpMetAP1
Burkholderia pseudomallei methionine aminopeptidase 1
- DMSO
dimethyl sulfoxide
- HMVEC
human microvascular endothelial cells
- Km
kanamycin
- MetAP
methionine aminopeptidase
- Met-Gly-Pro-AMC
methionine-glycine-proline-7-amino-4-methylcoumarin
- rhDPPIV
human DPPIV/CD26
Supporting Information Available
Experimental procedures for the synthesis and characterization of the compounds, the in vitro activity assay, the in vivo antibacterial assay, and the 1H NMR and 13C NMR spectra of the reported compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
We acknowledge Northern Illinois University for supporting this work. This project has been funded in part with funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Nos. HHSN272200700057C and HHSN272201200025C.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Limmathurotsakul D.; Peacock S. J. Melioidosis: A Clinical Overview. Br. Med. Bull. 2011, 99, 125–139. [DOI] [PubMed] [Google Scholar]
- Sarovich D. S.; Price E. P.; Von Schulze A. T.; Cook J. M.; Mayo M.; Watson L. M.; Richardson L.; Seymour M. L.; Tuanyok A.; Engelthaler D. M.; Pearson T.; Peacock S. J.; Currie B. J.; Keim P.; Wagner D. M. Characterization of Ceftazidime Resistance Mechanisms in Clinical Isolates of Burkholderia pseudomallei from Australia. PLoS One 2012, 7, e30789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam I. C.; See K. H.; Puthucheary S. D. Variations in Ceftazidime and Amoxicillin-Clavulanate Susceptibilities within a Clonal Infection of Burkholderia pseudomallei. J. Clin. Microbiol. 2009, 47, 1556–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarovich D. S.; Price E. P.; Limmathurotsakul D.; Cook J. M.; Von Schulze A. T.; Wolken S. R.; Keim P.; Peacock S. J.; Pearson T. Development of Ceftazidime Resistance in an Acute Burkholderia pseudomallei Infection. Infect. Drug Resist. 2012, 5, 129–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer H. P. Mechanisms of Antimicrobial Resistance in Burkholderia pseudomallei: Implications for Treatment of Melioidosis. Future Microbiol. 2012, 7, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglione C.; Boularot A.; Meinnel T. Protein N-Terminal Methionine Excision. Cell. Mol. Life Sci. 2004, 61, 1455–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Chai S. C.; Ye Q. Catalysis and Inhibition of Mycobacterium tuberculosis Methionine Aminopeptidase. J. Med. Chem. 2010, 53, 1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M.; BaMaung N. Y.; Fidanze S. D.; Erickson S. A.; Comess K. M.; Kalvin D.; Wang J.; Zhang Q.; Lou P.; Tucker-Garcia L.; Bouska J.; Bell R. L.; Lesniewski R.; Henkin J.; Sheppard G. S. Development of Sulfonamide Compounds As Potent Methionine Aminopeptidase Type II Inhibitors with Antiproliferative Properties. Bioorg. Med. Chem. Lett. 2006, 16, 3574–3577. [DOI] [PubMed] [Google Scholar]
- Oefner C.; Douangamath A.; D’Arcy A.; Häfeli S.; Mareque D.; Sweeney A. M.; Padilla J.; Pierau S.; Schulz H.; Thormann M.; Wadman S.; Dale G. E. The 1.15 Å Crystal Structure of the Staphylococcus aureus Methionyl-Aminopeptidae and Complexes with Triazole Based Inhibitors. J. Mol. Biol. 2003, 332, 13–21. [DOI] [PubMed] [Google Scholar]
- Huang Q.; Huang M.; Nan F.; Ye Q. Metalloform-Selective Inhibition: Synthesis and Structure–Activity Analysis of Mn(II)-Form-Selective Inhibitors of Escherichia coli Methionine Aminopeptidase. Bioorg. Med. Chem. Lett. 2005, 15, 5386–5391. [DOI] [PubMed] [Google Scholar]
- Shim J. S.; Matsui Y.; Bhat S.; Nacev B. A.; Xu J.; Bhange H. C.; Dhara S.; Han K. C.; Chong C. R.; Pomper M. G.; So A.; Liu J. O. Effect of Nitrooxline on Angiogenesis and Growth of Human Bladder Cancer. J. Natl. Cancer Inst. 2010, 102, 1855–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier C.; Prognon P.; Bourlioux P. Roles of Divalent Cations and pH in Mechanism of Action of Nitroxoline against Escherichia coli Strains. Antimicrob. Agents Chemother. 1995, 39, 707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Guo X.; Yi T.; Yoshimoto T.; Pei D. Two Continuous Spectrophotometric Assays for Methionine Aminopeptidase. Anal. Biochem. 2000, 280, 159–165. [DOI] [PubMed] [Google Scholar]
- Bhat S.; Shim J. S.; Zhang F.; Chong C. R.; Liu J. O. Substituted Oxines Inhibit Endothelial Cell Proliferation and Angiogenesis. Org. Biomol. Chem. 2012, 10, 2979–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. T.; Mantei R. A.; Kawai M.; Tedrow J. S.; Barnes D. M.; Wang J.; Zhang Q.; Lou P.; Garcia L. A.; Bouska J.; Yates M.; Park C.; Judge R. A.; Lesniewski R.; Sheppard G. S.; Bell R. L. Lead Optimization of Methionine Aminopeptidase-2 (MetAP2) Inhibitiors Containing Sulfonamides of 5,6-Disubstituted Anthranilic Acids. Bioorg. Med. Chem. Lett. 2007, 17, 2817–1822. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Kim H. S.; Chua H. H.; Lin C. H.; Sim S. H.; Lin D.; Derr A.; Engels R.; DeShazer D.; Birren B.; Nierman W. C.; Tan P. Genomic Patterns of Pathogen Evolution Revealed by Comparison of Burkholderia pseudomallei, the Causative Agent of Melioidosis, to Avirulent Burkholderia thailandensis. BMC Microbiol. 2006, 6, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M. S.; Garcia E. C.; Cotter P. A. The Burkholderia bcpAIOB Genes Define Unique Classes of Two-Partner Secretion and Contact Dependent Growth Inhibition Systems. PLoS Genet. 2012, 8, e1002877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer H. P.; Peacock S. J. Antimicrobial Drug-Selection Markers for Burkholderia pseudomallei and B. mallei. Emerging Infect. Dis. 2008, 14, 1689–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima T.; Schweizer H. P. The BpeAB-OprB Efflux Pump of Burkholderia pseudomallei 1026b Does Not Play a Role in Quorum Sensing, Virulence Factor Production, or Extrusion of Aminoglycosides but Is a Broad-Spectrum Drug Efflux System. Antimicrob. Agents Chemother. 2010, 54, 3113–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.